

Protein Aggregates and Aggrephagy in Myopathies


Abstract
1. Introduction
2. Protein Quality Control (PQC) System: Protein Misfolding and Aggrephagy
2.1. Protein Folding and Misfolding and Molecular Helpers
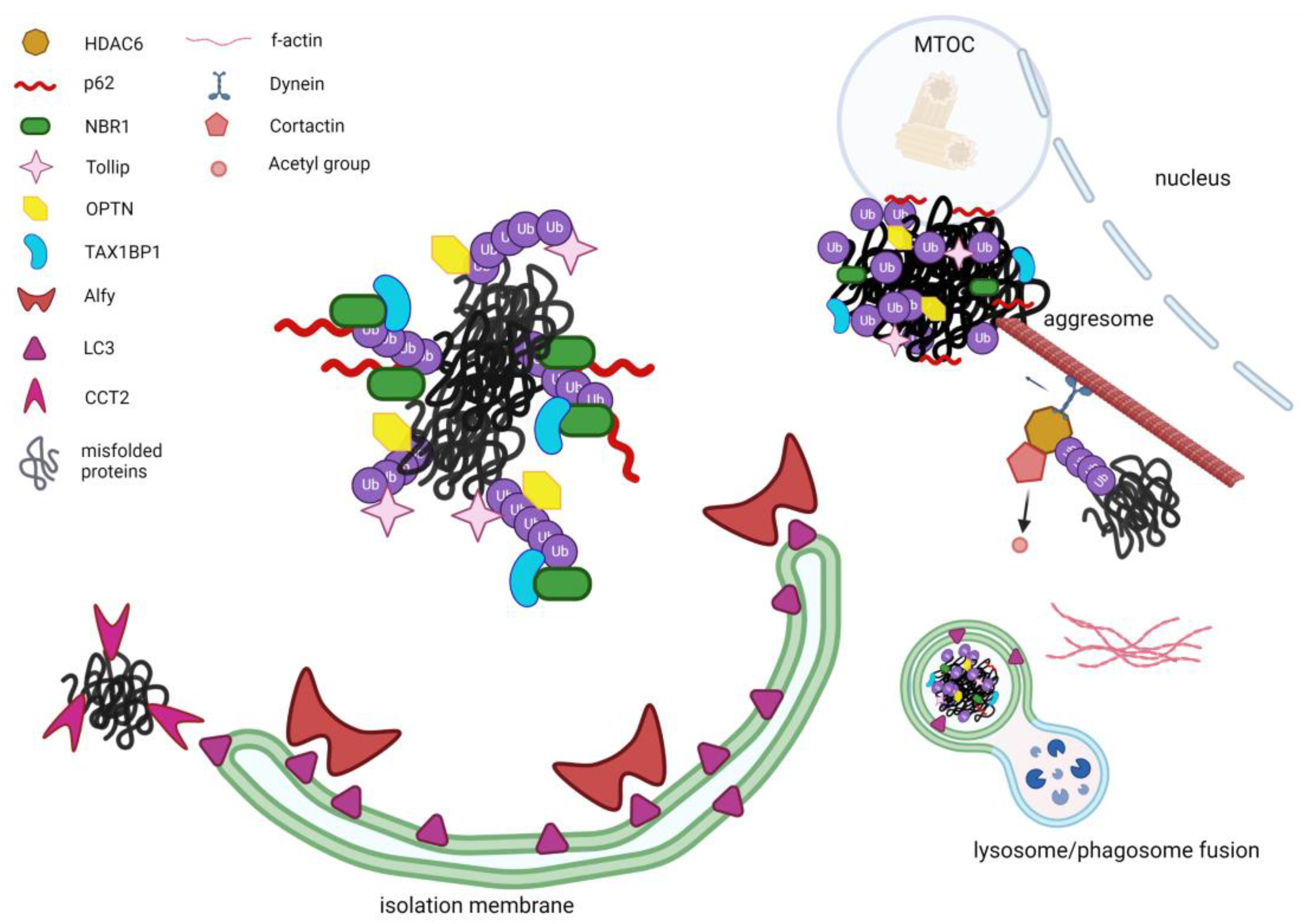
2.2. Alternative PQC: Autophagy and Aggrephagy
2.3. Key Proteins in Aggrephagy
2.3.1. HDAC6
2.3.2. P62/SQSTM1
2.3.3. NBR1 (Neighbor of BRCA1 Gene 1)
2.3.4. WDFY3 (Alfy)
2.3.5. TOLLIP (TOLL-Interacting Protein)
2.3.6. Optineurin
2.3.7. TAX1BP1 (Tax1-Binding Protein 1)
2.3.8. CCT2 (Chaperonin Containing TCP1 Subunit 2)


{kind=link}
{kind=link}
| Protein | Function/Structure | Main Functions in Aggrephagy | Interactors |
|---|---|---|---|
| HDAC6 | Microtubule-associate deacetylase. BUZ domain; DD1 and DD2 catalytic domains. |
|
|
| P62/SQSTM1 | Receptor for polyubiquitinated proteins. PB1 and ZZ domains; Two NLS and one NES sequences; LIR motif; C-terminal UBA domain. |
|
|
| NBR1 | Soluble selective autophagy receptor. PB1, ZZ and C-terminal UBA domains; LIR motif; FW and HA domains. |
|
|
| WDFY3 (ALFY) | Scaffold protein, evolutionarily conserved and expressed in all tissues. Five WD40 repeats PH-BEACH domain assemblage; C terminal PI3P-binding FYVE domain. |
|
|
| TOLLIP | Member of the CUET family of protein. Ubiquitin-binding CUE domain; LIR motif. |
|
|
| OPTINEURIN | Cytosolic protein ubiquitously expressed implies in cellular functions such as vesicle trafficking, cell division control, and autophagy. NEMO-like, LZ, UBD, CC and ZnF domains; LIR motif. |
|
|
| TAX1BP1 | Protein implies in regulation of cell death and inflammatory signalling pathways. N-terminal SKICH domain; Central oligomerization domain containing three CC regions; C-terminal UBD containing two zinc fingers. |
|
|
| CCT2 | Chaperonin subunit Implies in protein-folding and autophagy ATG8-interaction motif |
|
|
2.4. Aggrephagy Players as Therapeutic Targets
3. Aggrephagy Involvement in Muscle Diseases
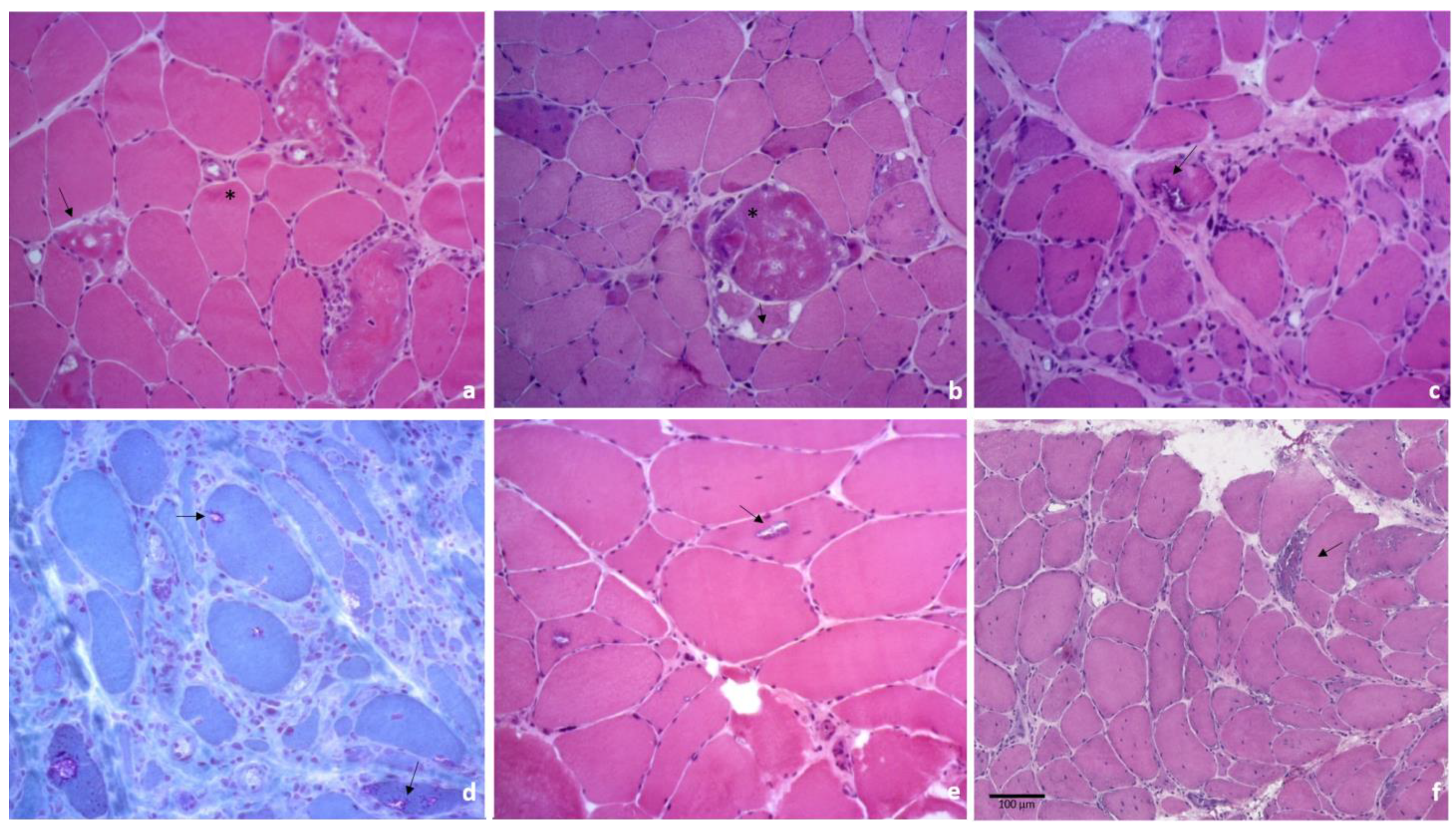
3.1. Myotilinopathies and Desminopathies
3.2. Sporadic Inclusion Body Myositis
3.3. Rigid Spine Syndrome with FHL1 Mutation
3.4. Oculopharyngeal Muscular Dystrophy
3.5. PLIN4-Related Myopathy
| Disease | Gene/ Inheritance | Age at Onset | Predominant Muscle Involvement | Systemic Involvement | Evolution, Loss of Ambulation | Creatin Kinase Level | Muscle Biopsy: Rimmed Vacuoles and Protein Aggregates |
|---|---|---|---|---|---|---|---|
| Myofibrillar myopathy [156,157,158,167] | MYOT/AD DES/AD | Adult Young adult | Distal Distal + limb-girdle | Rarely cardiomyopathy and peripheral neuropathy Cardiomyopathy | Slowly progressive Slowly progressive, LoA in later stages | Mild to highly elevated | Rimmed vacuoles and protein aggregates + |
| Inclusion body myopathy [174,177] | sporadic | Late adult | Quadriceps and/or finger flexors | Dysphagia, respiratory involvement | Progressive, LoA in later stages | Very variable, ranging from normal to up to 15 times UNL | Rimmed vacuoles and protein aggregates ++ |
| oculo-pharyngeal muscular dystrophy [186,191] | PABPN1 */AD | Late adult | Eyelid ptosis+ bulbar | Dysphagia, respiratory involvement, executive function deficits, rarely peripheral neuropathy | Slowly progressive | Slightly elevated in mild cases, higher severe cases | Rimmed vacuoles +/− |
| FHL1 myopathy [180] | FHL1 †/XLD | Young adult | Neck flexors and limb-girdle | Respiratory involvement, scoliosis, rigid spinel, rarely cardiomyopathy | Severe progressive course, LoA at early stages | Mildly elevated | Protein aggregates + |
| PLIN4 myopathy [8,189,190] | PLIN4 §/AD | Adult | Distal and limb-girdle | None | Slowly progressive, LoA at early stages | Normal to slightly elevated | Rimmed vacuoles and preotein aggregates + |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Margeta, M. Autophagy Defects in Skeletal Myopathies. Annu. Rev. Pathol. 2020, 15, 261–285. [Google Scholar] [CrossRef]
- Franco-Romero, A.; Sandri, M. Role of autophagy in muscle disease. Mol. Aspects. Med. 2021, 82, 101041. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K. Autophagic vacuolar myopathy: Danon disease and related myopathies. Neurol. Clin. Neurosci. 2022, 10, 273–278. [Google Scholar] [CrossRef]
