
The Transcriptomic Profiles of ESR1 and MMP3 Stratify the Risk of Biochemical Recurrence in Primary Prostate Cancer beyond Clinical Features


Abstract
1. Introduction
2. Results
2.1. Transcriptomic Markers Improve Preoperative Prediction of BCR among PRAD Patients
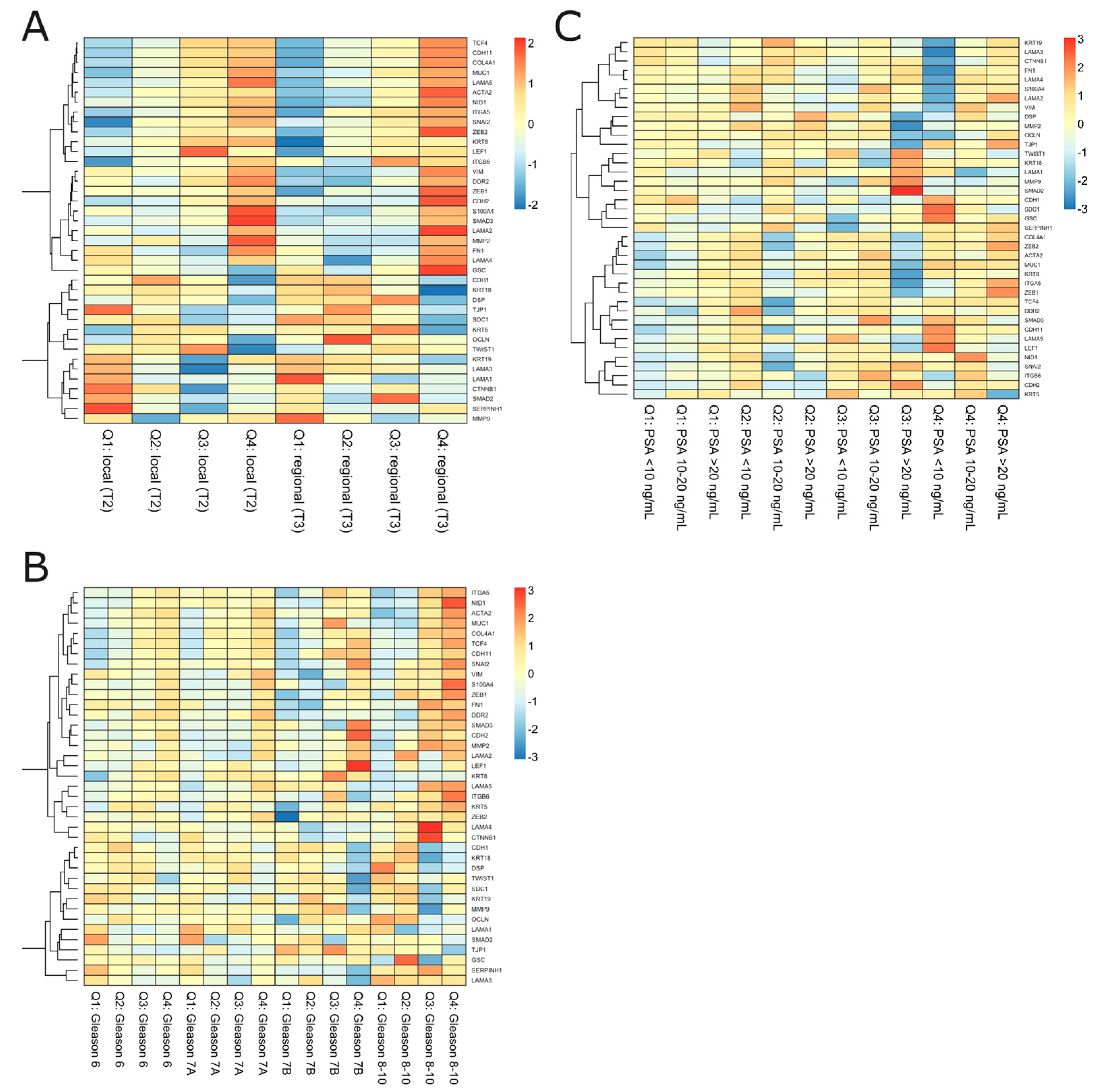
2.2. The Specific Combination of ESR1 and MMP3 Discriminates EMT-Related Potential for PRAD Recurrence
3. Discussion
4. Materials and Methods
4.1. Data Retrieval
4.2. BCR-Related Risk Group Classification
4.3. Identification of the Downstream Targets of AR, ESR1, and ESR2 Transcription Factors (TFs) with a Special Focus on the Markers of the EMT
4.4. Determination of Optimal Cutpoints of Gene Expression Stratifying BCR-Free Survival
4.5. Identification of Molecular Signature Altering BCR-Free Survival
4.6. Survival Analysis
4.7. Correspondence Analysis (CA)
4.8. Hierarchical Clustering by Stratum Combined with Clinical Parameters
4.9. Validation of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Jedroszka, D.; Orzechowska, M.; Hamouz, R.; Gorniak, K.; Bednarek, A.K. Markers of Epithelial-to-Mesenchymal Transition Reflect Tumor Biology According to Patient Age and Gleason Score in Prostate Cancer. PLoS ONE 2017, 12, e0188842. [Google Scholar] [CrossRef]
- Orzechowska, M.J.; Anusewicz, D.; Bednarek, A.K. Age- and Stage-Dependent Prostate Cancer Aggressiveness Associated with Differential Notch Signaling. Int. J. Mol. Sci. 2022, 24, 164. [Google Scholar] [CrossRef]
- Kensler, K.H.; Rebbeck, T.R. Cancer Progress and Priorities: Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 267–277. [Google Scholar] [CrossRef]
- Papachristodoulou, A.; Abate-Shen, C. Precision Intervention for Prostate Cancer: Re-Evaluating Who Is at Risk. Cancer Lett. 2022, 538, 215709. [Google Scholar] [CrossRef]
- Papachristodoulou, A.; Rodriguez-Calero, A.; Panja, S.; Margolskee, E.; Virk, R.K.; Milner, T.A.; Martina, L.P.; Kim, J.Y.; Di Bernardo, M.; Williams, A.B.; et al. NKX3.1 Localization to Mitochondria Suppresses Prostate Cancer Initiation. Cancer Discov. 2021, 11, 2316–2333. [Google Scholar] [CrossRef]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline Mutations in HOXB13 and Prostate-Cancer Risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef]
- Haffner, M.C.; Zwart, W.; Roudier, M.P.; True, L.D.; Nelson, W.G.; Epstein, J.I.; De Marzo, A.M.; Nelson, P.S.; Yegnasubramanian, S. Genomic and Phenotypic Heterogeneity in Prostate Cancer. Nat. Rev. Urol. 2021, 18, 79–92. [Google Scholar] [CrossRef]
- Tang, D.G. Understanding and Targeting Prostate Cancer Cell Heterogeneity and Plasticity. Semin. Cancer Biol. 2022, 82, 68–93. [Google Scholar] [CrossRef]
- Erickson, A.; Hayes, A.; Rajakumar, T.; Verrill, C.; Bryant, R.J.; Hamdy, F.C.; Wedge, D.C.; Woodcock, D.J.; Mills, I.G.; Lamb, A.D. A Systematic Review of Prostate Cancer Heterogeneity: Understanding the Clonal Ancestry of Multifocal Disease. Eur. Urol. Oncol. 2021, 4, 358–369. [Google Scholar] [CrossRef]
- Compérat, E.; Wasinger, G.; Oszwald, A.; Kain, R.; Cancel-Tassin, G.; Cussenot, O. The Genetic Complexity of Prostate Cancer. Genes 2020, 11, 1396. [Google Scholar] [CrossRef]
- Simmons, M.N.; Stephenson, A.J.; Klein, E.A. Natural History of Biochemical Recurrence after Radical Prostatectomy: Risk Assessment for Secondary Therapy. Eur. Urol. 2007, 51, 1175–1184. [Google Scholar] [CrossRef]
