
Mechanisms Limiting Renal Tissue Protection and Repair in Glomerulonephritis
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Oxidative Systems
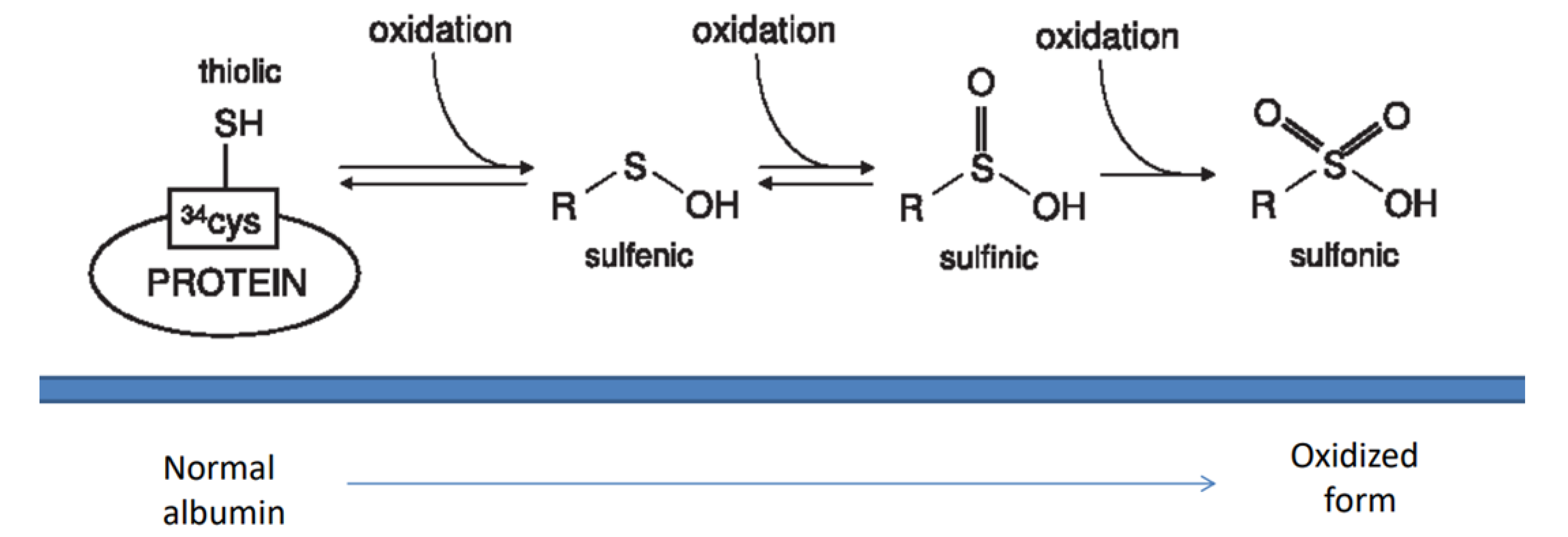
2.1. Oxidants and Antioxidants in Glomerulonephritis
2.2. Anti-SOD2 Antibodies
2.3. Anti GST Antibodies
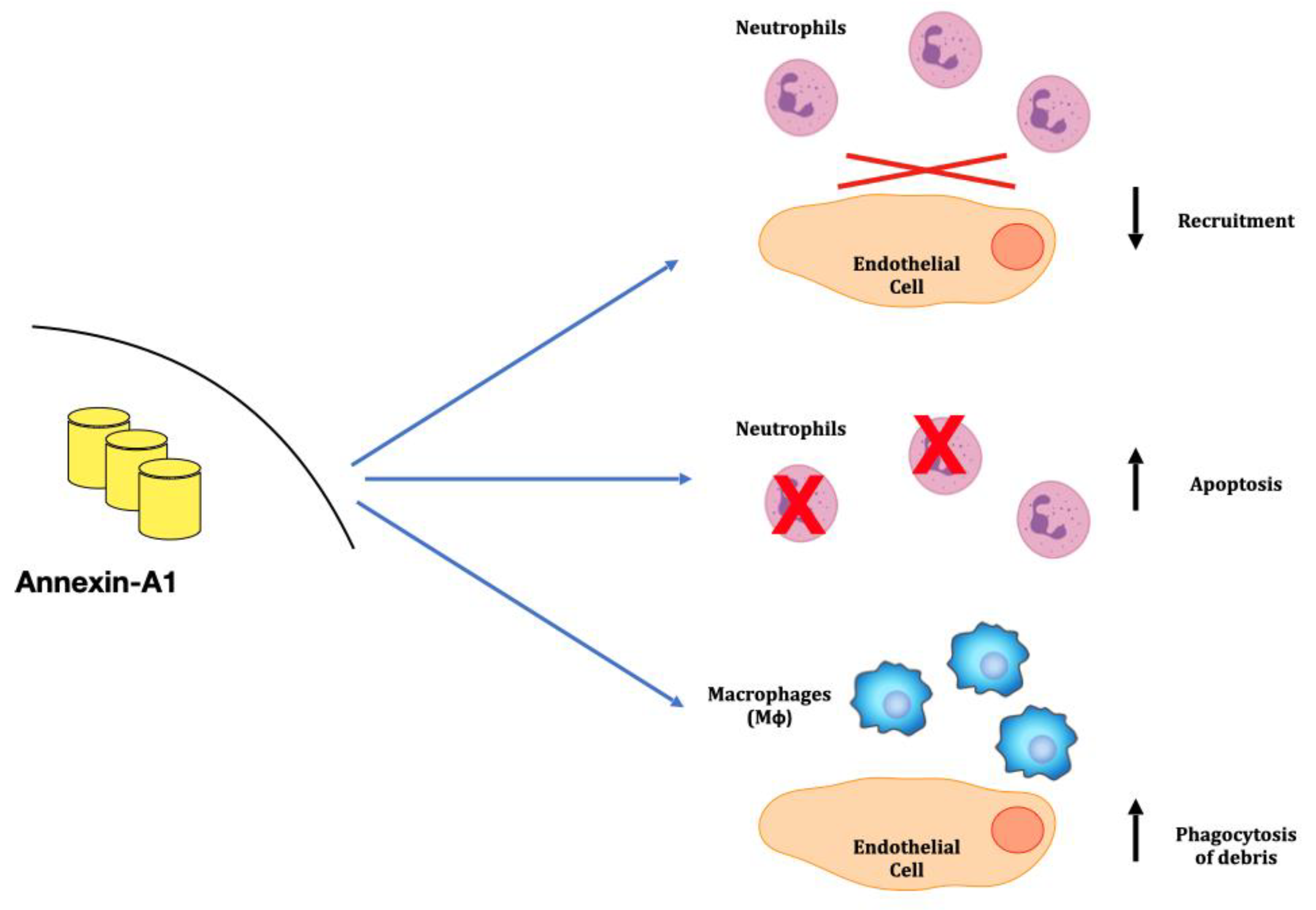
3. Anti-Inflammatory Mechanisms: The Annexins Family
3.1. Neutrophyl ANXA1 in Granulomatosis with Polyangiitis in Kidney
3.2. Annexin A1 in Rheumatoid Arthritis (RA) and in SLE
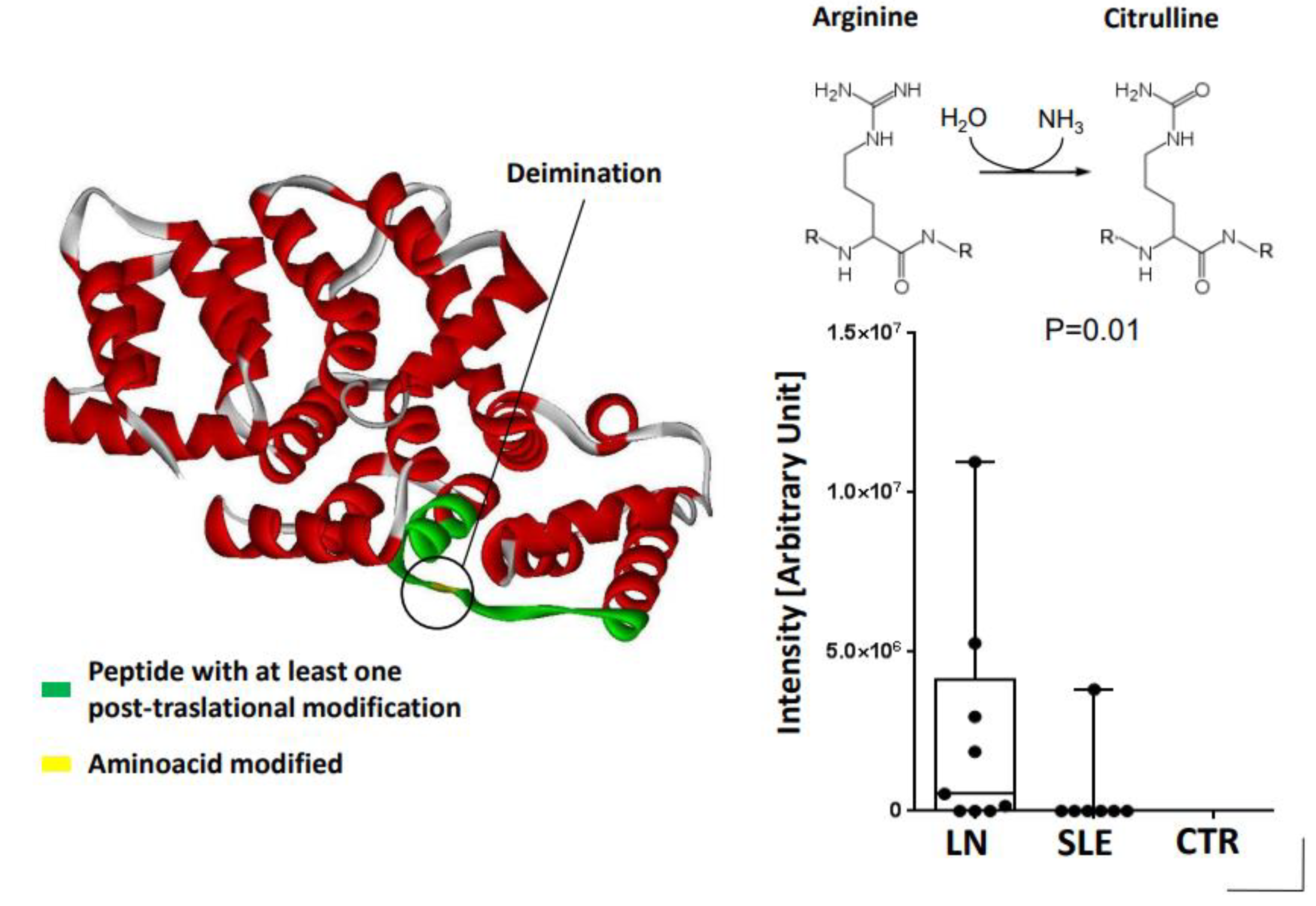
3.3. Anti-ANXA1 Antibodies in Lupus Nephritis (LN)
3.4. ANXA2 Antibodies in SLE, Antiphospholipid Syndrome and Nephrotic Syndrome
4. Macrophages, Tissue Infiltration and Repair
4.1. Macrophage Implication in Gn
4.2. Anti-Macrophage Antibodies in Gn
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Bruschi, M.; Petretto, A.; Vaglio, A.; Santucci, L.; Candiano, G.; Ghiggeri, G.M. Annexin A1 and Autoimmunity: From Basic Science to Clinical Applications. Int. J. Mol. Sci. 2018, 19, 1348. [Google Scholar] [CrossRef] [PubMed]
- Puthumana, J.; Thiessen-Philbrook, H.; Xu, L.; Coca, S.G.; Garg, A.X.; Himmelfarb, J.; Bhatraju, P.K.; Ikizler, T.A.; Siew, E.D.; Ware, L.B.; et al. Biomarkers of inflammation and repair in kidney disease progression. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Ghiggeri, G.M.; Seitz-Polski, B.; Justino, J.; Zaghrini, C.; Payré, C.; Brglez, V.; Dolla, G.; Sinico, A.; Scolari, F.; Vaglio, A.; et al. Multi-Autoantibody Signature and Clinical Outcome in Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2020, 15, 1762–1776. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Sinico, R.A.; Moroni, G.; Pratesi, F.; Migliorini, P.; Galetti, M.; Murtas, C.; Tincani, A.; Madaio, M.; Radice, A.; et al. Glomerular autoimmune multicomponents of human lupus nephritis in vivo: Alpha-enolase and annexin AI. J. Am. Soc. Nephrol. 2014, 25, 2483–2498. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Cavalli, A.; Moll, S.; Candiano, G.; Scapozza, L.; Patel, J.J.; Tan, J.C.; Lo, K.C.; Angeletti, A.; Ghiggeri, G.M.; et al. Discovery of anti-Formin-like 1 protein (FMNL1) antibodies in membranous nephropathy and other glomerular diseases. Sci. Rep. 2022, 12, 13659. [Google Scholar] [CrossRef] [PubMed]
- Ginevri, F.; Ghiggeri, G.M.; Candiano, G.; Oleggini, R.; Bertelli, R.; Piccardo, M.T.; Perfumo, F.; Gusmano, R. Peroxidative damage of the erythrocyte membrane in children with nephrotic syndrome. Pediatr. Nephrol. 1989, 3, 25–32. [Google Scholar] [CrossRef]
