


Integration of ATAC-Seq and RNA-Seq Analysis to Identify Key Genes in the Longissimus Dorsi Muscle Development of the Tianzhu White Yak

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
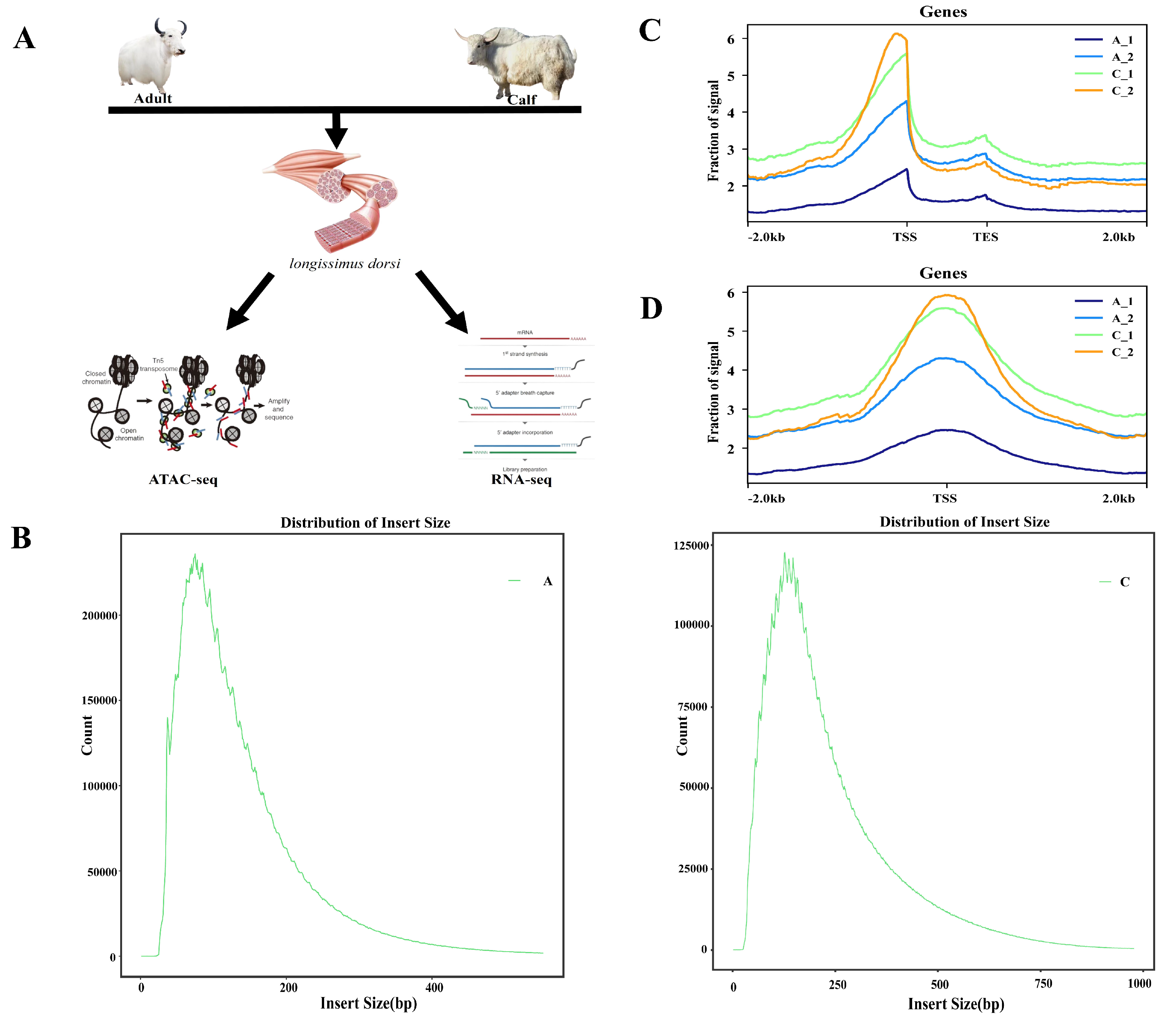
2.1. ATAC-Seq Quality Control of the White Yak Longissimus Dorsi Muscle
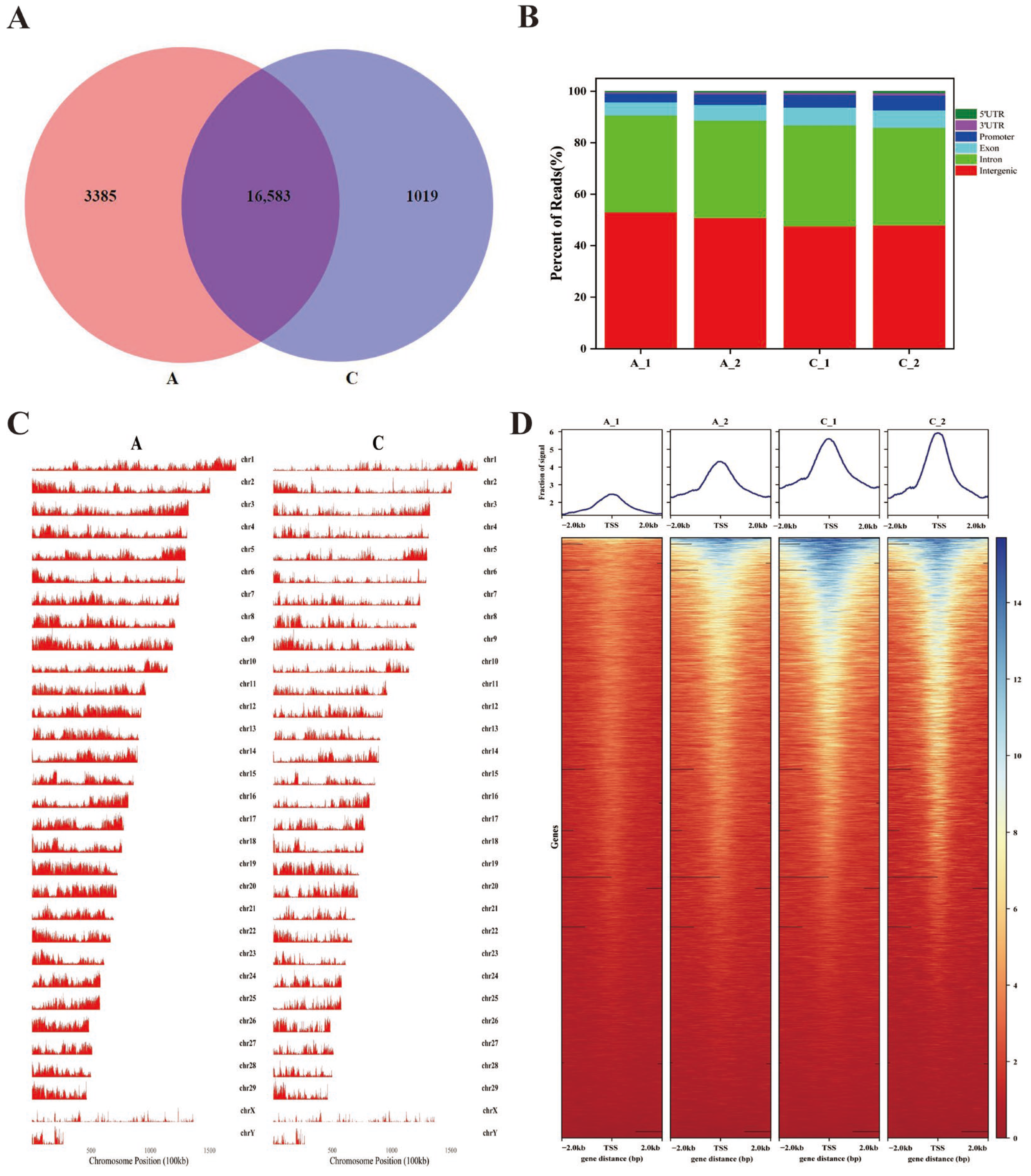
2.2. Chromatin Accessibility in the Longissimus Dorsi Muscle of the White Yak
2.3. RNA-Seq Data from Longissimus Dorsi Muscle of the White Yak
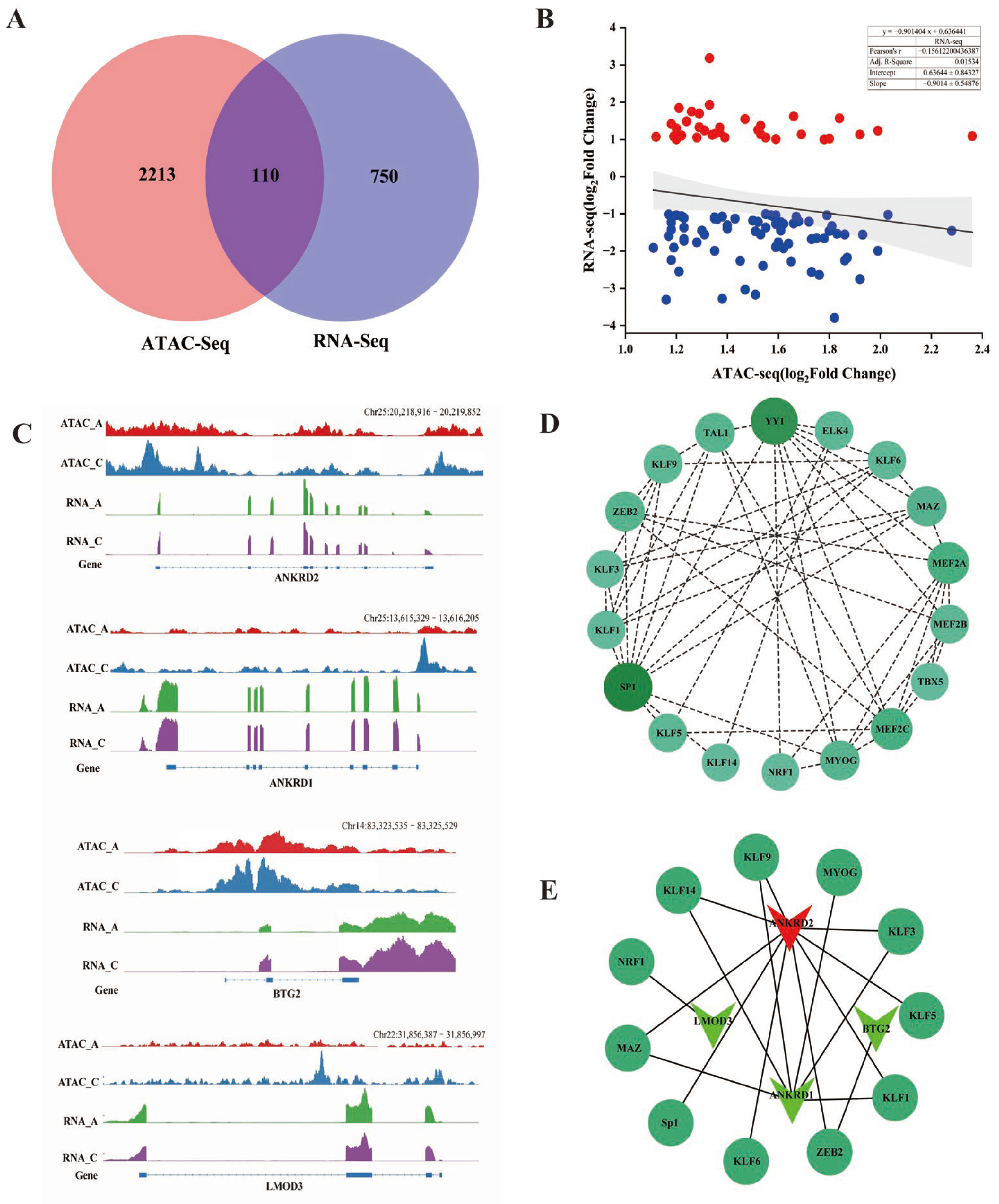
2.4. Combined Analysis of ATAC-Seq and RNA-Seq
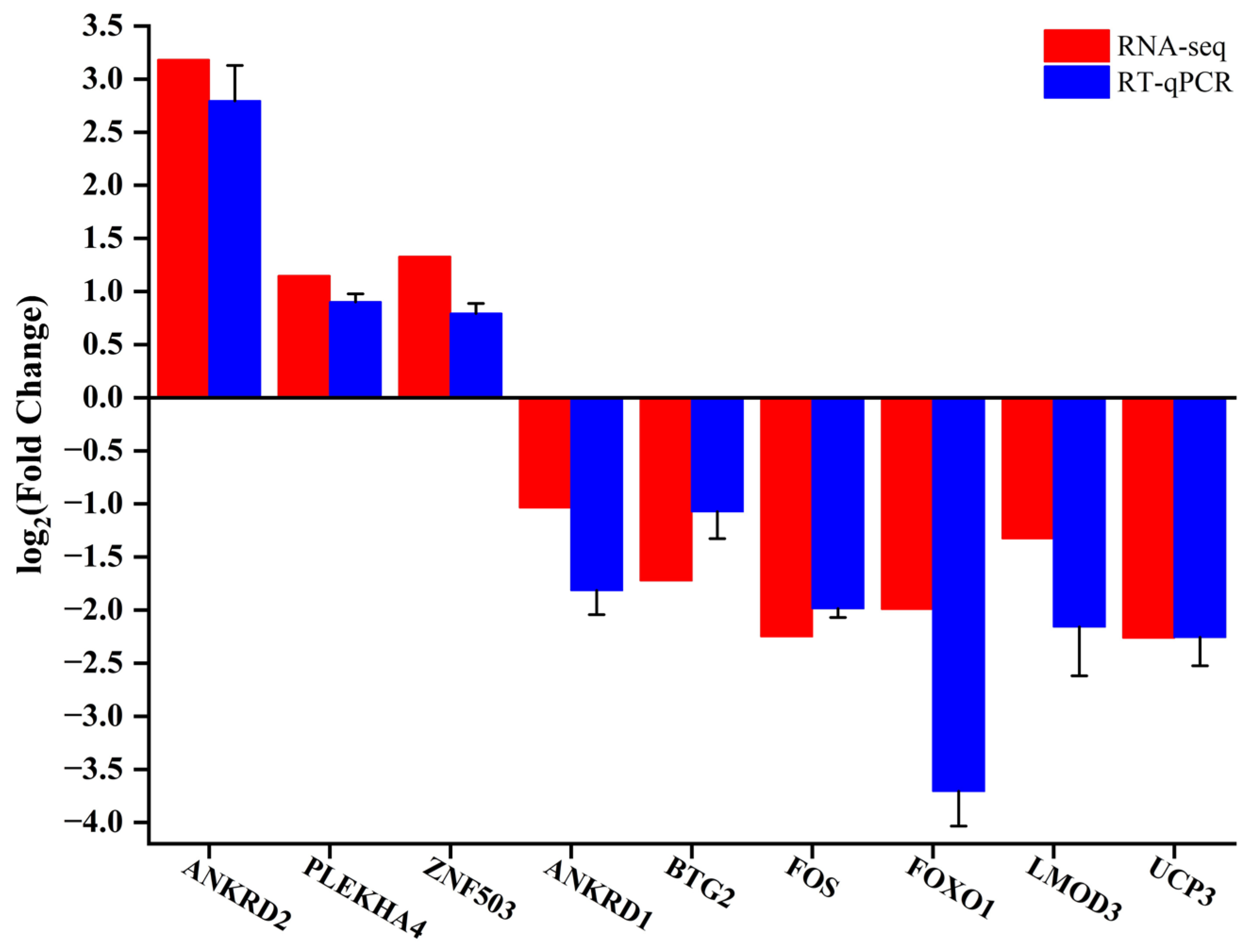
2.5. Validation of the Results by RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Collection of Samples
4.2. ATAC-Seq
4.3. RNA-Seq
4.4. Integration Analysis of ATAC-Seq and RNA-Seq
4.5. Gene Functional Annotation
4.6. RT-qPCR
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, D.; Ding, X.; Wang, S.; Wójcik, J.M.; Zhang, Y.; Tokarska, M.; Li, Y.; Wang, M.; Faruque, O.; Nielsen, R.; et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat. Ecol. Evol. 2018, 2, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, X.; Pei, J.; Chu, M.; Ding, X.; Wu, X.; Liang, C.; Yan, P. CircRNA Expression Profile during Yak Adipocyte Differentiation and Screen Potential circRNAs for Adipocyte Differentiation. Genes 2020, 11, 414. [Google Scholar] [CrossRef] [PubMed]
- Wiener, G.; Han, J.L.; Long, R.J. The Yak, 2nd ed.; Regional Office for Asia and the Pacifit of the Food and Agriculture Organization of the United Nations: Bangkok, Thailand, 2003; p. 460. [Google Scholar]
- Lu, H. The performance and reproductive capacity of yak. China Yak 1989, 3, 1–12. [Google Scholar]
- Huang, C.; Ge, F.; Ma, X.; Dai, R.; Dingkao, R.; Zhaxi, Z.; Burenchao, G.; Bao, P.; Wu, X.; Guo, X.; et al. Comprehensive Analysis of mRNA, lncRNA, circRNA, and miRNA Expression Profiles and Their ceRNA Networks in the Longissimus Dorsi Muscle of Cattle-Yak and Yak. Front. Genet. 2021, 12, 772557. [Google Scholar] [CrossRef]
- Cao, M.; Pei, J.; Xiong, L.; Guo, S.; Wang, X.; Kang, Y.; Guo, X. Analysis of Chromatin Openness in Testicle Tissue of Yak and Cattle-Yak. Int. J. Mol. Sci. 2022, 23, 15810. [Google Scholar] [CrossRef]