- Freedland, S.J.; Humphreys, E.B.; Mangold, L.A.; Eisenberger, M.; Dorey, F.J.; Walsh, P.C.; Partin, A.W. Risk of Prostate Cancer–Specific Mortality Following Biochemical Recurrence After Radical Prostatectomy. JAMA 2005, 294, 433. [Google Scholar] [CrossRef]
- Mottet, N.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer-2020 Update. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2021, 79, 243–262. [Google Scholar] [CrossRef]
- D’Amico, A.V.; Whittington, R.; Malkowicz, S.B.; Schultz, D.; Blank, K.; Broderick, G.A.; Tomaszewski, J.E.; Renshaw, A.A.; Kaplan, I.; Beard, C.J.; et al. Biochemical Outcome after Radical Prostatectomy, External Beam Radiation Therapy, or Interstitial Radiation Therapy for Clinically Localized Prostate Cancer. JAMA 1998, 280, 969–974. [Google Scholar] [CrossRef]
- Cooperberg, M.R.; Pasta, D.J.; Elkin, E.P.; Litwin, M.S.; Latini, D.M.; Du CHANE, J.; Carroll, P.R. The University of California, San Francisco Cancer of the Prostate Risk Assessment Score: A Straightforward and Reliable Preoperative Predictor of Disease Recurrence after Radical Prostatectomy. J. Urol. 2005, 173, 1938–1942. [Google Scholar] [CrossRef]
- Merriel, S.W.D.; Pocock, L.; Gilbert, E.; Creavin, S.; Walter, F.M.; Spencer, A.; Hamilton, W. Systematic Review and Meta-Analysis of the Diagnostic Accuracy of Prostate-Specific Antigen (PSA) for the Detection of Prostate Cancer in Symptomatic Patients. BMC Med. 2022, 20, 54. [Google Scholar] [CrossRef]
- Salman, J.W.; Schoots, I.G.; Carlsson, S.V.; Jenster, G.; Roobol, M.J. Prostate Specific Antigen as a Tumor Marker in Prostate Cancer: Biochemical and Clinical Aspects. In Advances in Cancer Biomarkers; Scatena, R., Ed.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2015; Volume 867, pp. 93–114. ISBN 978-94-017-7214-3. [Google Scholar]
- Mahal, B.A.; Aizer, A.A.; Efstathiou, J.A.; Nguyen, P.L. Association of Very Low Prostate-Specific Antigen Levels with Increased Cancer-Specific Death in Men with High-Grade Prostate Cancer: Low PSA Levels and Increased Cancer Mortality. Cancer 2016, 122, 78–83. [Google Scholar] [CrossRef]
- Pezaro, C.; Woo, H.H.; Davis, I.D. Prostate Cancer: Measuring PSA: Measuring PSA. Intern. Med. J. 2014, 44, 433–440. [Google Scholar] [CrossRef]
- Welch, H.G.; Albertsen, P.C. Reconsidering Prostate Cancer Mortality—The Future of PSA Screening. N. Engl. J. Med. 2020, 382, 1557–1563. [Google Scholar] [CrossRef]
- Stephenson, A.J.; Kattan, M.W.; Eastham, J.A.; Dotan, Z.A.; Bianco, F.J.; Lilja, H.; Scardino, P.T. Defining Biochemical Recurrence of Prostate Cancer after Radical Prostatectomy: A Proposal for a Standardized Definition. J. Clin. Oncol. 2006, 24, 3973–3978. [Google Scholar] [CrossRef]
- Toussi, A.; Stewart-Merrill, S.B.; Boorjian, S.A.; Psutka, S.P.; Thompson, R.H.; Frank, I.; Tollefson, M.K.; Gettman, M.T.; Carlson, R.E.; Rangel, L.J.; et al. Standardizing the Definition of Biochemical Recurrence after Radical Prostatectomy-What Prostate Specific Antigen Cut Point Best Predicts a Durable Increase and Subsequent Systemic Progression? J. Urol. 2016, 195, 1754–1759. [Google Scholar] [CrossRef]
- Boorjian, S.A.; Karnes, R.J.; Rangel, L.J.; Bergstralh, E.J.; Blute, M.L. Mayo Clinic Validation of the D’Amico Risk Group Classification for Predicting Survival Following Radical Prostatectomy. J. Urol. 2008, 179, 1354–1361. [Google Scholar] [CrossRef]