- Musante, L.; Candiano, G.; Petretto, A.; Bruschi, M.; Dimasi, N.; Caridi, G.; Pavone, B.; Del Boccio, P.; Galliano, M. Active focal segmental glomerulosclerosis is associated with massive oxidation of plasma albumin. J. Am. Soc. Nephrol. 2007, 18, 799–810. [Google Scholar] [CrossRef]
- Bona, N.; Pezzarini, E.; Balbi, B.; Daniele, S.M.; Rossi, M.F.; Monje, A.L.; Basiglio, C.; Pelusa, H.F.; Arriaga, S.M.M. Oxidative stress, inflammation and disease activity biomarkers in lupus nephropathy. Lupus 2020, 29, 311–323. [Google Scholar] [CrossRef]
- Apeland, T.; Ushakova, A.; Mansoor, M.A.; Furriol, J.; Jonsson, G.; Marti, H.-P. Association of redox and inflammation-related biomarkers with prognosis in IgA nephropathy: A prospective observational study. Free. Radic. Biol. Med. 2022, 188, 62–70. [Google Scholar] [CrossRef]
- Bruschi, M.; Candiano, G.; Santucci, L.; Ghiggeri, G.M. Oxidized albumin. The long way of a protein of uncertain function. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2013, 1830, 5473–5479. [Google Scholar] [CrossRef]
- Russell, T.M.; Richardson, D.R. The good Samaritan glutathione-S-transferase P1: An evolving relationship in nitric oxide metabolism mediat-ed by the direct interactions between multiple effector molecules. Redox Biol. 2023, 59, 102568. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.-Y.; Zheng, X.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug. Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Buelli, S.; Perico, L.; Galbusera, M.; Abbate, M.; Morigi, M.; Novelli, R.; Gagliardini, E.; Tentori, C.; Rottoli, D.; Sabadini, E.; et al. Mitochondrial-dependent Autoimmunity in Membranous Nephropathy of IgG4-related Disease. Ebiomedicine 2015, 2, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Tomas, N.M.; Hoxha, E.; Reinicke, A.T.; Fester, L.; Helmchen, U.; Gerth, J.; Bachmann, F.; Budde, K.; Koch-Nolte, F.; Zahner, G.; et al. Autoantibodies against thrombospondin type 1 domain–containing 7A induce membranous nephropathy. J. Clin. Investig. 2016, 126, 2519–2532. [Google Scholar] [CrossRef] [PubMed]