- Carton, F.; Di Francesco, D.; Fusaro, L.; Zanella, E.; Apostolo, C.; Oltolina, F.; Cotella, D.; Prat, M.; Boccafoschi, F. Myogenic Potential of Extracellular Matrix Derived from Decellularized Bovine Pericardium. Int. J. Mol. Sci. 2021, 22, 9406. [Google Scholar] [CrossRef]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The formation of skeletal muscle: From somite to limb. J. Anat. 2003, 202, 59–68. [Google Scholar] [CrossRef]
- Parakati, R.; DiMario, J.X. Repression of Myoblast Proliferation and Fibroblast Growth Factor Receptor 1 Promoter Activity by KLF10 Protein. J. Biol. Chem. 2013, 288, 13876–13884. [Google Scholar] [CrossRef]
- Chen, M. Circular RNA circMYBPC1 promotes skeletal muscle differentiation by targeting MyHC. Mol. Ther.-Nucleic Acids 2021, 24, 352–368. [Google Scholar] [CrossRef]
- Czerwinska, A.M.; Nowacka, J.; Aszer, M.; Gawrzak, S.; Archacka, K.; Fogtman, A.; Iwanicka-Nowicka, R.; Jańczyk-Ilach, K.; Koblowska, M.; Ciemerych, M.A.; et al. Cell cycle regulation of embryonic stem cells and mouse embryonic fibroblasts lacking functional Pax7. Cell Cycle 2016, 15, 2931–2942. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; La, Y.; Bao, P.; Chu, M.; Guo, X.; Wu, X.; Pei, J.; Ding, X.; Liang, C.; Yan, P. Regulatory Role of N6-Methyladenosine in Longissimus Dorsi Development in Yak. Front. Vet. Sci. 2022, 9, 757115. [Google Scholar] [CrossRef] [PubMed]
- Schübeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Jia, C.; Chu, M.; Fu, D.; Lei, Q.; Ding, X.; Wu, X.; Guo, X.; Pei, J.; Bao, P.; et al. Transcriptome and DNA Methylation Analyses of the Molecular Mechanisms Underlying with Longissimus dorsi Muscles at Different Stages of Development in the Polled Yak. Genes 2019, 10, 970. [Google Scholar] [CrossRef]
- Ming, H.; Sun, J.; Pasquariello, R.; Gatenby, L.; Herrick, J.R.; Yuan, Y.; Pinto, C.R.; Bondioli, K.R.; Krisher, R.L.; Jiang, Z. The landscape of accessible chromatin in bovine oocytes and early embryos. Epigenetics 2021, 16, 300–312. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Hu, X.; Zhang, Y.; Li, H.; Zhang, Q.; Cai, W.; Wang, Z.; Zhu, B.; Xu, L.; et al. Transcriptional states and chromatin accessibility during bovine myoblasts proliferation and myogenic differentiation. Cell Proliferat. 2022, 55, e13219. [Google Scholar] [CrossRef]