- Olivé, M.; Winter, L.; Fürst, D.O.; Schröder, R.; ENMC Workshop Study Group. 246th ENMC International Workshop: Protein aggregate myopathies 24-26 May 2019, Hoofddorp, The Netherlands. Neuromuscul Disord 2021, 31, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.C.; Goebel, H.H. Protein aggregate myopathies. Neurol. India 2005, 53, 273–279. [Google Scholar] [CrossRef]
- Malicdan, M.C.; Nishino, I. Autophagy in lysosomal myopathies. Brain Pathol. 2012, 22, 82–88. [Google Scholar] [CrossRef]
- Ruggieri, A.; Naumenko, S.; Smith, M.A.; Iannibelli, E.; Blasevich, F.; Bragato, C.; Gibertini, S.; Barton, K.; Vorgerd, M.; Marcus, K.; et al. Multiomic elucidation of a coding 99-mer repeat-expansion skeletal muscle disease. Acta Neuropathol. 2020, 140, 231–235. [Google Scholar] [CrossRef]
- Goebel, H.H.; Fardeau, M.; Olivé, M.; Schröder, R. 156th ENMC International Workshop: Desmin and protein aggregate myopathies, 9-11 November 2007, Naarden, The Netherlands. Neuromuscul. Disord. 2008, 18, 583–592. [Google Scholar] [CrossRef]
- Kundra, R.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Protein homeostasis of a metastable subproteome associated with Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5703–E5711. [Google Scholar] [CrossRef]
- Sandri, M.; Coletto, L.; Grumati, P.; Bonaldo, P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci. 2013, 126, 5325–5333. [Google Scholar] [CrossRef]
- Goebel, H.H.; Müller, H.D. Protein aggregate myopathies. Semin. Pediatr. Neurol. 2006, 13, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Goebel, H.H. Protein aggregate myopathies. Introduction. Brain Pathol. 2009, 19, 480–482. [Google Scholar] [CrossRef] [PubMed]
- Klinge, L.; Straub, V.; Neudorf, U.; Voit, T. Enzyme replacement therapy in classical infantile pompe disease: Results of a ten-month follow-up study. Neuropediatrics 2005, 36, 6–11. [Google Scholar] [CrossRef]
- Stevens, D.; Milani-Nejad, S.; Mozaffar, T. Pompe Disease: A Clinical, Diagnostic, and Therapeutic Overview. Curr. Treat. Options Neurol. 2022, 24, 573–588. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Liutkute, M.; Samatova, E.; Rodnina, M.V. Cotranslational Folding of Proteins on the Ribosome. Biomolecules 2020, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Jahn, T.R.; Radford, S.E. The Yin and Yang of protein folding. FEBS J. 2005, 272, 5962–5970. [Google Scholar] [CrossRef]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef]
- Trivedi, R.; Nagarajaram, H.A. Intrinsically Disordered Proteins: An Overview. Int. J. Mol. Sci. 2022, 23, 14050. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, G.G.; Hipp, M.S.; Hartl, F.U. Functional Modules of the Proteostasis Network. Cold Spring Harb. Perspect. Biol. 2020, 12, a033951. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress. Chaperones. 2009, 14, 105–111. [Google Scholar] [CrossRef]
- Brocchieri, L.; Conway de Macario, E.; Macario, A.J. hsp70 genes in the human genome: Conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC Evol. Biol. 2008, 8, 19. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm (2020) 2022, 3, e161. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef]
- Piette, B.L.; Alerasool, N.; Lin, Z.Y.; Lacoste, J.; Lam, M.H.Y.; Qian, W.W.; Tran, S.; Larsen, B.; Campos, E.; Peng, J.; et al. Comprehensive interactome profiling of the human Hsp70 network highlights functional differentiation of J domains. Mol. Cell 2021, 81, 2549–2565.e2548. [Google Scholar] [CrossRef]
- Mayer, M.P. Gymnastics of molecular chaperones. Mol. Cell 2010, 39, 321–331. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Behnke, J.; Feige, M.J.; Hendershot, L.M. BiP and its nucleotide exchange factors Grp170 and Sil1: Mechanisms of action and biological functions. J. Mol. Biol. 2015, 427, 1589–1608. [Google Scholar] [CrossRef] [PubMed]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Pezeshki, A.M.; Yu, X.; Guo, C.; Subjeck, J.R.; Wang, X.Y. The Endoplasmic Reticulum Chaperone GRP170: From Immunobiology to Cancer Therapeutics. Front. Oncol. 2014, 4, 377. [Google Scholar] [CrossRef]
