
FRA-1 as a Regulator of EMT and Metastasis in Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The AP-1 Family of Transcription Factors
2.1. FRA-1 Structure and Regulation
2.2. FRA-1 in Tumorigenesis
2.3. FRA-1 Controls Breast Cancer Cell Motility, Invasion, and Proliferation
3. FRA-1 Is Required for EMT and Gain of Stem-like Features
3.1. FRA-1 Is Both a Transcriptional Inducer and Target of Multiple EMT-TFs

3.2. The FRA-1 Target Genes in the Paracrine Control of EMT and Extracellular Proteolysis
3.3. The FRA-1-Regulated Genes as Therapeutic Targets
3.4. The Role of FRA-1-Repressed Genes in the Control of EMT
3.5. FRA-1 Effects on the Architecture of Target Promoters
3.6. Diagnostic and Prognostic Significance of the FRA-1 Oncoprotein and FRA-1-Derived Signatures in TNBC
4. Open Questions and Novel Perspectives
4.1. The Role of Long Non-Coding RNAs as Downstream Effectors of FRA-1
4.2. The In Vivo Analysis of Fra-1 Functions in Mouse Models of TNBC
4.3. FRA-1 in Tumor Microenvironment
4.4. FRA-1 in the Control of the Hybrid E/M (Partial EMT) Phenotypes
4.5. CTCs (Circulating Tumor Cells) and PDXs as Tools for In Vivo Studies on FRA-1 in Human Cancer
4.6. FOSL1/FRA-1 as a Target of Therapeutic Intervention in TNBC/Basal-like Breast Cancer
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Lawrence, T.S.; Rosenberg, S.A. Cancer: Principles & Practice of Oncology: Primer of the Molecular Biology of Cancer; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011. [Google Scholar]
- Van Dam, P.-J.; Van Laere, S. Molecular profiling in cancer research and personalized medicine. In Oxford Textbook of Cancer Biology; Pezzella, F., Tavassoli, M., Kerr, D.J., Eds.; Oxford University Press: Oxford, UK, 2019; pp. 347–362. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Feldker, N.; Ferrazzi, F.; Schuhwerk, H.; Widholz, S.A.; Guenther, K.; Frisch, I.; Jakob, K.; Kleemann, J.; Riegel, D.; Bönisch, U.; et al. Genome-wide cooperation of EMT transcription factor ZEB 1 with YAP and AP-1 in breast cancer. EMBO J. 2020, 39, e103209. [Google Scholar] [CrossRef] [PubMed]
- Desmet, C.J.; Gallenne, T.; Prieur, A.; Reyal, F.; Visser, N.L.; Wittner, B.S.; Smit, M.A.; Geiger, T.R.; Laoukili, J.; Iskit, S.; et al. Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 5139–5144. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Qiao, Y.; Jonsson, P.; Wang, J.; Xu, L.; Rouhi, P.; Sinha, I.; Cao, Y.; Williams, C.; Dahlman-Wright, K. Genome-wide Profiling of AP-1–Regulated Transcription Provides Insights into the Invasiveness of Triple-Negative Breast Cancer. Cancer Res. 2014, 74, 3983–3994. [Google Scholar] [CrossRef]
- Bejjani, F.; Tolza, C.; Boulanger, M.; Downes, D.; Romero, R.; Maqbool, M.A.; El Aabidine, A.Z.; Andrau, J.-C.; Lebre, S.; Brehelin, L.; et al. Fra-1 regulates its target genes via binding to remote enhancers without exerting major control on chromatin architecture in triple negative breast cancers. Nucleic Acids Res. 2021, 49, 2488–2508. [Google Scholar] [CrossRef]