- Wang, J.; Li, B.; Yang, X.; Liang, C.; Raza, S.H.A.; Pan, Y.; Zhang, K.; Zan, L. Integration of RNA-seq and ATAC-seq identifies muscle-regulated hub genes in cattle. Front. Vet. Sci. 2022, 9, 925590. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Bao, Q.; Gu, Y.; Liang, C.; Chu, M.; Guo, X.; Bao, P.; Yan, P. The Landscape of Accessible Chromatin during Yak Adipocyte Differentiation. Int. J. Mol. Sci. 2022, 23, 9960. [Google Scholar] [CrossRef]
- Chen, X.; Shen, Y.; Draper, W.; Buenrostro, J.D.; Litzenburger, U.; Cho, S.W.; Satpathy, A.T.; Carter, A.C.; Ghosh, R.P.; East-Seletsky, A.; et al. ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing. Nat. Methods 2016, 13, 1013–1020. [Google Scholar] [CrossRef]
- Bottaro, A.; Lansford, R.; Xu, L.; Zhang, J.; Rothman, P.; Alt, F.W. S region transcription per se promotes basal IgE class switch recombination but additional factors regulate the efficiency of the process. EMBO J. 1994, 13, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Schoenfelder, S.; Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Jason, D.; Buenrostro, P.G.G.L. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNdnA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Kissane, S. Protocol for assay of transposase accessible chromatin sequencing in non-model species. STAR Protoc. 2021, 2, 100341. [Google Scholar] [CrossRef] [PubMed]
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022, 17, 1518–1552. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, M.; Wei, X.; Wu, L.; Xu, J.; Dai, X.; Xia, J.; Cheng, M.; Yuan, Y.; Zhang, P.; et al. An ATAC-seq atlas of chromatin accessibility in mouse tissues. Sci. Data 2019, 6, 65. [Google Scholar] [CrossRef]
- Cai, S.; Hu, B.; Wang, X.; Liu, T.; Lin, Z.; Tong, X.; Xu, R.; Chen, M.; Duo, T.; Zhu, Q.; et al. Integrative single-cell RNA-seq and ATAC-seq analysis of myogenic differentiation in pig. BMC Biol. 2023, 21, 19. [Google Scholar] [CrossRef]
- Feng, L.; Si, J.; Yue, J.; Zhao, M.; Qi, W.; Zhu, S.; Mo, J.; Wang, L.; Lan, G.; Liang, J. The Landscape of Accessible Chromatin and Developmental Transcriptome Maps Reveal a Genetic Mechanism of Skeletal Muscle Development in Pigs. Int. J. Mol. Sci. 2023, 24, 6413. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wires RNA 2017, 8, e1364. [Google Scholar] [CrossRef]