- Basha, E.; O’Neill, H.; Vierling, E. Small heat shock proteins and α-crystallins: Dynamic proteins with flexible functions. Trends. Biochem. Sci. 2012, 37, 106–117. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Stan, G.; Lorimer, G.H.; Thirumalai, D. Friends in need: How chaperonins recognize and remodel proteins that require folding assistance. Front. Mol. Biosci. 2022, 9, 1071168. [Google Scholar] [CrossRef]
- Vijay-Kumar, S.; Bugg, C.E.; Wilkinson, K.D.; Cook, W.J. Three-dimensional structure of ubiquitin at 2.8 A resolution. Proc. Natl. Acad. Sci. USA 1985, 82, 3582–3585. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef]
- Hochstrasser, M. Evolution and function of ubiquitin-like protein-conjugation systems. Nat. Cell Biol. 2000, 2, E153–E157. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Reinstein, E.; Ciechanover, A. Narrative review: Protein degradation and human diseases: The ubiquitin connection. Ann. Intern. Med. 2006, 145, 676–684. [Google Scholar] [CrossRef]
- Dikic, I.; Wakatsuki, S.; Walters, K.J. Ubiquitin-binding domains—From structures to functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 659–671. [Google Scholar] [CrossRef]
- Weissman, A.M.; Shabek, N.; Ciechanover, A. The predator becomes the prey: Regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell Biol. 2011, 12, 605–620. [Google Scholar] [CrossRef]
- Ciechanover, A.; Stanhill, A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim. Biophys. Acta 2014, 1843, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef]
- Morris, J.R.; Garvin, A.J. SUMO in the DNA Double-Stranded Break Response: Similarities, Differences, and Cooperation with Ubiquitin. J. Mol. Biol. 2017, 429, 3376–3387. [Google Scholar] [CrossRef]
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The Roles of Ubiquitin in Mediating Autophagy. Cells 2020, 9, 2025. [Google Scholar] [CrossRef] [PubMed]
- Navon, A.; Ciechanover, A. The 26 S proteasome: From basic mechanisms to drug targeting. J. Biol. Chem. 2009, 284, 33713–33718. [Google Scholar] [CrossRef]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef]
- Huang, L.; Chen, C.H. Proteasome regulators: Activators and inhibitors. Curr. Med. Chem. 2009, 16, 931–939. [Google Scholar] [CrossRef]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Ciechanover, A.; Iwai, K. The ubiquitin system: From basic mechanisms to the patient bed. IUBMB Life 2004, 56, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat. Commun. 2020, 11, 477. [Google Scholar] [CrossRef]
- Tan, J.M.; Wong, E.S.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.P.; Ho, M.W.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef]
- Zheng, Q.; Su, H.; Tian, Z.; Wang, X. Proteasome malfunction activates macroautophagy in the heart. Am. J. Cardiovasc. Dis. 2011, 1, 214–226. [Google Scholar] [PubMed]
- Zhu, K.; Dunner, K.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Øverbye, A.; Fengsrud, M.; Seglen, P.O. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy 2007, 3, 300–322. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Eskelinen, E.L.; Deretic, V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes... wait, I’m confused. Autophagy 2014, 10, 549–551. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Zhang, S.; Mizushima, N. Autophagy genes in biology and disease. Nat. Rev. Genet. 2023. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef]
- Yuan, H.X.; Russell, R.C.; Guan, K.L. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2013, 9, 1983–1995. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Amin, A.G.; Cooper, J.B.; Das, K.; Schmidt, M.H.; Murali, R. Discrete signaling mechanisms of mTORC1 and mTORC2: Connected yet apart in cellular and molecular aspects. Adv. Biol. Regul. 2017, 64, 39–48. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR-Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9, 655731. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Autophagy in skeletal muscle. FEBS Lett. 2010, 584, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.P.; Franco-Romero, A.; Cefis, M.; Moamer, A.; Broering, F.E.; Milan, G.; Sartori, R.; Chaffer, T.J.; Dulac, M.; Marcangeli, V.; et al. MYTHO is a novel regulator of skeletal muscle autophagy and integrity. Nat. Commun. 2023, 14, 1199. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Papandreou, M.E.; Tavernarakis, N. Nucleophagy: From homeostasis to disease. Cell Death. Differ. 2019, 26, 630–639. [Google Scholar] [CrossRef]