- Lambert, A.W.; Weinberg, R.A. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer 2021, 21, 325–338. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Weinberg, R.A. The Biology of Cancer; W.W. Norton & Company: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Fougner, C.; Bergholtz, H.; Norum, J.H.; Sørlie, T. Re-definition of claudin-low as a breast cancer phenotype. Nat. Commun. 2020, 11, 1787. [Google Scholar] [CrossRef]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFβ/TNFα-Mediated Epithelial–Mesenchymal Transition Generates Breast Cancer Stem Cells with a Claudin-Low Phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the Interaction of BRD4 with Diacetylated Twist Suppresses Tumorigenesis in Basal-like Breast Cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, Z.; Lin, X.; Tian, L.; Su, Q.; An, P.; Li, W.; Wu, Y.; Du, J.; Shan, H.; et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020, 27, 255–268. [Google Scholar] [CrossRef]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein Kinase C α Is a Central Signaling Node and Therapeutic Target for Breast Cancer Stem Cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, H.; Zheng, L. Research progress on RNA-binding proteins in breast cancer. Front. Oncol. 2022, 12, 974523. [Google Scholar] [CrossRef]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, X.-J.; Sun, X.; Xia, T.-S.; Li, X.-X.; Shi, L.; Zhu, L.; Zhou, W.-B.; Wei, J.-F.; Ding, Q. RBM38 is involved in TGF-β-induced epithelial-to-mesenchymal transition by stabilising zonula occludens-1 mRNA in breast cancer. Br. J. Cancer 2017, 117, 675–684. [Google Scholar] [CrossRef]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 11–23. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E. AP-1—The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell. Signal. 2010, 22, 894–899. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Tulchinsky, E. FRA-1 as a driver of tumour heterogeneity: A nexus between oncogenes and embryonic signalling pathways in cancer. Oncogene 2015, 34, 4421–4428. [Google Scholar] [CrossRef] [PubMed]
- Talotta, F.; Casalino, L.; Verde, P. The nuclear oncoprotein Fra-1: A transcription factor knocking on therapeutic applications’ door. Oncogene 2020, 39, 4491–4506. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Xie, H.; Dou, Y.; Yuan, J.; Zeng, D.; Xiao, S. Expression and function of FRA1 protein in tumors. Mol. Biol. Rep. 2020, 47, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, V.V.; Khashukoeva, A.Z.; Evina, O.E.; Geppe, N.A.; Chebysheva, S.N.; Korsunskaya, I.M.; Tchepourina, E.; Mezentsev, A. Role of the Transcription Factor FOSL1 in Organ Development and Tumorigenesis. Int. J. Mol. Sci. 2022, 23, 1521. [Google Scholar] [CrossRef]
- Zippo, A.; De Robertis, A.; Serafini, R.; Oliviero, S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat. Cell Biol. 2007, 9, 932–944. [Google Scholar] [CrossRef]
- Zippo, A.; Serafini, R.; Rocchigiani, M.; Pennacchini, S.; Krepelova, A.; Oliviero, S. Histone Crosstalk between H3S10ph and H4K16ac Generates a Histone Code that Mediates Transcription Elongation. Cell 2009, 138, 1122–1136. [Google Scholar] [CrossRef]