- Mo, X.Y.; Lan, J.; Jiao, Q.Z.; Xiong, Y.Z.; Zuo, B.; Li, F.E.; Xu, D.Q.; Lei, M.G. Molecular characterization, expression pattern and association analysis of the porcine BTG2 gene. Mol. Biol. Rep. 2011, 38, 4389–4396. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.H.; Wang, A.; Dai, W.; Chen, S.; Ding, Y.; Sun, L.V. Lmod3 promotes myoblast differentiation and proliferation via the AKT and ERK pathways. Exp. Cell Res. 2020, 396, 112297. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, N.; Jasnic, J.; Novkovic, M.; Milosevic, E.; Boskovic, S.; Kojic, A.; Popic, K.; Stankovic, M.; Wang, Y.; Milenkovic, S.; et al. Cloning and expression profiling of muscle regulator ANKRD2 in domestic chicken Gallus gallus. Histochem. Cell Biol. 2020, 154, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, P.; Fan, S.; Zhai, B.; Li, S.; Li, H.; Zhang, Y.; Li, W.; Sun, G.; Han, R.; et al. miR-30a-3p can inhibit the proliferation and promote the differentiation of chicken primary myoblasts. Brit. Poult. Sci. 2022, 63, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Huang, Z.; Liu, H.; Zhang, Y.; Ren, F. Yak milk fat globules from the Qinghai-Tibetan Plateau: Membrane lipid composition and morphological properties. Food Chem. 2017, 245, 731–737. [Google Scholar] [CrossRef]
- Shi, B.; Shi, X.; Zuo, Z.; Zhao, S.; Zhao, Z.; Wang, J.; Zhou, H.; Luo, Y.; Hu, J.; Hickford, J.G.H. Identification of differentially expressed genes at different post-natal development stages of longissimus dorsi muscle in Tianzhu white yak. Gene 2022, 823, 146356. [Google Scholar] [CrossRef]
- Meng, G.; La, Y.; Bao, Q.; Wu, X.; Ma, X.; Huang, C.; Chu, M.; Liang, C.; Yan, P. Early Growth and Development and Nonlinear Model Fitting Analysis of Ashidan Yak. Animals 2023, 13, 1545. [Google Scholar] [CrossRef]
- Chai Shuncang, M.C. Nonlinear growth curve analysis of yak weight in Qinghai plateau. Chin. Qinghai J. Anim. Vet. Sci. 2018, 05, 44–46. [Google Scholar]
- Hampsey, M. A New Direction for Gene Loops. Science 2012, 338, 624–625. [Google Scholar] [CrossRef]
- Miao, W.; Ma, Z.; Tang, Z.; Yu, L.; Liu, S.; Huang, T.; Wang, P.; Wu, T.; Song, Z.; Zhang, H.; et al. Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Muscle of Luchuan and Duroc Pigs. Front. Nutr. 2021, 8, 742672. [Google Scholar] [CrossRef]