- Beese, C.J.; Brynjólfsdóttir, S.H.; Frankel, L.B. Selective Autophagy of the Protein Homeostasis Machinery: Ribophagy, Proteaphagy and ER-Phagy. Front. Cell Dev. Biol. 2019, 7, 373. [Google Scholar] [CrossRef]
- Shin, D.W. Lipophagy: Molecular Mechanisms and Implications in Metabolic Disorders. Mol. Cells 2020, 43, 686–693. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Kravic, B.; Meyer, H. Repair or Lysophagy: Dealing with Damaged Lysosomes. J. Mol. Biol. 2020, 432, 231–239. [Google Scholar] [CrossRef]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold. Des. 1998, 3, R9–R23. [Google Scholar] [CrossRef]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends. Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell. Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef]
- García-Mata, R.; Bebök, Z.; Sorscher, E.J.; Sztul, E.S. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 1999, 146, 1239–1254. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Yan, J. Interplay between HDAC6 and its interacting partners: Essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol. 2014, 33, 567–580. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef]
- Haack, T.B.; Ignatius, E.; Calvo-Garrido, J.; Iuso, A.; Isohanni, P.; Maffezzini, C.; Lönnqvist, T.; Suomalainen, A.; Gorza, M.; Kremer, L.S.; et al. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am. J. Hum. Genet. 2016, 99, 735–743. [Google Scholar] [CrossRef]
- Bucelli, R.C.; Arhzaouy, K.; Pestronk, A.; Pittman, S.K.; Rojas, L.; Sue, C.M.; Evilä, A.; Hackman, P.; Udd, B.; Harms, M.B.; et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 2015, 85, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Hocking, L.J.; Lucas, G.J.; Daroszewska, A.; Mangion, J.; Olavesen, M.; Cundy, T.; Nicholson, G.C.; Ward, L.; Bennett, S.T.; Wuyts, W.; et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum. Mol. Genet. 2002, 11, 2735–2739. [Google Scholar] [CrossRef]
- Pankiv, S.; Lamark, T.; Bruun, J.A.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J. Biol. Chem. 2010, 285, 5941–5953. [Google Scholar] [CrossRef] [PubMed]
- Isogai, S.; Morimoto, D.; Arita, K.; Unzai, S.; Tenno, T.; Hasegawa, J.; Sou, Y.S.; Komatsu, M.; Tanaka, K.; Shirakawa, M.; et al. Crystal structure of the ubiquitin-associated (UBA) domain of p62 and its interaction with ubiquitin. J. Biol. Chem. 2011, 286, 31864–31874. [Google Scholar] [CrossRef]
- Lim, J.; Lachenmayer, M.L.; Wu, S.; Liu, W.; Kundu, M.; Wang, R.; Komatsu, M.; Oh, Y.J.; Zhao, Y.; Yue, Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015, 11, e1004987. [Google Scholar] [CrossRef]
- Ciuffa, R.; Lamark, T.; Tarafder, A.K.; Guesdon, A.; Rybina, S.; Hagen, W.J.; Johansen, T.; Sachse, C. The selective autophagy receptor p62 forms a flexible filamentous helical scaffold. Cell Rep. 2015, 11, 748–758. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.S.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008, 283, 22847–22857. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, G.; Savova, A.; Danieli, A.; Romanov, J.; Tremel, S.; Ebner, M.; Peterbauer, T.; Sztacho, M.; Trapannone, R.; Tarafder, A.K.; et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 2018, 37, e98308. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell. 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Waters, S.; Marchbank, K.; Solomon, E.; Whitehouse, C.; Gautel, M. Interactions with LC3 and polyubiquitin chains link nbr1 to autophagic protein turnover. FEBS Lett. 2009, 583, 1846–1852. [Google Scholar] [CrossRef]