- Shu, L.; Chen, A.; Li, L.; Yao, L.; He, Y.; Xu, J.; Gu, W.; Li, Q.; Wang, K.; Zhang, T.; et al. NRG1 regulates Fra-1 transcription and metastasis of triple-negative breast cancer cells via the c-Myc ubiquitination as manipulated by ERK1/2-mediated Fbxw7 phosphorylation. Oncogene 2022, 41, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, Y.; Gao, J.; Zhang, T.; Li, S.; Luo, A.; Chen, H.; Ding, F.; Wang, X.; Liu, Z. MicroRNA-34 suppresses breast cancer invasion and metastasis by directly targeting Fra-1. Oncogene 2013, 32, 4294–4303. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, M.; Huang, J.; Li, Y.; Wang, S.; Harrington, C.A.; Qian, D.Z.; Sun, X.; Dai, M. microRNA-130a suppresses breast cancer cell migration and invasion by targeting FOSL1 and upregulating ZO-1. J. Cell Biochem. 2018, 119, 4945–4956. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.; Chen, C.; Liu, Y.; Si, Q.; Chuang, T.-H.; Li, N.; Gomez-Cabrero, A.; Reisfeld, R.A.; Xiang, R.; et al. MicroRNA-19a-3p inhibits breast cancer progression and metastasis by inducing macrophage polarization through downregulated expression of Fra-1 proto-oncogene. Oncogene 2014, 33, 3014–3023. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, B.G.; Jang, Y.; Kang, S.; Lee, J.H.; Cho, N.H. The stromal loss of miR-4516 promotes the FOSL1-dependent proliferation and malignancy of triple negative breast cancer. Cancer Lett. 2020, 469, 256–265. [Google Scholar] [CrossRef]
- Di Fazio, P. The epitranscriptome: At the crossroad of cancer prognosis. eBioMedicine 2021, 64, 103231. [Google Scholar] [CrossRef]
- Fan, D.; Liu, B.; Gu, X.; Zhang, Q.; Ye, Q.; Xi, X.; Xia, Y.; Wang, Q.; Wang, Z.; Wang, B.; et al. Potential Target Analysis of Triptolide Based on Transcriptome-Wide m6A Methylome in Rheumatoid Arthritis. Front. Pharmacol. 2022, 13, 843358. [Google Scholar] [CrossRef]
- Ramesh-Kumar, D.; Guil, S. The IGF2BP family of RNA binding proteins links epitranscriptomics to cancer. Semin. Cancer Biol. 2022, 86, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Hu, J.; Zhang, H.; Li, W.; Jia, M.; Zhang, X.; Wu, X.; Cheng, H.; Xiang, L.; Zhou, G. IMP3 expression is associated with epithelial-mesenchymal transition in breast cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 3008–3017. [Google Scholar] [PubMed]
- Zirkel, A.; Lederer, M.; Stöhr, N.; Pazaitis, N.; Hüttelmaier, S. IGF2BP1 promotes mesenchymal cell properties and migration of tumor-derived cells by enhancing the expression of LEF1 and SNAI2 (SLUG). Nucleic Acids Res. 2013, 41, 6618–6636. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, Q.; Zhang, G.; Mohammed, D.; Amadou, S.; Tan, G.; Zhang, X. HOXA11-AS1 Promotes PD-L1-Mediated Immune Escape and Metastasis of Hypopharyngeal Carcinoma by Facilitating PTBP1 and FOSL1 Association. Cancers 2022, 14, 3694. [Google Scholar] [CrossRef]
- Hou, P.; Li, L.; Chen, F.; Chen, Y.; Liu, H.; Li, J.; Bai, J.; Zheng, J. PTBP3-Mediated Regulation of ZEB1 mRNA Stability Promotes Epithelial–Mesenchymal Transition in Breast Cancer. Cancer Res. 2018, 78, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Basbous, J.; Chalbos, D.; Hipskind, R.; Jariel-Encontre, I.; Piechaczyk, M. Ubiquitin-Independent Proteasomal Degradation of Fra-1 Is Antagonized by Erk1/2 Pathway-Mediated Phosphorylation of a Unique C-Terminal Destabilizer. Mol. Cell. Biol. 2007, 27, 3936–3950. [Google Scholar] [CrossRef]