- Reese, A.C.; Pierorazio, P.M.; Han, M.; Partin, A.W. Contemporary Evaluation of the National Comprehensive Cancer Network Prostate Cancer Risk Classification System. Urology 2012, 80, 1075–1079. [Google Scholar] [CrossRef]
- Cimino, S.; Reale, G.; Castelli, T.; Favilla, V.; Giardina, R.; Russo, G.I.; Privitera, S.; Morgia, G. Comparison between Briganti, Partin and MSKCC Tools in Predicting Positive Lymph Nodes in Prostate Cancer: A Systematic Review and Meta-Analysis. Scand. J. Urol. 2017, 51, 345–350. [Google Scholar] [CrossRef]
- Horninger, W.; Berger, A.P.; Rogatsch, H.; Gschwendtner, A.; Steiner, H.; Niescher, M.; Klocker, H.; Bartsch, G. Characteristics of Prostate Cancers Detected at Low PSA Levels. Prostate 2004, 58, 232–237. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Papanikolaou, S.; Vourda, A.; Syggelos, S.; Gyftopoulos, K. Cell Plasticity and Prostate Cancer: The Role of Epithelial–Mesenchymal Transition in Tumor Progression, Invasion, Metastasis and Cancer Therapy Resistance. Cancers 2021, 13, 2795. [Google Scholar] [CrossRef]
- Rubicz, R.; Zhao, S.; April, C.; Wright, J.L.; Kolb, S.; Coleman, I.; Lin, D.W.; Nelson, P.S.; Ostrander, E.A.; Feng, Z.; et al. Expression of Cell Cycle-Regulated Genes and Prostate Cancer Prognosis in a Population-Based Cohort. Prostate 2015, 75, 1354–1362. [Google Scholar] [CrossRef]
- Pound, C.R.; Partin, A.W.; Eisenberger, M.A.; Chan, D.W.; Pearson, J.D.; Walsh, P.C. Natural History of Progression after PSA Elevation Following Radical Prostatectomy. JAMA 1999, 281, 1591–1597. [Google Scholar] [CrossRef]
- Boorjian, S.A.; Karnes, R.J.; Viterbo, R.; Rangel, L.J.; Bergstralh, E.J.; Horwitz, E.M.; Blute, M.L.; Buyyounouski, M.K. Long-Term Survival after Radical Prostatectomy versus External-Beam Radiotherapy for Patients with High-Risk Prostate Cancer. Cancer 2011, 117, 2883–2891. [Google Scholar] [CrossRef]
- Boorjian, S.A.; Thompson, R.H.; Siddiqui, S.; Bagniewski, S.; Bergstralh, E.J.; Karnes, R.J.; Frank, I.; Blute, M.L. Long-Term Outcome after Radical Prostatectomy for Patients with Lymph Node Positive Prostate Cancer in the Prostate Specific Antigen Era. J. Urol. 2007, 178, 864–870; discussion 870–871. [Google Scholar] [CrossRef]
- Han, M.; Partin, A.W.; Zahurak, M.; Piantadosi, S.; Epstein, J.I.; Walsh, P.C. Biochemical (Prostate Specific Antigen) Recurrence Probability Following Radical Prostatectomy for Clinically Localized Prostate Cancer. J. Urol. 2003, 169, 517–523. [Google Scholar] [CrossRef]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef]
- Berthold, D.R.; Pond, G.R.; Soban, F.; de Wit, R.; Eisenberger, M.; Tannock, I.F. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer: Updated Survival in the TAX 327 Study. J. Clin. Oncol. 2008, 26, 242–245. [Google Scholar] [CrossRef]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Pellegrino, F.; Coghi, A.; Lavorgna, G.; Cazzaniga, W.; Guazzoni, E.; Locatelli, I.; Villa, I.; Bolamperti, S.; Finocchio, N.; Alfano, M.; et al. A Mechanistic Insight into the Anti-Metastatic Role of the Prostate Specific Antigen. Transl. Oncol. 2021, 14, 101211. [Google Scholar] [CrossRef]
- Lilja, H.; Ulmert, D.; Vickers, A.J. Prostate-Specific Antigen and Prostate Cancer: Prediction, Detection and Monitoring. Nat. Rev. Cancer 2008, 8, 268–278. [Google Scholar] [CrossRef]
- Bonk, S.; Kluth, M.; Hube-Magg, C.; Polonski, A.; Soekeland, G.; Makropidi-Fraune, G.; Möller-Koop, C.; Witt, M.; Luebke, A.M.; Hinsch, A.; et al. Prognostic and Diagnostic Role of PSA Immunohistochemistry: A Tissue Microarray Study on 21,000 Normal and Cancerous Tissues. Oncotarget 2019, 10, 5439–5453. [Google Scholar] [CrossRef]