- Prunotto, M.; Carnevali, M.L.; Candiano, G.; Murtas, C.; Bruschi, M.; Corradini, E.; Trivelli, A.; Magnasco, A.; Petretto, A.; Santucci, L.; et al. Autoimmunity in Membranous Nephropathy Targets Aldose Reductase and SOD2. J. Am. Soc. Nephrol. 2010, 21, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, A.; Bruschi, M.; Moroni, G.; Sinico, R.A.; Franceschini, F.; Fredi, M.; Vaglio, A.; Cavagna, L.; Petretto, A.; Pratesi, F.; et al. Second Wave Antibodies in Autoimmune Renal Diseases: The Case of Lupus Nephritis. J. Am. Soc. Nephrol. 2021, 32, 3020–3023. [Google Scholar] [CrossRef]
- Beck, L.H.; Bonegio, R.G.; Lambeau, G.; Beck, D.M.; Powell, D.W.; Cummins, T.D.; Klein, J.B.; Salant, D.J. M-Type Phospholipase A2 Receptor as Target Antigen in Idiopathic Membranous Nephropathy. N. Engl. J. Med. 2009, 361, 11–21. [Google Scholar] [CrossRef]
- Aguilera, I.; Alvarez-Marquez, A.; Gentil, M.A.; Fernandez-Alonso, J.; Fijo, J.; Saez, C.; Wichmann, I.; Nuñez-Roldan, A. Anti-glutathione S-transferase T1 antibody-mediated rejection in C4d-positive renal allo-graft recipients. Nephrol. Dial. Transplant. 2008, 23, 2393–2398. [Google Scholar] [CrossRef]
- Comoli, P.; Cioni, M.; Ray, B.; Tagliamacco, A.; Innocente, A.; Caridi, G.; Bruschi, M.; Hariharan, J.; Fontana, I.; Trivelli, A.; et al. Anti-glutathione S-transferase theta 1 antibodies correlate with graft loss in non-sensitized pediatric kidney recipients. Front. Med. 2022, 9, 3559. [Google Scholar] [CrossRef]
- Creutz, C.; Pazoles, C.; Pollard, H. Identification and purification of an adrenal medullary protein (synexin) that causes calcium-dependent aggregation of isolated chromaffin granules. J. Biol. Chem. 1978, 253, 2858–2866. [Google Scholar] [CrossRef]
- Markoff, A.; Gerke, V. Expression and functions of annexins in the kidney. Am. J. Physiol. Physiol. 2005, 289, F949–F956. [Google Scholar] [CrossRef] [PubMed]
- McKanna, J.A.; Chuncharunee, A.; Munger, K.A.; Breyer, J.A.; Cohen, S.; Harris, R.C. Localization of p35 (annexin I, lipocortin I) in normal adult rat kidney and during recovery from ischemia. J. Cell. Physiol. 1992, 153, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; Gavins, F.N.E. Annexin 1: An Endogenous Anti-Inflammatory Protein. Physiology 2003, 18, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Gerke, V.; Moss, S.E. Annexins: From Structure to Function. Physiol. Rev. 2002, 82, 331–371. [Google Scholar] [CrossRef]
- Ferlazzo, V.; D’Agostino, P.; Milano, S.; Caruso, R.; Feo, S.; Cillari, E.; Parente, L. Anti-inflammatory effects of annexin-1: Stimulation of IL-10 release and inhibition of nitric oxide synthesis. Int. Immunopharmacol. 2003, 3, 1363–1369. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Vago, J.P.; Teixeira, M.M.; Sousa, L.P. Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J. Immunol. Res. 2016, 2016, 8239258. [Google Scholar] [CrossRef]
