
Genomic and Transcriptional Profiles of Kelch-like (klhl) Gene Family in Polyploid Carassius Complex

 ,
,  and
and
Abstract
1. Introduction
2. Results
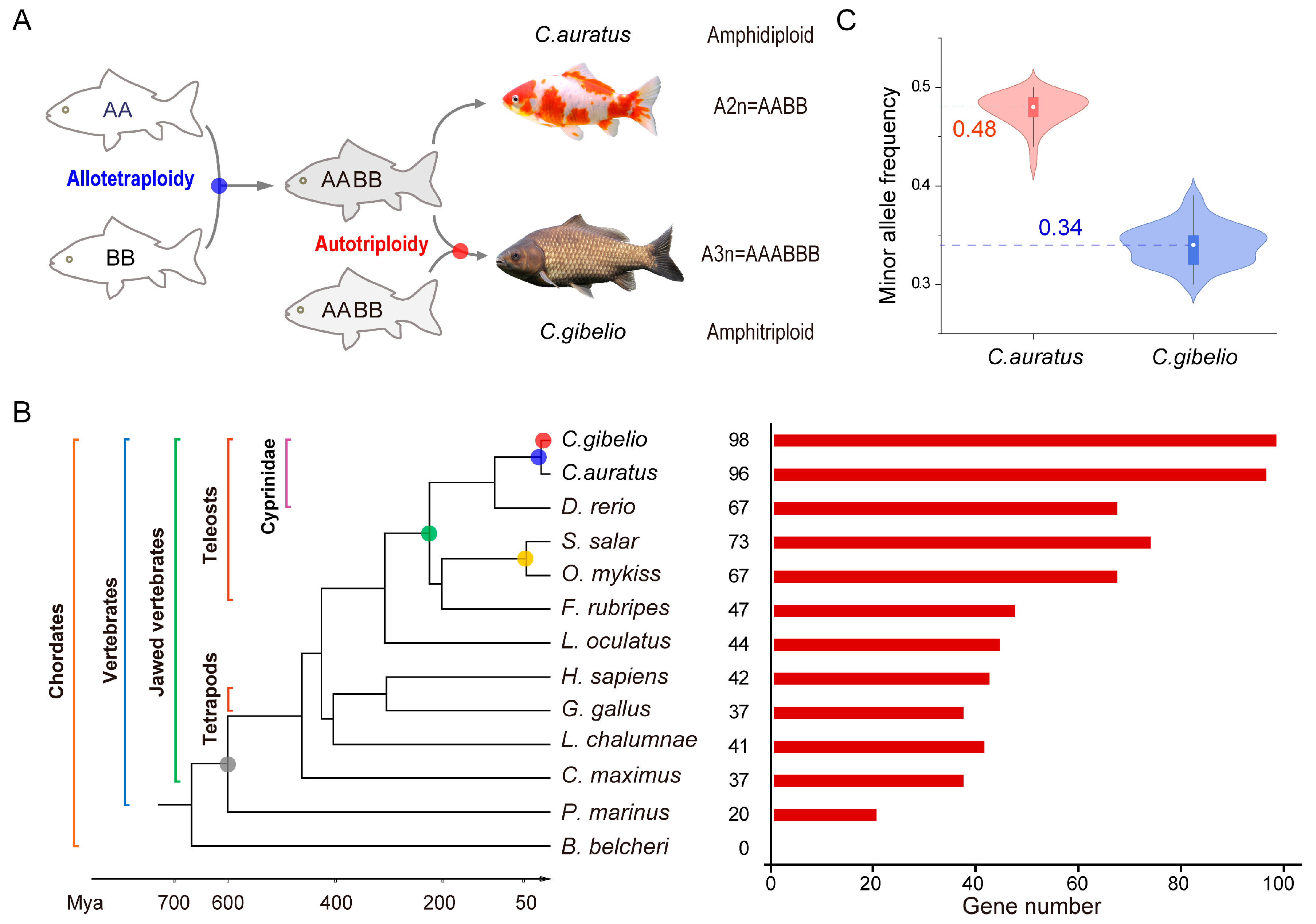
2.1. Genome-Wide Identification of klhl Genes
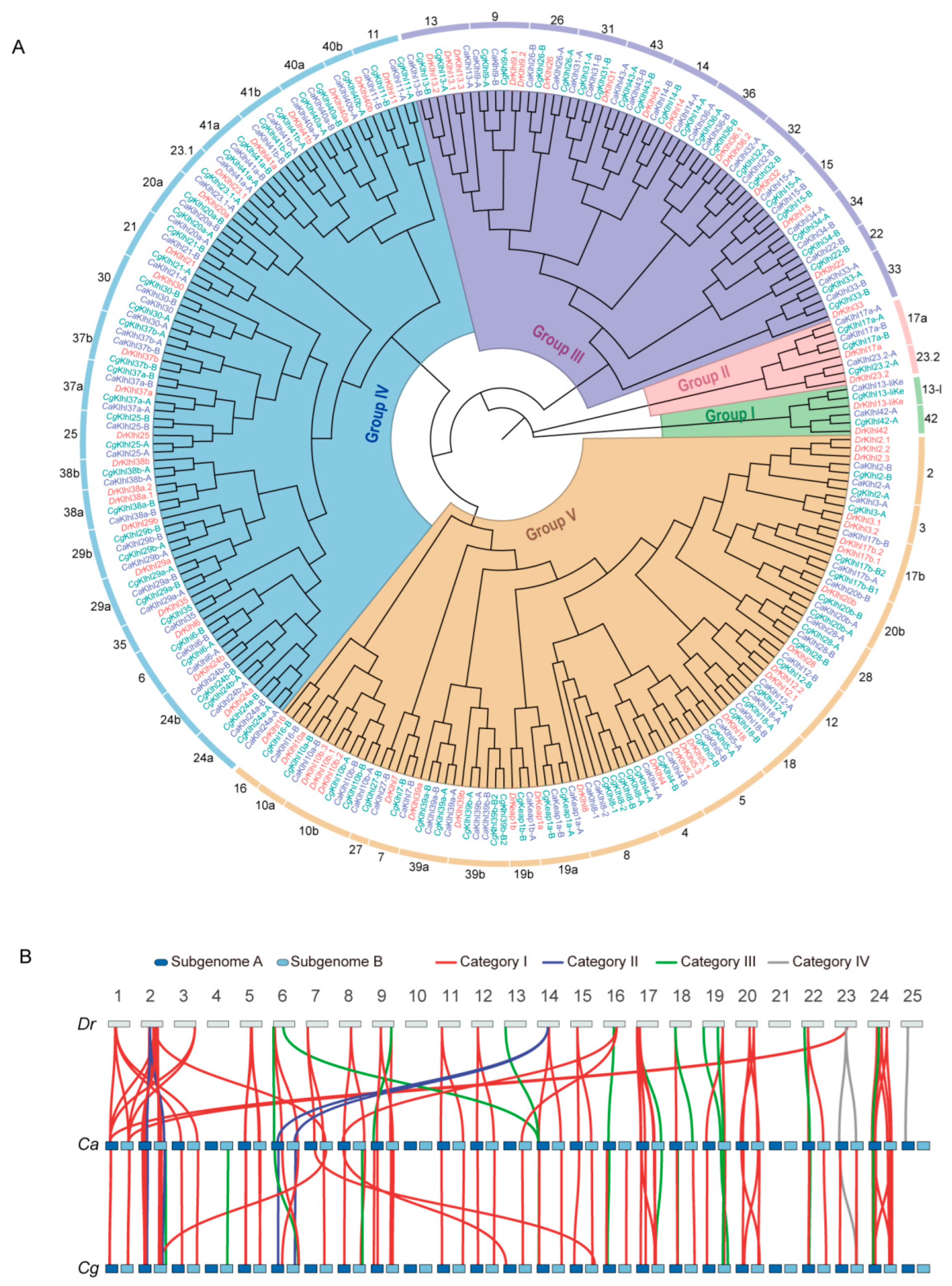
2.2. Evolution of klhl Gene Family in Carassius
2.3. Expression Atlas of klhl Genes in Carassius
2.4. Varied Expression Bias of klhl Homeolog Pairs in Carassius
2.5. klhl Expression in Response to Herpesvirus Infection in C. gibelio
2.6. klhl Alleles in C. gibelio
2.7. Bias Allelic Expression of Cgklhl Genes
3. Discussion
4. Materials and Methods
4.1. Identification and Characterization of klhl Family Genes
4.2. Phylogenetic Analysis
4.3. Synteny Analysis
4.4. RNA-seq Samples and Gene Expression Analysis
4.5. Polyploidy Evaluation and Allele Identification of Cgklhl Genes
4.6. Allele-Specific Expression Analysis
4.7. The Domains of Klhl Alleles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dhanoa, B.S.; Cogliati, T.; Satish, A.G.; A Bruford, E.; Friedman, J.S. Update on the Kelch-like (KLHL) gene family. Hum. Genom. 2013, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- A Gupta, V.; Beggs, A.H. Kelch proteins: Emerging roles in skeletal muscle development and diseases. Skelet. Muscle 2014, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, K.; Ingvarsdottir, K.; Bonasio, R.; Saraf, A.; Florens, L.; Washburn, M.P.; Tadros, S.; Green, M.R.; Busino, L. Loss of KLHL6 promotes diffuse large B-cell lymphoma growth and survival by stabilizing the mRNA decay factor roquin2. Nature 2018, 20, 586–596. [Google Scholar] [CrossRef]
- Shi, X.; Xiang, S.; Cao, J.; Zhu, H.; Yang, B.; He, Q.; Ying, M. Kelch-like proteins: Physiological functions and relationships with diseases. Pharmacol. Res. 2019, 148, 104404. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Alvarez, M.J.; Talos, F.; Dhruv, H.; Rieckhof, G.E.; Iyer, A.; Diefes, K.L.; Aldape, K.; Berens, M.; Shen, M.; et al. Identification of Causal Genetic Drivers of Human Disease through Systems-Level Analysis of Regulatory Networks. Cell 2014, 159, 402–414. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Louis-Dit-Picard, H.; International Consortium for Blood Pressure (ICBP); Barc, J.; Trujillano, D.; Miserey-Lenkei, S.; Bouatia-Naji, N.; Pylypenko, O.; Beaurain, G.; Bonnefond, A.; Sand, O.; et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012, 44, 456–460. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Amores, A.; Force, A.; Yan, Y.-L.; Joly, L.; Amemiya, C.; Fritz, A.; Ho, R.K.; Langeland, J.; Prince, V.; Wang, Y.-L.; et al. Zebrafish hox Clusters and Vertebrate Genome Evolution. Science 1998, 282, 1711–1714. [Google Scholar] [CrossRef]