- Ben Jemaa, A.; Bouraoui, Y.; Sallami, S.; Banasr, A.; Rais, N.B.; Ouertani, L.; Nouira, Y.; Horchani, A.; Oueslati, R. Co-Expression and Impact of Prostate Specific Membrane Antigen and Prostate Specific Antigen in Prostatic Pathologies. J. Exp. Clin. Cancer Res. 2010, 29, 171. [Google Scholar] [CrossRef]
- Papadopoulos, I.; Sivridis, E.; Giatromanolaki, A.; Koukourakis, M.I. Tumor Angiogenesis Is Associated with MUC1 Overexpression and Loss of Prostate-Specific Antigen Expression in Prostate Cancer. Clin. Cancer Res. 2001, 7, 1533–1538. [Google Scholar]
- Schweizer, M.T.; Yu, E.Y. Persistent Androgen Receptor Addiction in Castration-Resistant Prostate Cancer. J. Hematol. Oncol. 2015, 8, 128. [Google Scholar] [CrossRef]
- Huggins, C. Effect of orchiectomy and irradiation on cancer of the prostate. Ann. Surg. 1942, 115, 1192–1200. [Google Scholar] [CrossRef]
- Sharifi, N. Androgen Deprivation Therapy for Prostate Cancer. JAMA 2005, 294, 238. [Google Scholar] [CrossRef]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.-P. Loss and Revival of Androgen Receptor Signaling in Advanced Prostate Cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef]
- Bonkhoff, H.; Fixemer, T.; Hunsicker, I.; Remberger, K. Progesterone Receptor Expression in Human Prostate Cancer: Correlation with Tumor Progression. Prostate 2001, 48, 285–291. [Google Scholar] [CrossRef]
- Chen, R.; Yu, Y.; Dong, X. Progesterone Receptor in the Prostate: A Potential Suppressor for Benign Prostatic Hyperplasia and Prostate Cancer. J. Steroid Biochem. Mol. Biol. 2017, 166, 91–96. [Google Scholar] [CrossRef]
- Bonkhoff, H. Estrogen Receptor Signaling in Prostate Cancer: Implications for Carcinogenesis and Tumor Progression. Prostate 2018, 78, 2–10. [Google Scholar] [CrossRef]
- Friedman, A.E. Can a Single Model Explain Both Breast Cancer and Prostate Cancer? Biol. Med. Model 2007, 4, 28. [Google Scholar] [CrossRef]
- Bonkhoff, H.; Fixemer, T.; Hunsicker, I.; Remberger, K. Estrogen Receptor Expression in Prostate Cancer and Premalignant Prostatic Lesions. Am. J. Pathol. 1999, 155, 641–647. [Google Scholar] [CrossRef]
- Price, D.; Stein, B.; Sieber, P.; Tutrone, R.; Bailen, J.; Goluboff, E.; Burzon, D.; Bostwick, D.; Steiner, M. Toremifene for the Prevention of Prostate Cancer in Men with High Grade Prostatic Intraepithelial Neoplasia: Results of a Double-Blind, Placebo Controlled, Phase IIB Clinical Trial. J. Urol. 2006, 176, 965–970; discussion 970–971. [Google Scholar] [CrossRef]
- Ricke, W.A.; McPherson, S.J.; Bianco, J.J.; Cunha, G.R.; Wang, Y.; Risbridger, G.P. Prostatic Hormonal Carcinogenesis Is Mediated by in Situ Estrogen Production and Estrogen Receptor Alpha Signaling. FASEB J. 2008, 22, 1512–1520. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Bilancio, A.; Perillo, B.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer. Cancers 2019, 11, 1418. [Google Scholar] [CrossRef]
- Lafront, C.; Germain, L.; Weidmann, C.; Audet-Walsh, É. A Systematic Study of the Impact of Estrogens and Selective Estrogen Receptor Modulators on Prostate Cancer Cell Proliferation. Sci. Rep. 2020, 10, 4024. [Google Scholar] [CrossRef]