- Belguise, K.; Milord, S.; Galtier, F.; Moquet-Torcy, G.; Piechaczyk, M.; Chalbos, D. The PKCθ pathway participates in the aberrant accumulation of Fra-1 protein in invasive ER-negative breast cancer cells. Oncogene 2012, 31, 4889–4897. [Google Scholar] [CrossRef]
- Belguise, K.; Cherradi, S.; Sarr, A.; Boissière, F.; Boulle, N.; Simony-Lafontaine, J.; Choesmel-Cadamuro, V.; Wang, X.; Chalbos, D. PKCθ-induced phosphorylations control the ability of Fra-1 to stimulate gene expression and cancer cell migration. Cancer Lett. 2017, 385, 97–107. [Google Scholar] [CrossRef]
- Rattanasinchai, C.; Llewellyn, B.J.; Conrad, S.E.; Gallo, K.A. MLK3 regulates FRA-1 and MMPs to drive invasion and transendothelial migration in triple-negative breast cancer cells. Oncogenesis 2017, 6, e345. [Google Scholar] [CrossRef] [PubMed]
- Tulchinsky, E. Fos family members: Regulation, structure and role in oncogenic transformation. Histol. Histopathol. 2000, 15, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Milde-Langosch, K. The Fos family of transcription factors and their role in tumourigenesis. Eur. J. Cancer 2005, 41, 2449–2461. [Google Scholar] [CrossRef] [PubMed]
- Young, M.R.; Colburn, N.H. Fra-1 a target for cancer prevention or intervention. Gene 2006, 379, 1–11. [Google Scholar] [CrossRef]
- Verde, P.; Casalino, L.; Talotta, F.; Yaniv, M.; Weitzman, J.B. Deciphering AP-1 Function in Tumorigenesis: Fra-ternizing on Target Promoters. Cell Cycle 2007, 6, 2633–2639. [Google Scholar] [CrossRef]
- Casalino, L.; Talotta, F.; Cimmino, A.; Verde, P. The Fra-1/AP-1 Oncoprotein: From the “Undruggable” Transcription Factor to Therapeutic Targeting. Cancers 2022, 14, 1480. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; He, J.; Jin, X.; Liao, Q.; Chen, Z.; Peng, H.; Zhou, Y. FRA-1: A key factor regulating signal transduction of tumor cells and a potential target molecule for tumor therapy. Biomed. Pharmacother. 2022, 150, 113037. [Google Scholar] [CrossRef] [PubMed]
- Kustikova, O.; Kramerov, D.; Grigorian, M.; Berezin, V.; Bock, E.; Lukanidin, E.; Tulchinsky, E. Fra-1 Induces Morphological Transformation and Increases In Vitro Invasiveness and Motility of Epithelioid Adenocarcinoma Cells. Mol. Cell. Biol. 1998, 18, 7095–7105. [Google Scholar] [CrossRef]
- Zajchowski, D.A.; Bartholdi, M.F.; Gong, Y.; Webster, L.; Liu, H.L.; Munishkin, A.; Beauheim, C.; Harvey, S.; Ethier, S.P.; Johnson, P.H. Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res. 2001, 61, 5168–5178. [Google Scholar]
- Belguise, K.; Kersual, N.; Galtier, F.; Chalbos, D. FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene 2005, 24, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Dimitri, C.A.; Yoon, S.-O.; Dowdle, W.; Blenis, J. ERK2 but Not ERK1 Induces Epithelial-to-Mesenchymal Transformation via DEF Motif-Dependent Signaling Events. Mol. Cell 2010, 38, 114–127. [Google Scholar] [CrossRef]