- Rasmussen, N.L.; Kournoutis, A.; Lamark, T.; Johansen, T. NBR1: The archetypal selective autophagy receptor. J. Cell Biol. 2022, 221, e202208092. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Marchbank, K.; Waters, S.; Roberts, R.G.; Solomon, E.; Whitehouse, C.A. MAP1B Interaction with the FW Domain of the Autophagic Receptor Nbr1 Facilitates Its Association to the Microtubule Network. Int. J. Cell Biol. 2012, 2012, 208014. [Google Scholar] [CrossRef]
- Mardakheh, F.K.; Auciello, G.; Dafforn, T.R.; Rappoport, J.Z.; Heath, J.K. Nbr1 is a novel inhibitor of ligand-mediated receptor tyrosine kinase degradation. Mol. Cell Biol. 2010, 30, 5672–5685. [Google Scholar] [CrossRef]
- Turco, E.; Savova, A.; Gere, F.; Ferrari, L.; Romanov, J.; Schuschnig, M.; Martens, S. Reconstitution defines the roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation and autophagy initiation. Nat. Commun. 2021, 12, 5212. [Google Scholar] [CrossRef]
- Chauveau, C.; Bonnemann, C.G.; Julien, C.; Kho, A.L.; Marks, H.; Talim, B.; Maury, P.; Arne-Bes, M.C.; Uro-Coste, E.; Alexandrovich, A.; et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum. Mol. Genet. 2014, 23, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Xiang, F.; Yakovenko, A.; Vihola, A.; Hackman, P.; Rostkova, E.; Kristensen, J.; Brandmeier, B.; Franzen, G.; Hedberg, B.; et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science 2005, 308, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, H. Core myopathies—A short review. Acta Myol. 2020, 39, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Amburgey, K.; McNamara, N.; Bennett, L.R.; McCormick, M.E.; Acsadi, G.; Dowling, J.J. Prevalence of congenital myopathies in a representative pediatric united states population. Ann. Neurol. 2011, 70, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Birkeland, H.C.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 2004, 117, 4239–4251. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Øvervatn, A.; Stenmark, H.; Bjørkøy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef]
- Isakson, P.; Holland, P.; Simonsen, A. The role of ALFY in selective autophagy. Cell Death. Differ. 2013, 20, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, R.; Tiloca, C.; Pensato, V.; Corrado, L.; Ratti, A.; Ticozzi, N.; Corti, S.; Castellotti, B.; Mazzini, L.; Sorarù, G.; et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1239–1243. [Google Scholar] [CrossRef]
- Osawa, T.; Mizuno, Y.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Optineurin in neurodegenerative diseases. Neuropathology 2011, 31, 569–574. [Google Scholar] [CrossRef]
- Iida, A.; Hosono, N.; Sano, M.; Kamei, T.; Oshima, S.; Tokuda, T.; Kubo, M.; Nakamura, Y.; Ikegawa, S. Optineurin mutations in Japanese amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 233–235. [Google Scholar] [CrossRef]
- Rezaie, T.; Child, A.; Hitchings, R.; Brice, G.; Miller, L.; Coca-Prados, M.; Héon, E.; Krupin, T.; Ritch, R.; Kreutzer, D.; et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 2013, 126, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef]
- Newman, A.C.; Scholefield, C.L.; Kemp, A.J.; Newman, M.; McIver, E.G.; Kamal, A.; Wilkinson, S. TBK1 kinase addiction in lung cancer cells is mediated via autophagy of Tax1bp1/Ndp52 and non-canonical NF-κB signalling. PLoS ONE 2012, 7, e50672. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Shah, H.V.; Kanfer, G.; Pickrell, A.M.; Holtzclaw, L.A.; Ward, M.E.; Youle, R.J. Loss of TAX1BP1-Directed Autophagy Results in Protein Aggregate Accumulation in the Brain. Mol. Cell 2020, 80, 779–795.e710. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Suklabaidya, S.; Vo, M.T.; Choi, Y.B.; Harhaj, E.W. Multifaceted roles of TAX1BP1 in autophagy. Autophagy 2023, 19, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lu, C.; Chen, Y.; Li, S.; Ma, N.; Tao, X.; Li, Y.; Wang, J.; Zhou, M.; Yan, Y.B.; et al. CCT2 is an aggrephagy receptor for clearance of solid protein aggregates. Cell 2022, 185, 1325–1345.e1322. [Google Scholar] [CrossRef] [PubMed]