- Christoffels, A.; Koh, E.G.L.; Chia, J.-M.; Brenner, S.; Aparicio, S.; Venkatesh, B. Fugu Genome Analysis Provides Evidence for a Whole-Genome Duplication Early During the Evolution of Ray-Finned Fishes. Mol. Biol. Evol. 2004, 21, 1146–1151. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.-M.; Brunet, F.; Petit, J.-L.; Stange-Thomann, N.; Mauceli, E.; Bouneau, L.; Fischer, C.; Ozouf-Costaz, C.; Bernot, A.; et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 2004, 431, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sado, T.; Hirt, M.V.; Pasco-Viel, E.; Arunachalam, M.; Li, J.; Wang, X.; Freyhof, J.; Saitoh, K.; Simons, A.M.; et al. Phylogeny and polyploidy: Resolving the classification of cyprinine fishes (Teleostei: Cypriniformes). Mol. Phylogenet. Evol. 2015, 85, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Wu, J.; Cai, X.; Liang, J.; Freeling, M.; Wang, X. Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 2018, 4, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Mandakova, T.; A Lysak, M. Post-polyploid diploidization and diversification through dysploid changes. Curr. Opin. Plant Biol. 2018, 42, 55–65. [Google Scholar] [CrossRef]
- Van De Peer, Y.; Mizrachi, Y.V.D.P.E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Carretero-Paulet, L.; Van de Peer, Y. The evolutionary conundrum of whole-genome duplication. Am. J. Bot. 2020, 107, 1101–1105. [Google Scholar] [CrossRef]
- Feliner, G.N.; Casacuberta, J.; Wendel, J.F. Genomics of Evolutionary Novelty in Hybrids and Polyploids. Front. Genet. 2020, 11, 792. [Google Scholar] [CrossRef]
- Wendel, J.F.; Lisch, D.; Hu, G.; Mason, A.S. The long and short of doubling down: Polyploidy, epigenetics, and the temporal dynamics of genome fractionation. Curr. Opin. Genet. Dev. 2018, 49, 1–7. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.-Y.; Xu, W.-J.; Wang, K.; Bin Wu, B.; Xu, M.; Chen, Y.; Miao, L.-J.; Wang, Z.-W.; Li, Z.; et al. Comparative genome anatomy reveals evolutionary insights into a unique amphitriploid fish. Nat. Ecol. Evol. 2022, 6, 1354–1366. [Google Scholar] [CrossRef]
- Kuhl, H.; Du, K.; Schartl, M.; Kalous, L.; Stöck, M.; Lamatsch, D.K. Equilibrated evolution of the mixed auto-/allopolyploid haplotype-resolved genome of the invasive hexaploid Prussian carp. Nat. Commun. 2022, 13, 1–11. [Google Scholar] [CrossRef]
- Lin, Q.; Mei, J. Genomic characterization of an amphitriploid fish and insights into the evolutionary mechanisms of unisexual reproduction success. Water Biol. Secur. 2022, 1, 100066. [Google Scholar] [CrossRef]