- Lim, L.H.K.; Solito, E.; Russo-Marie, F.; Flower, R.J.; Perretti, M. Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: Effect of lipocortin 1. Proc. Natl. Acad. Sci. USA 1998, 95, 14535–14539. [Google Scholar] [CrossRef]
- Zouki, C.; Ouellet, S.; Filep, J.G. The anti-inflammatory peptides, antiflammins, regulate the expression of adhesion molecules on human leu-kocytes and prevent neutrophil adhesion to endothelial cells. FASEB J. 2000, 14, 572–580. [Google Scholar] [CrossRef]
- Scannell, M.; Flanagan, M.B.; Destefani, A.; Wynne, K.J.; Cagney, G.; Godson, C.; Maderna, P. Annexin-1 and Peptide Derivatives Are Released by Apoptotic Cells and Stimulate Phagocytosis of Apoptotic Neutrophils by Macrophages. J. Immunol. 2007, 178, 4595–4605. [Google Scholar] [CrossRef]
- Ricci, E.; Ronchetti, S.; Pericolini, E.; Gabrielli, E.; Cari, L.; Gentili, M.; Roselletti, E.; Migliorati, G.; Vecchiarelli, A.; Riccardi, C. Role of the glucocorticoid-induced leucine zipper gene in dexamethasone-induced inhibition of mouse neutrophil migration via control of annexin A1 expression. FASEB J. 2017, 31, 3054–3065. [Google Scholar] [CrossRef]
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.E.; Yona, S.; Rosignoli, G.; Young, R.E.; Nourshargh, S.; Flower, R.J.; Perretti, M. Annexin 1-deficient neutrophils exhibit enhanced transmigration in vivo and increased respon-siveness in vitro. J. Leukoc. Biol. 2005, 78, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Frey, O.; Petrow, P.K.; Gajda, M.; Siegmund, K.; Huehn, J.; Scheffold, A.; Hamann, A.; Radbruch, A.; Bräuer, R. The role of regulatory T cells in antigen-induced arthritis: Aggravation of arthritis after depletion and amelioration after transfer of CD4+CD25+ T cells. Thromb. Haemost. 2005, 7, R291–R301. [Google Scholar] [CrossRef]
- Mozaffari, M.S. Therapeutic Potential of Annexin A1 Modulation in Kidney and Cardiovascular Disorders. Cells 2021, 10, 3420. [Google Scholar] [CrossRef]
- Everts-Graber, J.; Martin, K.R.; Thieblemont, N.; Mocek, J.; Roccabianca, A.; Chafey, P.; Le Gall, M.; Tacnet-Delorme, P.; Reutelingsperger, C.P.; Naccache, J.-M.; et al. Proteomic analysis of neutrophils in ANCA-associated vasculitis reveals a dysregulation in proteinase 3-associated proteins such as annexin-A1 involved in apoptotic cell clearance. Kidney Int. 2019, 96, 397–408. [Google Scholar] [CrossRef]