- Venclovas, Z.; Jievaltas, M.; Milonas, D. Significance of Time Until PSA Recurrence After Radical Prostatectomy Without Neo- or Adjuvant Treatment to Clinical Progression and Cancer-Related Death in High-Risk Prostate Cancer Patients. Front. Oncol. 2019, 9, 1286. [Google Scholar] [CrossRef]
- Fossati, N.; Willemse, P.-P.M.; Van den Broeck, T.; van den Bergh, R.C.N.; Yuan, C.Y.; Briers, E.; Bellmunt, J.; Bolla, M.; Cornford, P.; De Santis, M.; et al. The Benefits and Harms of Different Extents of Lymph Node Dissection During Radical Prostatectomy for Prostate Cancer: A Systematic Review. Eur. Urol. 2017, 72, 84–109. [Google Scholar] [CrossRef]
- Mattei, A.; Fuechsel, F.G.; Bhatta Dhar, N.; Warncke, S.H.; Thalmann, G.N.; Krause, T.; Studer, U.E. The Template of the Primary Lymphatic Landing Sites of the Prostate Should Be Revisited: Results of a Multimodality Mapping Study. Eur. Urol. 2008, 53, 118–125. [Google Scholar] [CrossRef]
- Sowalsky, A.G.; Kissick, H.T.; Gerrin, S.J.; Schaefer, R.J.; Xia, Z.; Russo, J.W.; Arredouani, M.S.; Bubley, G.J.; Sanda, M.G.; Li, W.; et al. Gleason Score 7 Prostate Cancers Emerge through Branched Evolution of Clonal Gleason Pattern 3 and 4. Clin. Cancer Res. 2017, 23, 3823–3833. [Google Scholar] [CrossRef]
- Lavery, H.J.; Droller, M.J. Do Gleason Patterns 3 and 4 Prostate Cancer Represent Separate Disease States? J. Urol. 2012, 188, 1667–1675. [Google Scholar] [CrossRef]
- Kishan, A.U.; Romero, T.; Alshalalfa, M.; Liu, Y.; Tran, P.T.; Nickols, N.G.; Ye, H.; Sajed, D.; Rettig, M.B.; Reiter, R.E.; et al. Transcriptomic Heterogeneity of Gleason Grade Group 5 Prostate Cancer. Eur. Urol. 2020, 78, 327–332. [Google Scholar] [CrossRef]
- Woodcock, D.J.; Riabchenko, E.; Taavitsainen, S.; Kankainen, M.; Gundem, G.; Brewer, D.S.; Ellonen, P.; Lepistö, M.; Golubeva, Y.A.; Warner, A.C.; et al. Prostate Cancer Evolution from Multilineage Primary to Single Lineage Metastases with Implications for Liquid Biopsy. Nat. Commun. 2020, 11, 5070. [Google Scholar] [CrossRef]
- Vendramin, R.; Litchfield, K.; Swanton, C. Cancer Evolution: Darwin and Beyond. EMBO J. 2021, 40, e108389. [Google Scholar] [CrossRef]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/MiR-200 Feedback Loop Controls Notch Signalling in Cancer Cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef]
- Gunnarsson, O.; Schelin, S.; Brudin, L.; Carlsson, S.; Damber, J.-E. Triple Treatment of High-Risk Prostate Cancer. A Matched Cohort Study with up to 19 Years Follow-up Comparing Survival Outcomes after Triple Treatment and Treatment with Hormones and Radiotherapy. Scand. J. Urol. 2019, 53, 102–108. [Google Scholar] [CrossRef]
- Deng, Q.; Tang, D.G. Androgen Receptor and Prostate Cancer Stem Cells: Biological Mechanisms and Clinical Implications. Endocr. Relat. Cancer 2015, 22, T209–T220. [Google Scholar] [CrossRef]
- Kolmykov, S.; Yevshin, I.; Kulyashov, M.; Sharipov, R.; Kondrakhin, Y.; Makeev, V.J.; Kulakovskiy, I.V.; Kel, A.; Kolpakov, F. GTRD: An Integrated View of Transcription Regulation. Nucleic Acids Res. 2021, 49, D104–D111. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Ogluszka, M.; Orzechowska, M.; Jedroszka, D.; Witas, P.; Bednarek, A.K. Evaluate Cutpoints: Adaptable Continuous Data Distribution System for Determining Survival in Kaplan-Meier Estimator. Comput. Methods Programs Biomed. 2019, 177, 133–139. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using “ggplot2”. 2021. Available online: https://cran.r-project.org/web/packages/survminer/survminer.pdf (accessed on 10 October 2022).