- Gui, J.; Zhou, L. Genetic basis and breeding application of clonal diversity and dual reproduction modes in polyploid Carassius auratus gibelio. Sci. China Life Sci. 2010, 53, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Vetešník, L.; Papoušek, I.; Halačka, K.; Lusková, V.; Mendel, J. Morphometric and genetic analysis of Carassius auratus complex from an artificial wetland in Morava River floodplain, Czech Republic. Fish. Sci. 2007, 73, 817–822. [Google Scholar] [CrossRef]
- Yu, P.; Wang, Y.; Yang, W.-T.; Li, Z.; Zhang, X.-J.; Zhou, L.; Gui, J.-F. Upregulation of the PPAR signaling pathway and accumulation of lipids are related to the morphological and structural transformation of the dragon-eye goldfish eye. Sci. China Life Sci. 2021, 64, 1031–1049. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Z.W.; Wang, Y.; Gui, J.F. Crucian carp and gibel carp culture. In Aquaculture in China: Success Stories and Modern Trends; Gui, J.F., Tang, Q.S., Li, Z.J., Liu, J.S., De Silva, S.S., Eds.; Wiley: Oxford, UK, 2018. [Google Scholar]
- Zhou, L.; Gui, J. Natural and artificial polyploids in aquaculture. Aquac. Fish. 2017, 2, 103–111. [Google Scholar] [CrossRef]
- Gui, J.F.; Zhou, L.; Yin, Z.; Hu, W.; Tong, J.G.; Sun, Y.H.; Xiao, W.H. Genetic Breeding in Aquaculture; Science Press: Beijing, China, 2021. [Google Scholar]
- Ding, M.; Li, X.-Y.; Zhu, Z.-X.; Chen, J.-H.; Lu, M.; Shi, Q.; Wang, Y.; Li, Z.; Zhao, X.; Wang, T.; et al. Genomic anatomy of male-specific microchromosomes in a gynogenetic fish. PLoS Genet. 2021, 17, e1009760. [Google Scholar] [CrossRef]
- Li, X.-Y.; Mei, J.; Ge, C.-T.; Liu, X.-L.; Gui, J.-F. Sex determination mechanisms and sex control approaches in aquaculture animals. Sci. China Life Sci. 2022, 65, 1091–1122. [Google Scholar] [CrossRef]
- Marlétaz, F.; Firbas, P.N.; Maeso, I.; Tena, J.J.; Bogdanovic, O.; Perry, M.; Wyatt, C.D.R.; de la Calle-Mustienes, E.; Bertrand, S.; Burguera, D.; et al. Amphioxus functional genomics and the origins of vertebrate gene regulation. Nature 2018, 564, 64–70. [Google Scholar] [CrossRef]
- Smith, J.J.; Timoshevskaya, N.; Ye, C.; Holt, C.; Keinath, M.C.; Parker, H.J.; Cook, M.E.; Hess, J.E.; Narum, S.R.; Lamanna, F.; et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet. 2018, 50, 270–277. [Google Scholar] [CrossRef]
- Smith, J.J.; Kuraku, S.; Holt, C.; Sauka-Spengler, T.; Jiang, N.; Campbell, M.S.; Yandell, M.D.; Manousaki, T.; Meyer, A.; E Bloom, O.; et al. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat. Genet. 2013, 45, 415–421. [Google Scholar] [CrossRef]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef]