- Conway, O.; Akpinar, H.A.; Rogov, V.V.; Kirkin, V. Selective Autophagy Receptors in Neuronal Health and Disease. J. Mol. Biol. 2020, 432, 2483–2509. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Wu, H.; Chen, Y.; Zhang, J.; Zheng, M.; Chen, G.; Li, L.; Lu, Q. The Therapeutic and Pathogenic Role of Autophagy in Autoimmune Diseases. Front. Immunol. 2018, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, M.E.; Tavernarakis, N. Selective Autophagy as a Potential Therapeutic Target in Age-Associated Pathologies. Metabolites 2021, 11, 588. [Google Scholar] [CrossRef] [PubMed]
- Kocak, M.; Ezazi Erdi, S.; Jorba, G.; Maestro, I.; Farrés, J.; Kirkin, V.; Martinez, A.; Pless, O. Targeting autophagy in disease: Established and new strategies. Autophagy 2022, 18, 473–495. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Y.; Shi, Y.; Zhang, Z.; Huang, C.; He, W.; Wang, C.; Shen, H.M. Autophagy in health and disease: From molecular mechanisms to therapeutic target. MedComm (2020) 2022, 3, e150. [Google Scholar] [CrossRef]
- Saunders, R.N.; Metcalfe, M.S.; Nicholson, M.L. Rapamycin in transplantation: A review of the evidence. Kidney Int. 2001, 59, 3–16. [Google Scholar] [CrossRef]
- Bhaoighill, M.N.; Dunlop, E.A. Mechanistic target of rapamycin inhibitors: Successes and challenges as cancer therapeutics. Cancer Drug Resist 2019, 2, 1069–1085. [Google Scholar] [CrossRef]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Su, Z.; Fei, H.; Liu, X.; Pan, Q. The novel mTOR inhibitor Torin-2 induces autophagy and downregulates the expression of UHRF1 to suppress hepatocarcinoma cell growth. Oncol. Rep. 2015, 34, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [PubMed]
- Kalid, O.; Gotliv, I.; Levy-Apter, E.; Beker, D.F.; Cherniavsky-Lev, M.; Rotem, E.; Miron, N. PTX80, a novel compound targeting the autophagy receptor p62/SQSTM1 for treatment of cancer. Chem. Biol. Drug. Des. 2022, 100, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Cho, W.J.; Najy, A.J.; Saliganan, A.D.; Pham, T.; Rakowski, J.; Loughery, B.; Ji, C.H.; Sakr, W.; Kim, S.; et al. p62/SQSTM1-induced caspase-8 aggresomes are essential for ionizing radiation-mediated apoptosis. Cell Death. Dis. 2021, 12, 997. [Google Scholar] [CrossRef]
- Dagvadorj, A.; Olivé, M.; Urtizberea, J.A.; Halle, M.; Shatunov, A.; Bönnemann, C.; Park, K.Y.; Goebel, H.H.; Ferrer, I.; Vicart, P.; et al. A series of West European patients with severe cardiac and skeletal myopathy associated with a de novo R406W mutation in desmin. J. Neurol. 2004, 251, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Ohno, K.; Engel, A.G. Myofibrillar myopathy: Clinical, morphological and genetic studies in 63 patients. Brain 2004, 127, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D. Myofibrillar myopathies. Neuromuscul. Disord. 2011, 21, 161–171. [Google Scholar] [CrossRef]
- Hedberg-Oldfors, C.; Meyer, R.; Nolte, K.; Abdul Rahim, Y.; Lindberg, C.; Karason, K.; Thuestad, I.J.; Visuttijai, K.; Geijer, M.; Begemann, M.; et al. Loss of supervillin causes myopathy with myofibrillar disorganization and autophagic vacuoles. Brain 2020, 143, 2406–2420. [Google Scholar] [CrossRef]
- Dafsari, H.S.; Kocaturk, N.M.; Daimagüler, H.S.; Brunn, A.; Dötsch, J.; Weis, J.; Deckert, M.; Cirak, S. Bi-allelic mutations in uncoordinated mutant number-45 myosin chaperone B are a cause for congenital myopathy. Acta Neuropathol. Commun. 2019, 7, 211. [Google Scholar] [CrossRef]
- Winter, L.; Goldmann, W.H. Biomechanical characterization of myofibrillar myopathies. Cell Biol. Int. 2015, 39, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Straussberg, R.; Schottmann, G.; Sadeh, M.; Gill, E.; Seifert, F.; Halevy, A.; Qassem, K.; Rendu, J.; van der Ven, P.F.; Stenzel, W.; et al. Kyphoscoliosis peptidase (KY) mutation causes a novel congenital myopathy with core targetoid defects. Acta Neuropathol. 2016, 132, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Gundesli, H.; Talim, B.; Korkusuz, P.; Balci-Hayta, B.; Cirak, S.; Akarsu, N.A.; Topaloglu, H.; Dincer, P. Mutation in exon 1f of PLEC, leading to disruption of plectin isoform 1f, causes autosomal-recessive limb-girdle muscular dystrophy. Am. J. Hum. Genet. 2010, 87, 834–841. [Google Scholar] [CrossRef]