- Hajjar, K.A.; Hamel, N.M. Identification and characterization of human endothelial cell membrane binding sites for tissue plasminogen acti-vator and urokinase. J. Biol. Chem. 1990, 265, 2908–2916. [Google Scholar] [CrossRef]
- Luo, M.; Flood, E.C.; Almeida, D.; Yan, L.; Berlin, D.A.; Heerdt, P.M.; Hajjar, K.A. Annexin A2 supports pulmonary microvascular integrity by linking vascular endothelial cadherin and protein tyrosine phosphatases. J. Exp. Med. 2017, 214, 2535–2545. [Google Scholar] [CrossRef]
- Laumonnier, Y.; Syrovets, T.; Burysek, L.; Simmet, T. Identification of the annexin A2 heterotetramer as a receptor for the plasmin-induced signaling in human peripheral monocytes. Blood 2006, 107, 3342–3349. [Google Scholar] [CrossRef]
- Brownstein, C.; Deora, A.B.; Jacovina, A.T.; Weintraub, R.; Gertler, M.; Khan, K.M.F.; Falcone, D.J.; Hajjar, K.A. Annexin II mediates plasminogen-dependent matrix invasion by human monocytes: Enhanced expression by macrophages. Blood 2004, 103, 317–324. [Google Scholar] [CrossRef]
- He, S.; Li, X.; Li, R.; Fang, L.; Sun, L.; Wang, Y.; Wu, M. Annexin A2 Modulates ROS and Impacts Inflammatory Response via IL-17 Signaling in Polymicrobial Sepsis Mice. PLOS Pathog. 2016, 12, e1005743. [Google Scholar] [CrossRef]
- Ling, Q.; Jacovina, A.T.; Deora, A.; Febbraio, M.; Simantov, R.; Silverstein, R.L.; Hempstead, B.; Mark, W.H.; Hajjar, K.A. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J. Clin. Investig. 2004, 113, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Hall, P.; Hutchinson, P.; Yang, Y.H. Regulation of Annexin I in Rheumatoid Synovial Cells by Glucocorticoids and Interleukin-1. Mediat. Inflamm. 2006, 2006, 73835. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Moroni, G.; Sinico, R.A.; Franceschini, F.; Fredi, M.; Vaglio, A.; Cavagna, L.; Petretto, A.; Pratesi, F.; Migliorini, P.; et al. Neutrophil Extracellular Traps in the Autoimmunity Context. Front. Med. 2021, 8, 614829. [Google Scholar] [CrossRef] [PubMed]
- Goulding, N.J.; Jefferiss, C.M.; Pan, L.; Rigby, W.F.; Guyre, P.M. Specific binding of lipocortin-1 (annexin I) to monocytes and neutrophils is de-creased in rheumatoid arthritis. Arthritis Rheum. 1992, 35, 1395–1397. [Google Scholar] [CrossRef] [PubMed]