- Shin, S.; Blenis, J. ERK2/Fra1/ZEB pathway induces epithelial-to-mesenchymal transition. Cell Cycle 2010, 9, 2483–2484. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Wilson, M.M.; Weinberg, R.A.; Lees, J.A.; Guen, V.J. Emerging Mechanisms by which EMT Programs Control Stemness. Trends Cancer 2020, 6, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Wu, F.; Hardy, K.; Li, J.; Tu, W.J.; McCuaig, R.; Harris, J.; Khanna, K.K.; Attema, J.; Gregory, P.A.; et al. Chromatinized Protein Kinase C-θ Directly Regulates Inducible Genes in Epithelial to Mesenchymal Transition and Breast Cancer Stem Cells. Mol. Cell. Biol. 2014, 34, 2961–2980. [Google Scholar] [CrossRef] [PubMed]
- Boulding, T.; McCuaig, R.D.; Tan, A.; Hardy, K.; Wu, F.; Dunn, J.; Kalimutho, M.; Sutton, C.R.; Forwood, J.K.; Bert, A.G.; et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci. Rep. 2018, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhu, G.; Li, Y.; Padia, R.N.; Dong, Z.; Pan, Z.K.; Liu, K.; Huang, S. Extracellular Signal–Regulated Kinase Signaling Pathway Regulates Breast Cancer Cell Migration by Maintaining slug Expression. Cancer Res. 2009, 69, 9228–9235. [Google Scholar] [CrossRef] [PubMed]
- Bakiri, L.; Macho-Maschler, S.; Custic, I.; Niemiec, J.; Guío-Carrión, A.; Hasenfuss, S.C.; Eger, A.; Müller, M.; Beug, H.; Wagner, E.F. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death Differ. 2015, 22, 336–350. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.-C. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial–mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
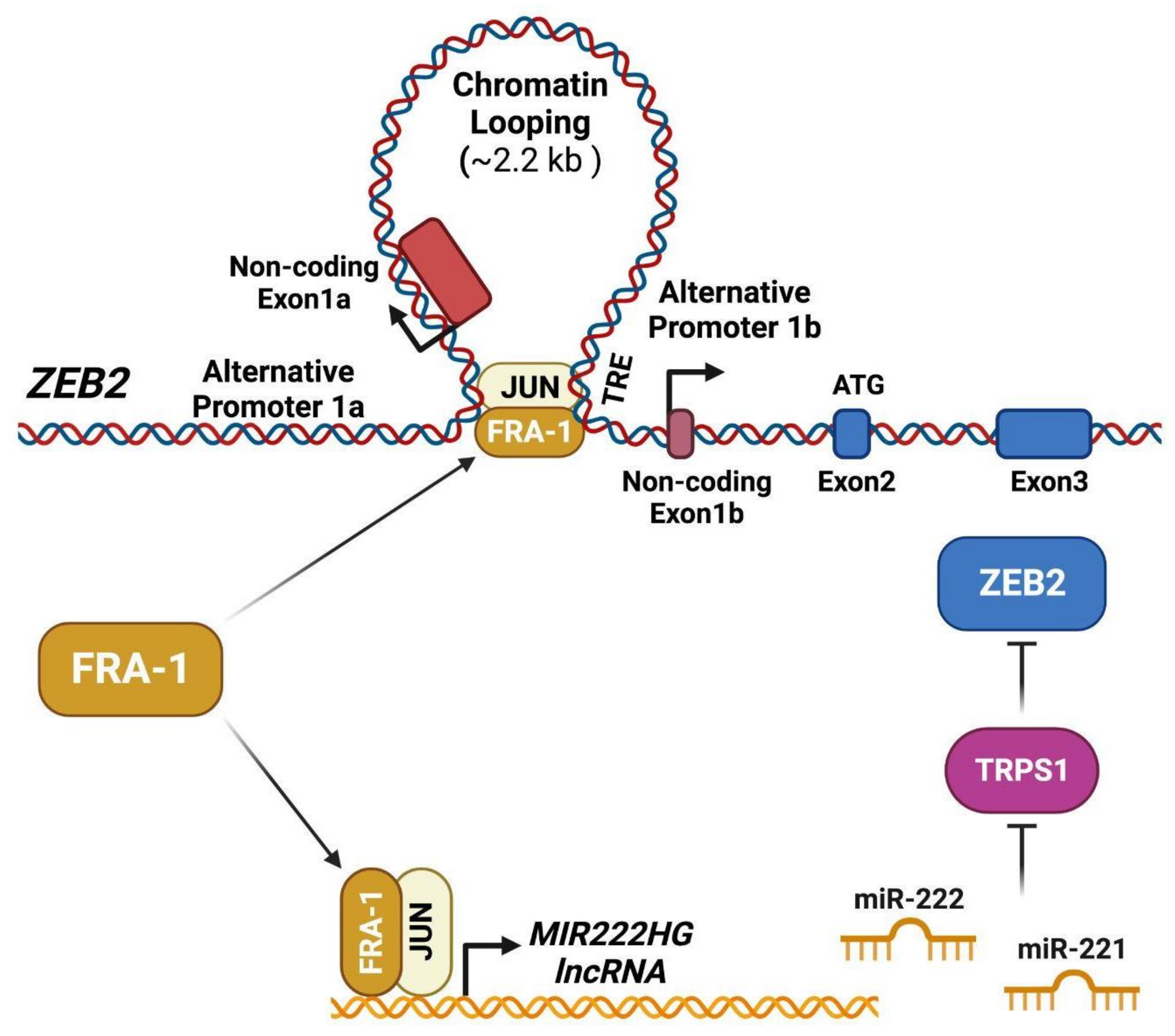
- Qiao, Y.; Shiue, C.-N.; Zhu, J.; Zhuang, T.; Jonsson, P.; Wright, A.P.H.; Zhao, C.; Dahlman-Wright, K. AP-1-mediated chromatin looping regulates ZEB2 transcription: New insights into TNFα-induced epithelial-mesenchymal transition in triple-negative breast cancer. Oncotarget 2015, 6, 7804–7814. [Google Scholar] [CrossRef]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.-J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. TRPS1 Targeting by miR-221/222 Promotes the Epithelial-to-Mesenchymal Transition in Breast Cancer. Sci. Signal. 2011, 4, ra41. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Ndlovu, M.N.; Van Lint, C.; Van Wesemael, K.; Callebert, P.; Chalbos, D.; Haegeman, G.; Vanden Berghe, W. Hyperactivated NF-κB and AP-1 Transcription Factors Promote Highly Accessible Chromatin and Constitutive Transcription across the Interleukin-6 Gene Promoter in Metastatic Breast Cancer Cells. Mol. Cell. Biol. 2009, 29, 5488–5504. [Google Scholar] [CrossRef]
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010, 20, 701–712. [Google Scholar] [CrossRef]
- Luo, Y.P.; Zhou, H.; Krueger, J.; Kaplan, C.; Liao, D.; Markowitz, D.; Liu, C.; Chen, T.; Chuang, T.-H.; Xiang, R.; et al. The role of proto-oncogene Fra-1 in remodeling the tumor microenvironment in support of breast tumor cell invasion and progression. Oncogene 2010, 29, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Moquet-Torcy, G.; Tolza, C.; Piechaczyk, M.; Jariel-Encontre, I. Transcriptional complexity and roles of Fra-1/AP-1 at the uPA/Plau locus in aggressive breast cancer. Nucleic Acids Res. 2014, 42, 11011–11024. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Qian, L.; Song, S.; Chen, L.; Zhang, Y.; Yuan, G.; Zhang, H.; Xia, Q.; Hu, M.; Yu, M.; et al. Fra-1 and Stat3 synergistically regulate activation of human MMP-9 gene. Mol. Immunol. 2008, 45, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Moirangthem, A.; Bondhopadhyay, B.; Mukherjee, M.; Bandyopadhyay, A.; Mukherjee, N.; Konar, K.; Bhattacharya, S.; Basu, A. Simultaneous knockdown of uPA and MMP9 can reduce breast cancer progression by increasing cell-cell adhesion and modulating EMT genes. Sci. Rep. 2016, 6, 21903. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Leupold, J.H.; Asangani, I.; Maurer, G.D.; Lengyel, E.; Post, S.; Allgayer, H. Src Induces Urokinase Receptor Gene Expression and Invasion/Intravasation via Activator Protein-1/p-c-Jun in Colorectal Cancer. Mol. Cancer Res. 2007, 5, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.D.; Jo, M.; Montel, V.; Takimoto, S.; Gonias, S.L. uPAR induces epithelial–mesenchymal transition in hypoxic breast cancer cells. J. Cell Biol. 2007, 178, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Vial, E.; Sahai, E.; Marshall, C.J. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell 2003, 4, 67–79. [Google Scholar] [CrossRef]
- Annis, M.G.; Ouellet, V.; Rennhack, J.P.; L’Esperance, S.; Rancourt, C.; Mes-Masson, A.-M.; Andrechek, E.R.; Siegel, P.M. Integrin-uPAR signaling leads to FRA-1 phosphorylation and enhanced breast cancer invasion. Breast Cancer Res. 2018, 20, 9. [Google Scholar] [CrossRef]
- Duffy, M.J.; McGowan, P.M.; Harbeck, N.; Thomssen, C.; Schmitt, M. uPA and PAI-1 as biomarkers in breast cancer: Validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014, 16, 428. [Google Scholar] [CrossRef]
- Sundqvist, A.; Zieba, A.; Vasilaki, E.; Herrera Hidalgo, C.; Söderberg, O.; Koinuma, D.; Miyazono, K.; Heldin, C.-H.; Landegren, U.; Ten Dijke, P.; et al. Specific interactions between Smad proteins and AP-1 components determine TGFβ-induced breast cancer cell invasion. Oncogene 2013, 32, 3606–3615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, J.; Fang, H.; Tang, L.; Chen, W.; Sun, Q.; Zhang, Q.; Yang, F.; Sun, Z.; Cao, L.; et al. Endothelial cells promote triple-negative breast cancer cell metastasis via PAI-1 and CCL5 signaling. FASEB J. 2018, 32, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Resmini, G.; Rizzo, S.; Franchin, C.; Zanin, R.; Penzo, C.; Pegoraro, S.; Ciani, Y.; Piazza, S.; Arrigoni, G.; Sgarra, R.; et al. HMGA1 regulates the Plasminogen activation system in the secretome of breast cancer cells. Sci. Rep. 2017, 7, 11768. [Google Scholar] [CrossRef] [PubMed]
- Tolza, C.; Bejjani, F.; Evanno, E.; Mahfoud, S.; Moquet-Torcy, G.; Gostan, T.; Maqbool, M.A.; Kirsh, O.; Piechaczyk, M.; Jariel-Encontre, I. AP-1 Signaling by Fra-1 Directly Regulates HMGA1 Oncogene Transcription in Triple-Negative Breast Cancers. Mol. Cancer Res. 2019, 17, 1999–2014. [Google Scholar] [CrossRef]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef]