- O’Grady, G.L.; Best, H.A.; Sztal, T.E.; Schartner, V.; Sanjuan-Vazquez, M.; Donkervoort, S.; Abath Neto, O.; Sutton, R.B.; Ilkovski, B.; Romero, N.B.; et al. Variants in the Oxidoreductase PYROXD1 Cause Early-Onset Myopathy with Internalized Nuclei and Myofibrillar Disorganization. Am. J. Hum. Genet. 2016, 99, 1086–1105. [Google Scholar] [CrossRef] [PubMed]
- De Bleecker, J.L.; Engel, A.G.; Ertl, B.B. Myofibrillar myopathy with abnormal foci of desmin positivity. II. Immunocytochemical analysis reveals accumulation of multiple other proteins. J. Neuropathol. Exp. Neurol. 1996, 55, 563–577. [Google Scholar] [CrossRef]
- Ferrer, I.; Martín, B.; Castaño, J.G.; Lucas, J.J.; Moreno, D.; Olivé, M. Proteasomal expression, induction of immunoproteasome subunits, and local MHC class I presentation in myofibrillar myopathy and inclusion body myositis. J. Neuropathol. Exp. Neurol. 2004, 63, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Olivé, M.; van Leeuwen, F.W.; Janué, A.; Moreno, D.; Torrejón-Escribano, B.; Ferrer, I. Expression of mutant ubiquitin (UBB+1) and p62 in myotilinopathies and desminopathies. Neuropathol. Appl. Neurobiol. 2008, 34, 76–87. [Google Scholar] [CrossRef]
- Dubowitz, V.; Sewry, C.; Oldfors, A. Muscle Biopsy A Practical Approach, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Machado, P.M.; Dimachkie, M.M.; Barohn, R.J. Sporadic inclusion body myositis: New insights and potential therapy. Curr. Opin. Neurol. 2014, 27, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Dimachkie, M.M. Idiopathic inflammatory myopathies. J. Neuroimmunol. 2011, 231, 32–42. [Google Scholar] [CrossRef]
- Dalakas, M.C. Mechanisms of disease: Signaling pathways and immunobiology of inflammatory myopathies. Nat. Clin. Pract. Rheumatol. 2006, 2, 219–227. [Google Scholar] [CrossRef]
- Brady, S.; Squier, W.; Sewry, C.; Hanna, M.; Hilton-Jones, D.; Holton, J.L. A retrospective cohort study identifying the principal pathological features useful in the diagnosis of inclusion body myositis. BMJ Open 2014, 4, e004552. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.R.; Group, E.I.W. 188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December 2011, Naarden, The Netherlands. Neuromuscul. Disord. 2013, 23, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.A. Inclusion body myositis: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 2019, 15, 257–272. [Google Scholar] [CrossRef]
- Nogalska, A.; Terracciano, C.; D’Agostino, C.; King Engel, W.; Askanas, V. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 2009, 118, 407–413. [Google Scholar] [CrossRef]
- Nogalska, A.; D’Agostino, C.; Terracciano, C.; Engel, W.K.; Askanas, V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am. J. Pathol. 2010, 177, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, C.; Nogalska, A.; Cacciottolo, M.; Engel, W.K.; Askanas, V. Abnormalities of NBR1, a novel autophagy-associated protein, in muscle fibers of sporadic inclusion-body myositis. Acta Neuropathol. 2011, 122, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Nicot, A.S.; Lo Verso, F.; Ratti, F.; Pilot-Storck, F.; Streichenberger, N.; Sandri, M.; Schaeffer, L.; Goillot, E. Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy 2014, 10, 1036–1053. [Google Scholar] [CrossRef] [PubMed]
- Britson, K.A.; Yang, S.Y.; Lloyd, T.E. New Developments in the Genetics of Inclusion Body Myositis. Curr. Rheumatol. Rep. 2018, 20, 26. [Google Scholar] [CrossRef]
- Sabatelli, P.; Castagnaro, S.; Tagliavini, F.; Chrisam, M.; Sardone, F.; Demay, L.; Richard, P.; Santi, S.; Maraldi, N.M.; Merlini, L.; et al. Aggresome-Autophagy Involvement in a Sarcopenic Patient with Rigid Spine Syndrome and a p.C150R Mutation in FHL1 Gene. Front. Aging Neurosci. 2014, 6, 215. [Google Scholar] [CrossRef]
- Schessl, J.; Columbus, A.; Hu, Y.; Zou, Y.; Voit, T.; Goebel, H.H.; Bönnemann, C.G. Familial reducing body myopathy with cytoplasmic bodies and rigid spine revisited: Identification of a second LIM domain mutation in FHL1. Neuropediatrics 2010, 41, 43–46. [Google Scholar] [CrossRef]