- Kretz, C.C.; Norpo, M.; Abeler-Dörner, L.; Linke, B.; Haust, M.; Edler, L.; Krammer, P.H.; Kuhn, A. Anti-annexin 1 antibodies: A new diagnostic marker in the serum of patients with discoid lupus erythematosus. Exp. Dermatol. 2010, 19, 919–921. [Google Scholar] [CrossRef]
- Bonanni, A.; Vaglio, A.; Bruschi, M.; Sinico, R.A.; Cavagna, L.; Moroni, G.; Franceschini, F.; Allegri, L.; Pratesi, F.; Migliorini, P.; et al. Multi-antibody composition in lupus nephritis: Isotype and antigen specificity make the difference. Autoimmun. Rev. 2015, 14, 692–702. [Google Scholar] [CrossRef]
- Bruschi, M.; Moroni, G.; Sinico, R.A.; Franceschini, F.; Fredi, M.; Vaglio, A.; Cavagna, L.; Petretto, A.; Pratesi, F.; Migliorini, P.; et al. Serum IgG2 antibody multicomposition in systemic lupus erythematosus and lupus nephritis (Part 1): Cross-sectional analysis. Rheumatology 2020, 60, 3176–3188. [Google Scholar] [CrossRef]
- Bruschi, M.; Angeletti, A.; Kajana, X.; Moroni, G.; Sinico, R.A.; Fredi, M.; Vaglio, A.; Cavagna, L.; Pratesi, F.; Migliorini, P.; et al. Evidence for charge-based mimicry in anti dsDNA antibody generation. J. Autoimmun. 2022, 132, 102900. [Google Scholar] [CrossRef]
- Cesarman-Maus, G.C.; Ríos-Luna, N.P.; Deora, A.B.; Huang, B.; Villa, R.; Cravioto, M.D.C.; Alarcón-Segovia, D.; Sánchez-Guerrero, J.; Hajjar, K.A. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood 2006, 107, 4375–4382. [Google Scholar] [CrossRef]
- Ao, W.; Zheng, H.; Chen, X.W.; Shen, Y.; Yang, C.D. Anti-annexin II antibody is associated with thrombosis and/or pregnancy morbidity in an-tiphospholipid syndrome and systemic lupus erythematosus with thrombosis. Rheumatol. Int. 2011, 31, 865–869. [Google Scholar] [CrossRef]
- Yung, S.; Cheung, K.F.; Zhang, Q.; Chan, T.M. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J. Am. Soc. Nephrol. 2010, 21, 1912–1927. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zhang, Y.; Zhuang, J.; Bi, Y.; Xu, H.; Shen, Q.; Liu, J.; Fu, H.; Wang, J.; Feng, C.; et al. The important roles and molecular mechanisms of annexin A2 autoantibody in children with nephrotic syndrome. Ann. Transl. Med. 2021, 9, 1452. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, F.; Castiglione, A.; Colasanti, G.; di Belgioioso, G.B.; Bertoli, S.; D’Amico, G.; Nava, S. The detection of monocytes in human glomerulonephritis. Kidney Int. 1985, 28, 513–519. [Google Scholar] [CrossRef]