- Berthelot, C.; Brunet, F.; Chalopin, D.; Juanchich, A.; Bernard, M.; Noël, B.; Bento, P.; Da Silva, C.; Labadie, K.; Alberti, A.; et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 2014, 5, 3657. [Google Scholar] [CrossRef]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Ashman, T.-L.; Soltis, P.S.; Soltis, D.E. Polyploidy: An evolutionary and ecological force in stressful times. Plant Cell 2021, 33, 11–26. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Gui, J.-F. Diversity, evolutionary contribution and ecological roles of aquatic viruses. Sci. China Life Sci. 2018, 61, 1486–1502. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.-T.; Chen, Z.-Y.; Deng, Y.-S.; Gui, J.-F.; Zhang, Q.-Y. Complete genome sequence and architecture of crucian carp Carassius auratus herpesvirus (CaHV). Arch. Virol. 2016, 161, 3577–3581. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.-X.; Wang, Y.; Zhang, Q.-Y.; Mou, C.-Y.; Li, Z.; Deng, Y.-S.; Zhou, L.; Gui, J.-F. Distinct herpesvirus resistances and immune responses of three gynogenetic clones of gibel carp revealed by comprehensive transcriptomes. BMC Genom. 2017, 18, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Burns, V.E.; Kerppola, T.K. Keap1 moderates the transcription of virus induced genes through G9a-GLP and NFκB p50 recruitment. Immunology 2022, 167, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Kung, Y.-A.; Hung, C.-T.; Chien, K.-Y.; Shih, S.-R. Control of the negative IREStrans-acting factor KHSRP by ubiquitination. Nucleic Acids Res. 2016, 45, 271–287. [Google Scholar] [CrossRef]
- Robles-Espinoza, C.D.; Mohammadi, P.; Bonilla, X.; Gutierrez-Arcelus, M. Allele-specific expression: Applications in cancer and technical considerations. Curr. Opin. Genet. Dev. 2020, 66, 10–19. [Google Scholar] [CrossRef]
- Bogart, J.P.; Bi, K.; Fu, J.; Noble, D.W.; Niedzwiecki, J. Unisexual salamanders (genus Ambystoma) present a new reproductive mode for eukaryotes. Genome 2007, 50, 119–136. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Maere, S.; Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009, 10, 725–732. [Google Scholar] [CrossRef]
- Soltis, P.S.; E Soltis, D. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 2016, 30, 159–165. [Google Scholar] [CrossRef]