- Selcen, D.; Bromberg, M.B.; Chin, S.S.; Engel, A.G. Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. Neurology 2011, 77, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Victor, M.; Hayes, R.; Adams, R.D. Oculopharyngeal muscular dystrophy. A familial disease of late life characterized by dysphagia and progressive ptosis of the evelids. N. Engl. J. Med. 1962, 267, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Anvar, S.Y.; Raz, Y.; Verway, N.; van der Sluijs, B.; Venema, A.; Goeman, J.J.; Vissing, J.; van der Maarel, S.M.; ‘t Hoen, P.A.; van Engelen, B.G.; et al. A decline in PABPN1 induces progressive muscle weakness in oculopharyngeal muscle dystrophy and in muscle aging. Aging 2013, 5, 412–426. [Google Scholar] [CrossRef]
- Beaulieu, Y.B.; Kleinman, C.L.; Landry-Voyer, A.M.; Majewski, J.; Bachand, F. Polyadenylation-dependent control of long noncoding RNA expression by the poly(A)-binding protein nuclear 1. PLoS Genet. 2012, 8, e1003078. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Yang, K.; Lin, M.T.; Zheng, F.Z.; Chen, L.; Ding, Y.L.; Ye, Z.X.; Lin, X.; Wang, N.; Wang, Z.Q. The phenotypic and genotypic features of Chinese patients with oculopharyngeal muscular dystrophy. Ann. Clin. Transl. Neurol. 2023, 10, 426–439. [Google Scholar] [CrossRef]
- Askanas, V.; Serdaroglu, P.; Engel, W.K.; Alvarez, R.B. Immunolocalization of ubiquitin in muscle biopsies of patients with inclusion body myositis and oculopharyngeal muscular dystrophy. Neurosci. Lett. 1991, 130, 73–76. [Google Scholar] [CrossRef]
- Abu-Baker, A.; Messaed, C.; Laganiere, J.; Gaspar, C.; Brais, B.; Rouleau, G.A. Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2003, 12, 2609–2623. [Google Scholar] [CrossRef]
- Yang, K.; Zeng, Y.H.; Qiu, Y.S.; Lin, F.; Chen, H.Z.; Jin, M.; Chen, L.; Zheng, F.Z.; Ding, Y.L.; Cao, C.Y.; et al. Expanding the phenotype and genotype spectra of PLIN4-associated myopathy with rimmed ubiquitin-positive autophagic vacuolation. Acta Neuropathol. 2022, 143, 733–735. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, M.; Zhang, W.; Gang, Q.; Xie, Z.; Xu, J.; Zhou, C.; Wang, D.; Meng, L.; Lv, H.; et al. Subsarcolemmal and cytoplasmic p62 positivity and rimmed vacuoles are distinctive for PLIN4-myopathy. Ann. Clin. Transl. Neurol. 2022, 9, 1813–1819. [Google Scholar] [CrossRef]
- Tankink, M.; Horlings, C.G.C.; Voermans, N.; van der Sluijs, B.; Kessels, R.P.C.; van Engelen, B.; Raaphorst, J. Behavioural Impairment and Frontotemporal Dementia in Oculopharyngeal Muscular Dystrophy. J. Neuromuscul. Dis. 2022, 9, 129–135. [Google Scholar] [CrossRef]
- Olivé, M.; Goldfarb, L.G.; Shatunov, A.; Fischer, D.; Ferrer, I. Myotilinopathy: Refining the clinical and myopathological phenotype. Brain 2005, 128, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- van der Klooster, Z.J.; Sepehrkhouy, S.; Dooijes, D.; Te Rijdt, W.P.; Schuiringa, F.S.A.M.; Lingeman, J.; van Tintelen, J.P.; Harakalova, M.; Goldschmeding, R.; Suurmeijer, A.J.H.; et al. P62-positive aggregates are homogenously distributed in the myocardium and associated with the type of mutation in genetic cardiomyopathy. J. Cell Mol. Med. 2021, 25, 3160–3166. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Su, H.; Ranek, M.J.; Wang, X. Autophagy and p62 in cardiac proteinopathy. Circ. Res. 2011, 109, 296–308. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibertini, S.; Ruggieri, A.; Cheli, M.; Maggi, L. Protein Aggregates and Aggrephagy in Myopathies. Int. J. Mol. Sci. 2023, 24, 8456. https://doi.org/10.3390/ijms24098456
Gibertini S, Ruggieri A, Cheli M, Maggi L. Protein Aggregates and Aggrephagy in Myopathies. International Journal of Molecular Sciences. 2023; 24(9):8456. https://doi.org/10.3390/ijms24098456
Chicago/Turabian StyleGibertini, Sara, Alessandra Ruggieri, Marta Cheli, and Lorenzo Maggi. 2023. "Protein Aggregates and Aggrephagy in Myopathies" International Journal of Molecular Sciences 24, no. 9: 8456. https://doi.org/10.3390/ijms24098456
APA StyleGibertini, S., Ruggieri, A., Cheli, M., & Maggi, L. (2023). Protein Aggregates and Aggrephagy in Myopathies. International Journal of Molecular Sciences, 24(9), 8456. https://doi.org/10.3390/ijms24098456