- Nolasco, F.E.; Cameron, J.S.; Hartley, B.; Coelho, A.; Hildreth, G.; Reuben, R. Intraglomerular T cells and monocytes in nephritis: Study with mono-clonal antibodies. Kidney Int. 1987, 31, 1160–1166. [Google Scholar] [CrossRef]
- Bolton, W.K.; Innes, D.J., Jr.; Sturgill, B.C.; Kaiser, D.L. T-cells and macrophages in rapidly progressive glomerulonephritis: Clinicopathologic correla-tions. Kidney Int. 1987, 32, 869–876. [Google Scholar] [CrossRef]
- Moll, S.; Angeletti, A.; Scapozza, L.; Cavalli, A.; Ghiggeri, G.M.; Prunotto, M. Glomerular Macrophages in Human Auto- and Allo-Immune Nephritis. Cells 2021, 10, 603. [Google Scholar] [CrossRef]
- Bohle, A.; Wehrmann, M.; Bogenschutz, O.; Batz, C.; Vog, W.; Schmitt, H.; Müller, C.A.; Müller, G.A. The long-term prognosis of the primary glomerulonephritides. A morphological and clinical analysis of 1747 cases. Pathol. Res. Pract. 1992, 188, 908–924. [Google Scholar] [CrossRef]
- Rodríguez-Iturbe, B.; Pons, H.; Herrera-Acosta, J.; Johnson, R.J. Role of immunocompetent cells in nonimmune renal diseases. Kidney Int. 2001, 59, 1626–1640. [Google Scholar] [CrossRef]
- Wang, Y.; Harris, D.C. Macrophages in Renal Disease. J. Am. Soc. Nephrol. 2011, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Cailhier, J.F.; Partolina, M.; Vuthoori, S.; Wu, S.; Ko, K.; Watson, S.; Savill, J.; Hughes, J.; Lang, R.A. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peri-toneal inflammation. J. Immunol. 2005, 174, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.-I.; Santos, K.D.L.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F.; et al. In Vivo Depletion of CD11c+ Dendritic Cells Abrogates Priming of CD8+ T Cells by Exogenous Cell-Associated Antigens. Immunity 2002, 17, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.J.; Chaves, L.D.; Chang, A.; Jacob, A.; Ritchie, M.; Quigg, R.J. CD11b is protective in complement-mediated immune complex glomeru-lonephritis. Kidney Int. 2015, 87, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, G.; Lee, V.; Ouyang, L.; Chang, D.; Mahajan, D.; Coombs, J.; Alexander, S.; Harris, D. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007, 72, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wang, Y.; Zheng, D.; Sun, Y.; Wang, Y.; Lee, V.W.S.; Zheng, G.; Tan, T.K.; Ince, J.; Alexander, S.; et al. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J. Am. Soc. Nephrol. 2010, 21, 933–942. [Google Scholar] [CrossRef]