- Inoue, J.; Sato, Y.; Sinclair, R.; Tsukamoto, K.; Nishida, M. Rapid genome reshaping by multiple-gene loss after whole-genome duplication in teleost fish suggested by mathematical modeling. Proc. Natl. Acad. Sci. USA 2015, 112, 14918–14923. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Xu, J.; Liu, G.; Chen, L.; Zhou, Z.; Peng, W.; Jiang, Y.; Zhao, Z.; Jia, Z.; Sun, Y.; et al. The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat. Commun. 2019, 10, 4625. [Google Scholar] [CrossRef] [PubMed]
- Kon, T.; Omori, Y.; Fukuta, K.; Wada, H.; Watanabe, M.; Chen, Z.; Iwasaki, M.; Mishina, T.; Matsuzaki, S.-I.S.; Yoshihara, D.; et al. The Genetic Basis of Morphological Diversity in Domesticated Goldfish. Curr. Biol. 2020, 30, 2260–2274.e6. [Google Scholar] [CrossRef]
- Kondrashov, F.A. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc. R. Soc. B Boil. Sci. 2012, 279, 5048–5057. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Chai, J.; Wen, Y.; Tao, M.; Lin, G.; Liu, X.; Ren, L.; Chen, Z.; Wu, S.; Li, S.; et al. From asymmetrical to balanced genomic diversification during rediploidization: Subgenomic evolution in allotetraploid fish. Sci. Adv. 2020, 6, z7677. [Google Scholar] [CrossRef]
- Li, J.-T.; Wang, Q.; Yang, M.-D.H.; Li, Q.-S.; Cui, M.-S.; Dong, Z.-J.; Wang, H.-W.; Yu, J.-H.; Zhao, Y.-J.; Yang, C.-R.; et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat. Genet. 2021, 53, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Soltis, P.S.; Marchant, D.B.; Van de Peer, Y.; E Soltis, D. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 2015, 35, 119–125. [Google Scholar] [CrossRef]
- Butlin, R. The costs and benefits of sex: New insights from old asexual lineages. Nat. Rev. Genet. 2002, 3, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Li, Z.; Zhu, Z.-Y.; Peng, F.; Wang, Y.; Li, X.-Y.; Wang, Z.-W.; Zhang, X.-J.; Zhou, L.; Gui, J.-F. Changes in Ploidy Drive Reproduction Transition and Genomic Diversity in a Polyploid Fish Complex. Mol. Biol. Evol. 2022, 39. [Google Scholar] [CrossRef]
- Cunha, C.; Doadrio, I.; Abrantes, J.; Coelho, M.M. The evolutionary history of the allopolyploid Squalius alburnoides (Cyprinidae) complex in the northern Iberian Peninsula. Heredity 2010, 106, 100–112. [Google Scholar] [CrossRef][Green Version]
- Christiansen, D.G. Gamete types, sex determination and stable equilibria of all-hybrid populations of diploid and triploid edible frogs (Pelophylax esculentus). BMC Evol. Biol. 2009, 9, 135. [Google Scholar] [CrossRef]