- Segalés, J.; Perdiguero, E.; Muñoz-Cánoves, P. Regulation of Muscle Stem Cell Functions: A Focus on the p38 MAPK Signaling Pathway. Front. Cell Dev. Biol. 2016, 4, 91. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Yoo, G.D.; Heo, W.; Oh, H.T.; Park, J.; Shin, S.; Do, Y.; Jeong, M.G.; Hwang, E.S.; Hong, J.H. TAZ stimulates exercise-induced muscle satellite cell activation via Pard3–p38 MAPK–TAZ signalling axis. J. Cachexia Sarcopenia Muscle 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Teng, Y.; Wang, J.; Yu, M.; Li, Y.; Zheng, H. Rap1 promotes proliferation and migration of vascular smooth muscle cell via the ERK pathway. Pathol.-Res. Pract. 2018, 214, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, J.; Wu, T.; Zhao, K.; Wu, X.; Shi, J.; Sun, W.; Kong, X. Multi-Omics Analysis to Examine Gene Expression and Metabolites from Multisite Adipose-Derived Mesenchymal Stem Cells. Front. Genet. 2021, 12, 627347. [Google Scholar] [CrossRef]
- Angione, A.R.; Jiang, C.; Pan, D.; Wang, Y.; Kuang, S. PPARδ regulates satellite cell proliferation and skeletal muscle regeneration. Skelet. Muscle 2011, 1, 33. [Google Scholar] [CrossRef]
- Bloise, F.F.; Oliveira, T.S.; Cordeiro, A.; Ortiga-Carvalho, T.M. Thyroid Hormones Play Role in Sarcopenia and Myopathies. Front. Physiol. 2018, 9, 560. [Google Scholar] [CrossRef]
- Flavia, F.; Bloise, A.C.T.M. Role of thyroid hormone in skeletal muscle physiology. J. Endocrinol. 2018, 236, R57–R68. [Google Scholar] [CrossRef]
- Sindoni, A.; Rodolico, C.; Pappalardo, M.A.; Portaro, S.; Benvenga, S. Hypothyroid myopathy: A peculiar clinical presentation of thyroid failure. Review of the literature. Rev. Endocr. Metab. Disord. 2016, 17, 499–519. [Google Scholar] [CrossRef]
- Leonardi, R.; Jackowski, S. Biosynthesis of Pantothenic Acid and Coenzyme A. EcoSal Plus 2007, 2. [Google Scholar] [CrossRef]
- Biglou, S.G.; Bendena, W.G.; Chin-Sang, I. An overview of the insulin signaling pathway in model organisms Drosophila melanogaster and Caenorhabditis elegans. Peptides 2021, 145, 170640. [Google Scholar] [CrossRef]
- Reynolds, N.; O’Shaughnessy, A.; Hendrich, B. Transcriptional repressors: Multifaceted regulators of gene expression. Development 2013, 140, 505–512. [Google Scholar] [CrossRef]