- Kostov, B.; Bécue-Bertaut, M.; Husson, F. Multiple Factor Analysis for Contingency Tables in the FactoMineR Package. 1ères Rencontres R. 2013, 5, 29. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. Artic. 2008, 25, 1–18. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cutpoint | n < cp | n > cp | HR (95% CI) | p-Value | rmean (sermean) | n0 | nevents | Endtime | |
|---|---|---|---|---|---|---|---|---|---|
| AR | 560.1 | 278 | 219 | 1.46 (0.917–2.32) | 0.109 | ||||
| ESR1 | 137.3 | 145 | 352 | 1.87 (1.06–3.31) | 0.029 | ||||
| Low (fav) | 3551.66 (275.68) | 122 | 15 | 4604 | |||||
| High (unfav) | 2295.26 (154.35) | 298 | 57 | 3524 | |||||
| ESR2 | 11 | 246 | 251 | 1.45 (0.903–2.33) | 0.122 | ||||
| PGR | 46.91 | 61 | 436 | 0.466 (0.267–0.813) | 0.006 | ||||
| Low (unfav) | 1905.99 (286.66) | 44 | 15 | 3487 | |||||
| High (fav) | 3180.63 (203.53) | 376 | 57 | 4604 | |||||
| NR3C1 | 528.5 | 87 | 410 | 0.687 (0.406–1.16) | 0.16 | ||||
| KLK2 | 138,600 | 276 | 221 | 0.677 (0.418–1.1) | 0.11 | ||||
| KLK3 | 253,100 | 123 | 374 | 0.423 (0.265–0.677) | 0.0002 | ||||
| Low (unfav) | 1742.94 (207.64) | 104 | 30 | 3253 | |||||
| High (fav) | 3442.65 (182.64) | 316 | 42 | 4604 | |||||
| FOLH1 | 19,080 | 360 | 137 | 1.6 (0.986–2.58) | 0.054 | ||||
| Low (fav) | 3231.79 (212.73) | 308 | 46 | 4604 | |||||
| High (unfav) | 2210.95 (224.19) | 112 | 26 | 3524 | |||||
| CDH1 | 9554 | 151 | 346 | 0.545 (0.342–0.868) | 0.009 | ||||
| Low (unfav) | 2119.8 (202.9) | 118 | 32 | 3524 | |||||
| High (fav) | 3315.46 (234.69) | 302 | 40 | 4604 | |||||
| VIM | 11,340 | 407 | 90 | 1.85 (1.11–3.07) | 0.017 | ||||
| Low (fav) | 3298.58 (192.62) | 345 | 51 | 4604 | |||||
| High (unfav) | 1474.64 (120.12) | 75 | 21 | 2211 | |||||
| CTNNB1 | 8689 | 321 | 176 | 1.39 (0.863–2.25) | 0.172 | ||||
| MMP2 | 3529 | 419 | 78 | 1.46 (0.834–2.54) | 0.184 | ||||
| MMP3 | 0.4285 | 91 | 405 | 2.93 (1.18–7.27) | 0.015 | ||||
| Low (unfav) | 3999.75 (302.74) | 76 | 5 | 4604 | |||||
| High (fav) | 2341.89 (134.3) | 344 | 67 | 3524 | |||||
| MMP9 | 238.6 | 294 | 203 | 1.43 (0.902–2.28) | 0.126 | ||||
| PSA | 6.9 | 224 | 283 | 1.93 (1.15–3.25) | 0.012 | ||||
| Low (fav) | 3500.86 (252.03) | 178 | 20 | 4604 | |||||
| High (unfav) | 2258.85 (155.49) | 242 | 52 | 3487 |
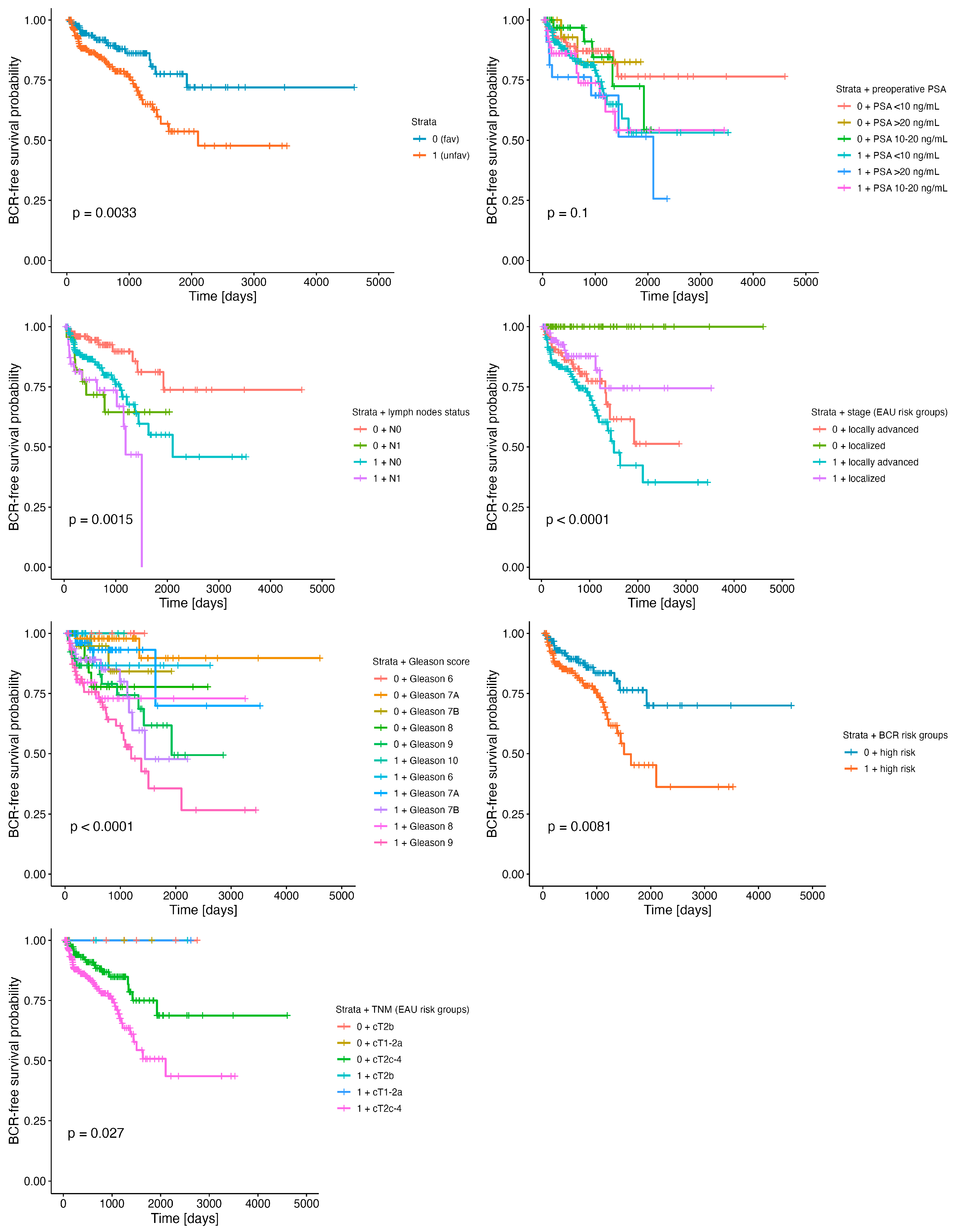
| Stratum | Clinical Variable | n0 | nevents | rmean (sermean) | Endtime | HR (95% CI) | p-Value | 2-Year BCR-Free Rate (95% CI) | 5-Year BCR-Free Rate (95% CI) |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 169 | 19 | 3602.05 (239.49) | 4604 | ref | 89% (84–95%) | 78% (67–90%) | ||
| 1 | 251 | 53 | 2198.3 (167.05) | 3524 | 2.2 (1.3–3.6) | 0.003 | 81% (75–87%) | 54% (42–68%) | |
| Lymph node status (pN) | |||||||||
| 0 | pN0 | 114 | 10 | 3707.84 (291.41) | 4606 | ref | 93% (87–99%) | 81% (69–96%) | |
| pN1 | 24 | 7 | 1443.27 (184.91) | 2040 | 3.17 (1.2–8.3) | 0.02 | 72% (55–94%) | 64% (46–91%) | |
| 1 | pN0 | 167 | 33 | 2202.1 (212.38) | 3524 | 2.58 (1.27–5.2) | 0.009 | 83% (76–90%) | 55% (41–74%) |
| pN1 | 44 | 13 | 1070.09 (99.5) | 1506 | 4.65 (2.02–10.7) | <0.001 | 74% (60–91%) | - | |
| Gleason score | |||||||||
| 0 | 6 | 18 | 0 | 1436 (-) | 1436 | - | - | - | |
| 7A | 56 | 2 | 4244.59 (269.05) | 4604 | ref | 98% (94–100%) | 90% (75–100%) | ||
| 7B | 28 | 2 | 1714.53 (139.46) | 1925 | 2.91 (0.41–20.7) | 0.3 | 95% (85–100%) | 84% (65–100%) | |