- Bogart, J.; Bi, K. Genetic and Genomic Interactions of Animals with Different Ploidy Levels. Cytogenet. Genome Res. 2013, 140, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Nembaware, V.; Lupindo, B.; Schouest, K.; Spillane, C.; Scheffler, K.; Seoighe, C. Genome-wide survey of allele-specific splicing in humans. BMC Genom. 2008, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hug, N.; Longman, D.; Cáceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.-H.; Wang, Y.; Li, Z.; Yu, Z.-X.; Li, X.-Y.; Tong, J.-F.; Wang, Z.-W.; Zhang, X.-J.; Zhou, L.; Gui, J.-F. Functional Divergence of Multiple Duplicated Foxl2 Homeologs and Alleles in a Recurrent Polyploid Fish. Mol. Biol. Evol. 2021, 38, 1995–2013. [Google Scholar] [CrossRef] [PubMed]
- Mou, C.-Y.; Wang, Y.; Zhang, Q.-Y.; Gao, F.-X.; Li, Z.; Tong, J.-F.; Zhou, L.; Gui, J.-F. Differential interferon system gene expression profiles in susceptible and resistant gynogenetic clones of gibel carp challenged with herpesvirus CaHV. Dev. Comp. Immunol. 2018, 86, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2011, 40, D302–D305. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Gene Name | Gene Number | Biased Retention/Loss of Paralogs | Biased Retention/Loss of Homeologs |
|---|---|---|---|---|
| I | klhl20, klhl24, klhl29, klhl37, klhl39, klhl41 | 6 | No | No |
| II | klhl10, keap1, klhl38, klhl40 | 4 | No | Yes |
| III | klhl2, klhl4, klhl5, klhl6, klhl9, klhl11, klhl12, klhl13, klhl14, klhl15, klhl18, klhl21, klhl25, klhl26, klhl28, klhl30, klhl31, klhl32, klhl33, klhl34, klhl36, klhl43 | 22 | Yes | No |
| VI | klhl3, klhl7, klhl13-like, klhl16, klhl22, klhl23.1, klhl23.2, klhl27, klhl35, klhl42 | 10 | Yes | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, F.; Zhou, L.; Lu, W.; Gan, R.; Lu, M.; Li, Z.; Zhang, X.; Wang, Y.; Gui, J. Genomic and Transcriptional Profiles of Kelch-like (klhl) Gene Family in Polyploid Carassius Complex. Int. J. Mol. Sci. 2023, 24, 8367. https://doi.org/10.3390/ijms24098367
Peng F, Zhou L, Lu W, Gan R, Lu M, Li Z, Zhang X, Wang Y, Gui J. Genomic and Transcriptional Profiles of Kelch-like (klhl) Gene Family in Polyploid Carassius Complex. International Journal of Molecular Sciences. 2023; 24(9):8367. https://doi.org/10.3390/ijms24098367
Chicago/Turabian StylePeng, Fang, Li Zhou, Weijia Lu, Ruihai Gan, Meng Lu, Zhi Li, Xiaojuan Zhang, Yang Wang, and Jianfang Gui. 2023. "Genomic and Transcriptional Profiles of Kelch-like (klhl) Gene Family in Polyploid Carassius Complex" International Journal of Molecular Sciences 24, no. 9: 8367. https://doi.org/10.3390/ijms24098367
APA StylePeng, F., Zhou, L., Lu, W., Gan, R., Lu, M., Li, Z., Zhang, X., Wang, Y., & Gui, J. (2023). Genomic and Transcriptional Profiles of Kelch-like (klhl) Gene Family in Polyploid Carassius Complex. International Journal of Molecular Sciences, 24(9), 8367. https://doi.org/10.3390/ijms24098367