- Alhaji, S.Y.; Ngai, S.C.; Abdullah, S. Silencing of transgene expression in mammalian cells by DNA methylation and histone modifications in gene therapy perspective. Biotechnol. Genet. Eng. Rev. 2018, 35, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Goodell, M.A. The push and pull of DNA methylation. Science 2021, 372, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Ryall, J.G. DNA Methylation in Skeletal Muscle Stem Cell Specification, Proliferation, and Differentiation. Stem Cells Int. 2016, 2016, 5725927. [Google Scholar] [CrossRef] [PubMed]
- Jasnic-Savovic, J.; Nestorovic, A.; Savic, S.; Karasek, S.; Vitulo, N.; Valle, G.; Faulkner, G.; Radojkovic, D.; Kojic, S. Profiling of skeletal muscle Ankrd2 protein in human cardiac tissue and neonatal rat cardiomyocytes. Histochem. Cell Biol. 2015, 143, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lei, M.; Xiong, Y. Molecular Characterization and Different Expression Patterns of the Muscle Ankyrin Repeat Protein (MARP) Family During Porcine Skeletal Muscle Development in vitro and in vivo. Anim. Biotechnol. 2011, 22, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.L.; Beishline, K.; Flashner, S.; Azizkhan-Clifford, J. DSB repair pathway choice is regulated by recruitment of 53BP1 through cell cycle-dependent regulation of Sp1. Cell Rep. 2021, 34, 108840. [Google Scholar] [CrossRef]
- Swift, M.L.; Beishline, K.; Azizkhan-Clifford, J. Sp1-dependent recruitment of the histone acetylase p300 to DSBs facilitates chromatin remodeling and recruitment of the NHEJ repair factor Ku70. DNA Repair 2021, 105, 103171. [Google Scholar] [CrossRef]
- Flashner, S.; Swift, M.; Sowash, A.; Fahmy, A.N.; Azizkhan-Clifford, J. Transcription factor Sp1 regulates mitotic chromosome assembly and segregation. Chromosoma 2022, 131, 175–191. [Google Scholar] [CrossRef]
- Chen, F.; Zhou, J.; Li, Y.; Zhao, Y.; Yuan, J.; Cao, Y.; Wang, L.; Zhang, Z.; Zhang, B.; Wang, C.C.; et al. YY 1 regulates skeletal muscle regeneration through controlling metabolic reprogramming of satellite cells. EMBO J. 2019, 38, e99727. [Google Scholar] [CrossRef]
- Liu, N.; Nelson, B.R.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 4109–4114. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Dyar, K.A.; Calabria, E. Skeletal muscle mass is controlled by the MRF4–MEF2 axis. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, L.; Jiang, Z.; Zhao, G.; Hassan, H.M.; Sun, L.; Fan, S.; Zhou, Z.; Zhang, L.; Wang, T. A new hypoglycemic mechanism of catalpol revealed by enhancing MyoD/MyoG-mediated myogenesis. Life Sci. 2018, 209, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Raza, S.H.A.; Wei, D.; Yaping, S.; Chao, J.; Jin, W.; Almohaimeed, H.M.; A Batarfi, M.; Assiri, R.; Aggad, W.S.; et al. Roles of MEF2A and MyoG in the transcriptional regulation of bovine LATS2 gene. Res. Vet. Sci. 2022, 152, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Latchman, D.S. Transcription Factors: An Overview. Int. J. Biochem. Cell Biol. 1997, 29, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, B.; Chen, H.; Yin, Q.; Liu, Y.; Xiang, Y.; Zhang, B.; Liu, B.; Wang, Q.; Xia, W.; et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 2016, 534, 652–657. [Google Scholar] [CrossRef]
- Guo, J.; Gao, J.; Liu, Z. HISAT2 Parallelization Method Based on Spark Cluster. J. Phys. Conf. Ser. 2022, 2179, 12038. [Google Scholar] [CrossRef]
- Bo, Y.Y.; Liang, L.D.; Hua, Y.J.; Zhao, Z.; Yao, M.S.; Shan, L.B.; Liang, C.Z. High-purity DNA extraction from animal tissue using picking in the TRIzol-based method. Biotechniques 2021, 70, 186–190. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Clean Ratio | Q20 | Q30 |
|---|---|---|---|---|---|---|---|
| A_1 | 128,541,756 | 19,281,263,400 | 127,569,914 | 13,604,132,565 | 99.24% | 98.36% | 95.01% |
| A_2 | 89,333,144 | 13,399,971,600 | 88,641,850 | 11,625,646,313 | 99.23% | 98.76% | 95.87% |
| C_1 | 135,034,832 | 20,255,224,800 | 133,887,694 | 17,651,791,047 | 99.15% | 98.63% | 95.46% |
| C_2 | 77,318,490 | 11,597,773,500 | 77,040,194 | 9,515,338,581 | 99.64% | 99.00% | 96.47% |
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Clean Ratio | Q20 | Q30 |
|---|---|---|---|---|---|---|---|
| A_1 | 78,265,742 | 11,739,861,300 | 77,683,168 | 11,626,880,356 | 99.26% | 98.25% | 94.88% |
| A_2 | 53,208,116 | 7,981,217,400 | 52,829,996 | 7,906,568,770 | 99.29% | 98.24% | 94.80% |
| A_3 | 121,439,410 | 18,215,911,500 | 120,564,720 | 18,063,425,239 | 99.28% | 98.23% | 94.76% |
| C_1 | 102,222,314 | 15,333,347,100 | 101,316,102 | 15,149,604,968 | 99.11% | 98.10% | 94.56% |
| C_2 | 36,264,690 | 5,439,703,500 | 35,877,070 | 5,370,251,031 | 98.93% | 98.19% | 94.77% |
| C_3 | 101,895,462 | 15,284,319,300 | 101,061,120 | 15,133,341,000 | 99.18% | 98.17% | 94.70% |