| 8 | 26 | 4 | 2086.41 (217.07) | 2576 | 5.77 (1.05–31.6) | 0.043 | 78% (61–100%) | 78% (61–100%) | |
| 9 | 41 | 11 | 1933.1 (218.99) | 2859 | 7.42 (1.64–33.5) | 0.009 | 79% (66–95%) | 62% (44–87%) | |
| 1 | 6 | 21 | 2 | 2336.27 (186.78) | 2620 | 2.56 (0.36–18.2) | 0.3 | 87% (71–100%) | 87% (71–100%) |
| 7A | 65 | 4 | 2863.35 (395.79) | 3524 | 2.43 (0.44–13.3) | 0.3 | 93% (86–100%) | 70% (39–100%) | |
| 7B | 54 | 11 | 1555.68 (155.16) | 2211 | 7.43 (1.65–33.6) | 0.009 | 85% (74–97%) | 48% (27–85%) | |
| 8 | 33 | 7 | 2439.79 (274.56) | 3253 | 9.05 (1.88–43.7) | 0.006 | 73% (57–94%) | 73% (57–94%) | |
| 9 | 75 | 29 | 1582.5 (230.84) | 3447 | 13.7 (3.27–57.4) | <0.001 | 69% (58–82%) | 36% (21–61%) | |
| 10 | 3 | 0 | 1065 (-) | 1065 | - | - | - | ||
| TNM (EAU risk groups for BCR) | |||||||||
| 0 | cT1-2a | 5 | 0 | 1815 (-) | 1815 | - | - | - | |
| cT2b | 6 | 0 | 2753 (-) | 2753 | - | - | - | ||
| cT2c-4 | 156 | 19 | 3489.94 (264.72) | 4604 | ref | 88% (82–95%) | 75% (63–89%) | ||
| 1 | cT1-2a | 2 | 0 | 2620 (-) | 2620 | - | - | - | |
| cT2b | 3 | 0 | 2553 (-) | 2553 | - | - | - | ||
| cT2c-4 | 243 | 53 | 2110.36 (179.47) | 3524 | 2.05 (1.21–3.46) | 0.008 | 80% (74–86%) | 51% (39–67%) | |
| Stage (EAU risk groups for BCR) | |||||||||
| 0 | Localized | 70 | 0 | 4604 (-) | 4604 | - | - | - | |
| Locally advanced | 97 | 19 | 1982.45 (175.46) | 2859 | ref | 83% (74–92%) | 62% (46–83%) | ||
| 1 | Localized | 83 | 9 | 2817.88 (241.55) | 3524 | 1.5 (0.87–2.57) | 0.14 | 88% (79–97%) | 74% (58–96%) |
| Locally advanced | 165 | 44 | 1870.44 (195.09) | 3447 | 0.61 (0.28–1.36) | 0.2 | 77% (70–85%) | 42% (28–64%) | |
| Preoperative PSA groups | |||||||||
| 0 | <10 ng/mL | 108 | 12 | 3707.59 (273.11) | 4604 | ref | 87% (80–95%) | 77% (63–94%) | |
| 10–20 ng/mL | 19 | 2 | 1628.96 (149.93) | 2056 | 1.03 (0.36–2.93) | 0.9 | 97% (91–100%) | 72% (50–100%) | |
| >20 ng/mL | 36 | 5 | 1739.72 (127.6) | 1860 | 0.82 (0.18–3.69) | 0.8 | 83% (63–100%) | 83% (63–100%) | |
| 1 | <10 ng/mL | 171 | 31 | 2301.4 (212.54) | 3524 | 1.79 (0.92–3.49) | 0.088 | 83% (76–90%) | 53% (38–75%) |
| 10–20 ng/mL | 49 | 12 | 2206.48 (291.11) | 2364 | 2.23 (1–4.97) | 0.049 | 74% (60–91%) | 54% (36–82%) | |
| >20 ng/mL | 24 | 8 | 1495.88 (225.37) | 3447 | 2.83 (1.16–6.94) | 0.023 | 76% (60–91%) | 51% (27–98%) | |
| n = 497 | |
|---|---|
| Age [years], median (range) | 61 (41–78) |
| Preoperative PSA [ng/mL], median (range) | 7.5 (0.7–107) |
| PSA (EAU risk groups for BCR) | |
| <10 ng/mL | 329 |
| 10–20 ng/mL | 99 |
| >20 ng/mL | 54 |
| NA | 15 |
| TNM (EAU risk groups for BCR) | |
| cT1-2a | 13 |
| cT2b | 10 |
| cT2c-4 | 467 |
| NA | 7 |
| Stage (EAU risk groups for BCR) | |
| Localized | 187 |
| Locally advanced | 303 |
| NA | 7 |
| Lymph node status (pN) | |
| N0 | 326 |
| N1 | 80 |
| NA | 91 |
| EAU risk groups for BCR | |
| Low risk | 2 |
| Intermediate risk | 11 |
| High risk | 391 |
| NA | 93 |
| Tumor status | |
| Tumor free | 288 |
| With tumor | 54 |
| NA | 155 |
| Vital status | |
| Alive | 489 |
| Dead | 8 |
| Laterality | |
| Bilateral | 432 |
| Left | 19 |
| Right | 38 |
| NA | 13 |
| Zone of origin | |
| Central zone | 4 |
| Overlapping/multiple zones | 127 |
| Peripheral zone | 137 |
| Transition zone | 8 |
| NA | 221 |
| Gleason score | |
| 6 | 45 |
| 7A (3 + 4) | 146 |
| 7B (4 + 3) | 101 |
| 8 | 64 |
| 9 | 137 |
| 10 | 4 |
| Primary pattern | |
| 3 | 198 |
| 4 | 250 |
| 5 | 49 |
| Secondary pattern | |
| 3 | 152 |
| 4 | 235 |
| 5 | 110 |