- Lu, J.; Cao, Q.; Zheng, D.; Sun, Y.; Wang, C.; Yu, X.; Wang, Y.; Lee, V.W.; Zheng, G.; Tan, T.K.; et al. Discrete functions of M 2a and M 2c macrophage subsets determine their relative efficacy in treating chronic kidney disease. Kidney Int. 2013, 84, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Petretto, A.; Santucci, L.; Vaglio, A.; Pratesi, F.; Migliorini, P.; Bertelli, R.; Lavarello, C.; Bartolucci, M.; Candiano, G.; et al. Neutrophil Extracellular Traps protein composition is specific for patients with Lupus nephritis and includes methyl-oxidized alphaenolase (methionine sulfoxide 93). Sci. Rep. 2019, 9, 7934. [Google Scholar] [CrossRef]
- Bello-Gamboa, A.; Velasco, M.; Moreno, S.; Herranz, G.; Ilie, R.; Huetos, S.; Dávila, S.; Sánchez, A.; De La Serna, J.B.; Calvo, V.; et al. Actin reorganization at the centrosomal area and the immune synapse regulates polarized secretory traffic of multivesicular bodies in T lymphocytes. J. Extracell. Vesicles 2020, 9, 1759926. [Google Scholar] [CrossRef]
- Kumari, S.; Colin-York, H.; Irvine, D.J.; Fritzsche, M. Not All T Cell Synapses Are Built the Same Way. Trends Immunol. 2019, 40, 977–980. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angeletti, A.; Bruschi, M.; Kajana, X.; Spinelli, S.; Verrina, E.; Lugani, F.; Caridi, G.; Murtas, C.; Candiano, G.; Prunotto, M.; et al. Mechanisms Limiting Renal Tissue Protection and Repair in Glomerulonephritis. Int. J. Mol. Sci. 2023, 24, 8318. https://doi.org/10.3390/ijms24098318
Angeletti A, Bruschi M, Kajana X, Spinelli S, Verrina E, Lugani F, Caridi G, Murtas C, Candiano G, Prunotto M, et al. Mechanisms Limiting Renal Tissue Protection and Repair in Glomerulonephritis. International Journal of Molecular Sciences. 2023; 24(9):8318. https://doi.org/10.3390/ijms24098318
Chicago/Turabian StyleAngeletti, Andrea, Maurizio Bruschi, Xuliana Kajana, Sonia Spinelli, Enrico Verrina, Francesca Lugani, Gialuca Caridi, Corrado Murtas, Giovanni Candiano, Marco Prunotto, and et al. 2023. "Mechanisms Limiting Renal Tissue Protection and Repair in Glomerulonephritis" International Journal of Molecular Sciences 24, no. 9: 8318. https://doi.org/10.3390/ijms24098318
APA StyleAngeletti, A., Bruschi, M., Kajana, X., Spinelli, S., Verrina, E., Lugani, F., Caridi, G., Murtas, C., Candiano, G., Prunotto, M., & Ghiggeri, G. M. (2023). Mechanisms Limiting Renal Tissue Protection and Repair in Glomerulonephritis. International Journal of Molecular Sciences, 24(9), 8318. https://doi.org/10.3390/ijms24098318