| Primer | Sequence (5′–3′) | Product Size |
|---|---|---|
| ANKRD2-F | CCTGAGAGTCCGTCCTTAC | 141 bp |
| ANKRD2-R | CCGTTTCTTCTGCTTGCGT | |
| PLEKHA4-F | GGCAATGCTCTCAGAAGGGA | 118 bp |
| PLEKHA4-R | AATGGCCAGAGAGGACGAAC | |
| ZNF503-F | CGCTCTCTGGAAATAGCTCCG | 122 bp |
| ZNF503-R | CTTGATGGGCAGGCGGTTAG | |
| ANKRD1-F | AGAAGAAAGGCAGTGGGGATG | 121 bp |
| ANKRD1-R | ACAAAGTGGACCGGAAGTGT | |
| BTG2-F | GCATCCGCATCAACCACAAG | 217 bp |
| BTG2-R | TTCTTGCAGGTGAGAAGCCC | |
| FOS-F | TACAGCCCACCCTAGTCTCC | 71 bp |
| FOS-R | AGTAGGGACTCCATAGGGGTG | |
| FOXO1-F | ACCCCACAAGGTTTCCGATG | 90 bp |
| FOXO1-R | AGTGTCCCCTCTCTTTCCAAC | |
| LMOD3-F | CCACCTTGTCCCCAGAAGAG | 129 bp |
| LMOD3-R | GGTCGAAGTTCCCTGTTGGT | |
| UCP3-F | CGGACCACTCCAGCATCATT | 186 bp |
| UCP3-R | CTTCCTCTCTGGCGATGGTC | |
| GAPDH-F | CCACGAGAAGTATAACAACACC | 120 bp |
| GAPDH-R | GTCATAAGTCCCTCCACGAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Chen, Z.; Bai, Y.; Wei, Y.; Guo, D.; Liu, Z.; Niu, Y.; Shi, B.; Zhang, X.; Cai, Y.; et al. Integration of ATAC-Seq and RNA-Seq Analysis to Identify Key Genes in the Longissimus Dorsi Muscle Development of the Tianzhu White Yak. Int. J. Mol. Sci. 2024, 25, 158. https://doi.org/10.3390/ijms25010158
Li J, Chen Z, Bai Y, Wei Y, Guo D, Liu Z, Niu Y, Shi B, Zhang X, Cai Y, et al. Integration of ATAC-Seq and RNA-Seq Analysis to Identify Key Genes in the Longissimus Dorsi Muscle Development of the Tianzhu White Yak. International Journal of Molecular Sciences. 2024; 25(1):158. https://doi.org/10.3390/ijms25010158
Chicago/Turabian StyleLi, Jingsheng, Zongchang Chen, Yanbin Bai, Yali Wei, Dashan Guo, Zhanxin Liu, Yanmei Niu, Bingang Shi, Xiaolan Zhang, Yuan Cai, and et al. 2024. "Integration of ATAC-Seq and RNA-Seq Analysis to Identify Key Genes in the Longissimus Dorsi Muscle Development of the Tianzhu White Yak" International Journal of Molecular Sciences 25, no. 1: 158. https://doi.org/10.3390/ijms25010158
APA StyleLi, J., Chen, Z., Bai, Y., Wei, Y., Guo, D., Liu, Z., Niu, Y., Shi, B., Zhang, X., Cai, Y., Zhao, Z., Hu, J., Wang, J., Liu, X., Li, S., & Zhao, F. (2024). Integration of ATAC-Seq and RNA-Seq Analysis to Identify Key Genes in the Longissimus Dorsi Muscle Development of the Tianzhu White Yak. International Journal of Molecular Sciences, 25(1), 158. https://doi.org/10.3390/ijms25010158