| BCR status | |
| No | 351 |
| Yes | 78 |
| NA | 68 |
| GTRD ID | No. of Mapped Target Genes | |
|---|---|---|
| AR | P10275 | 14,390 |
| ESR1 | P03372 | 15,405 |
| ESR2 | Q92731 | 1057 |
 | ||
| Gene Name | Marker of | |
|---|---|---|
| CDH1 | Cadherin 1, E-cadherin | Epithelial state |
| COL4A1 | Collagen type IV α-1 chain | |
| DSP | Desmoplakin | |
| KRT18 | Keratin 18 | |
| KRT19 | Keratin 19 | |
| KRT5 | Keratin 5 | |
| LAMA1 | Laminin subunit α-1 | |
| LAMA2 | Laminin subunit α-2 | |
| LAMA3 | Laminin subunit α-3 | |
| LAMA4 | Laminin subunit α-4 | |
| LAMA5 | Laminin subunit α-5 | |
| MUC1 | Mucin 1 | |
| NID1 | Nidogen 1 | |
| OCLN | Occludin | |
| TJP1 | Tight junction protein 1 | |
| ACTA2 | Actin 2, α 2 | Mesenchymal state |
| CDH11 | Cadherin 11 | |
| CDH2 | Cadherin 2, N-cadherin | |
| CTNNB1 | Catenin β 1 | |
| DDR2 | Discoidin domain receptor tyrosine kinase 2 | |
| FN1 | Fibronectin 1 | |
| GSC | Goosecoid homeobox | |
| ITGA5 | Integrin subunit α 5 | |
| ITGB6 | Integrin subunit β 6 | |
| KRT8 | Keratin 8 | |
| LEF1 | Lymphoid enhancer binding factor 1 | |
| MMP2 | Matrix metalloproteinase 2, Gelatinase A | |
| MMP3 | Matrix metalloproteinase 3, Stromelysin 1 | |
| MMP9 | Matrix metalloproteinase 9, Gelatinase B | |
| S100A4 | S100 calcium binding protein A4 | |
| SDC1 | Syndecan 1 | |
| SERPINH1 | Serpin family H member 1 | |
| SMAD2 | SMAD family member 2 | |
| SMAD3 | SMAD family member 3 | |
| SNAI2 | Snail family transcriptional repressor 2 | |
| TCF3 | Transcription factor 3 | |
| TCF4 | Transcription factor 4 | |
| TWIST1 | Twist family bHLH transcription factor 1 | |
| VIM | Vimentin | |
| ZEB1 | Zinc finger E-box binding homeobox 1 | |
| ZEB2 | Zinc finger E-box binding homeobox 2 |
| n = 503 | |
|---|---|
| Age group (years) | |
| 35–49 | 67 |
| 50–54 | 90 |
| 55–59 | 123 |
| 60–64 | 145 |
| 65–69 | 53 |
| 70–74 | 25 |
| Preoperative PSA for EAU BCR risk groups | |
| <10 ng/mL | 369 |
| 10–20 ng/mL | 72 |
| >20 ng/mL | 29 |
| NA | 33 |
| Stage | |
| Local | 338 |
| Regional | 165 |
| Gleason score | |
| 6 | 239 |
| 7A (3 + 4) | 184 |
| 7B (4 + 3) | 40 |
| 8–10 | 40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olczak, M.; Orzechowska, M.J.; Bednarek, A.K.; Lipiński, M. The Transcriptomic Profiles of ESR1 and MMP3 Stratify the Risk of Biochemical Recurrence in Primary Prostate Cancer beyond Clinical Features. Int. J. Mol. Sci. 2023, 24, 8399. https://doi.org/10.3390/ijms24098399
Olczak M, Orzechowska MJ, Bednarek AK, Lipiński M. The Transcriptomic Profiles of ESR1 and MMP3 Stratify the Risk of Biochemical Recurrence in Primary Prostate Cancer beyond Clinical Features. International Journal of Molecular Sciences. 2023; 24(9):8399. https://doi.org/10.3390/ijms24098399
Chicago/Turabian StyleOlczak, Michał, Magdalena Julita Orzechowska, Andrzej K. Bednarek, and Marek Lipiński. 2023. "The Transcriptomic Profiles of ESR1 and MMP3 Stratify the Risk of Biochemical Recurrence in Primary Prostate Cancer beyond Clinical Features" International Journal of Molecular Sciences 24, no. 9: 8399. https://doi.org/10.3390/ijms24098399
APA StyleOlczak, M., Orzechowska, M. J., Bednarek, A. K., & Lipiński, M. (2023). The Transcriptomic Profiles of ESR1 and MMP3 Stratify the Risk of Biochemical Recurrence in Primary Prostate Cancer beyond Clinical Features. International Journal of Molecular Sciences, 24(9), 8399. https://doi.org/10.3390/ijms24098399