- Dhar, A.; Hu, J.; Reeves, R.; Resar, L.M.; Colburn, N.H. Dominant-negative c-Jun (TAM67) target genes: HMGA1 is required for tumor promoter-induced transformation. Oncogene 2004, 23, 4466–4476. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Pegoraro, S.; Ros, G.; Piazza, S.; Sommaggio, R.; Ciani, Y.; Rosato, A.; Sgarra, R.; Del Sal, G.; Manfioletti, G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013, 4, 1293–1308. [Google Scholar] [CrossRef]
- Zanin, R.; Pegoraro, S.; Ros, G.; Ciani, Y.; Piazza, S.; Bossi, F.; Bulla, R.; Zennaro, C.; Tonon, F.; Lazarevic, D.; et al. HMGA1 promotes breast cancer angiogenesis supporting the stability, nuclear localization and transcriptional activity of FOXM1. J. Exp. Clin. Cancer Res. 2019, 38, 313. [Google Scholar] [CrossRef]
- Sayan, A.E.; Stanford, R.; Vickery, R.; Grigorenko, E.; Diesch, J.; Kulbicki, K.; Edwards, R.; Pal, R.; Greaves, P.; Jariel-Encontre, I.; et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2012, 31, 1493–1503. [Google Scholar] [CrossRef]
- Gjerdrum, C.; Tiron, C.; Høiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef]
- Leconet, W.; Chentouf, M.; Du Manoir, S.; Chevalier, C.; Sirvent, A.; Aït-Arsa, I.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Theillet, C.; et al. Therapeutic Activity of Anti-AXL Antibody against Triple-Negative Breast Cancer Patient-Derived Xenografts and Metastasis. Clin. Cancer Res. 2017, 23, 2806–2816. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Zhang, Y.; Wang, J.; Yu, Q.; Peng, X.; Zou, J.; Zhou, L.; Tan, L.; Duan, Y.; Zhou, Y.; et al. Discovery of 3-Aminopyrazole Derivatives as New Potent and Orally Bioavailable AXL Inhibitors. J. Med. Chem. 2022, 65, 15374–15390. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef] [PubMed]
- Illiano, M.; Sapio, L.; Salzillo, A.; Capasso, L.; Caiafa, I.; Chiosi, E.; Spina, A.; Naviglio, S. Forskolin improves sensitivity to doxorubicin of triple negative breast cancer cells via Protein Kinase A-mediated ERK1/2 inhibition. Biochem. Pharmacol. 2018, 152, 104–113. [Google Scholar] [CrossRef]
- Walia, V.; Ding, M.; Kumar, S.; Nie, D.; Premkumar, L.S.; Elble, R.C. hCLCA2 Is a p53-Inducible Inhibitor of Breast Cancer Cell Proliferation. Cancer Res. 2009, 69, 6624–6632. [Google Scholar] [CrossRef] [PubMed]
- Walia, V.; Yu, Y.; Cao, D.; Sun, M.; McLean, J.R.; Hollier, B.G.; Cheng, J.; Mani, S.A.; Rao, K.; Premkumar, L.; et al. Loss of breast epithelial marker hCLCA2 promotes epithelial-to-mesenchymal transition and indicates higher risk of metastasis. Oncogene 2012, 31, 2237–2246. [Google Scholar] [CrossRef]
- Ramena, G.; Yin, Y.; Yu, Y.; Walia, V.; Elble, R.C. CLCA2 Interactor EVA1 Is Required for Mammary Epithelial Cell Differentiation. PLoS ONE 2016, 11, e0147489. [Google Scholar] [CrossRef]
- Cornelissen, L.M.; Drenth, A.P.; Van Der Burg, E.; De Bruijn, R.; Pritchard, C.E.J.; Huijbers, I.J.; Zwart, W.; Jonkers, J. TRPS1 acts as a context-dependent regulator of mammary epithelial cell growth/differentiation and breast cancer development. Genes Dev. 2020, 34, 179–193. [Google Scholar] [CrossRef]
- Wang, H.; Tan, Z.; Hu, H.; Liu, H.; Wu, T.; Zheng, C.; Wang, X.; Luo, Z.; Wang, J.; Liu, S.; et al. microRNA-21 promotes breast cancer proliferation and metastasis by targeting LZTFL1. BMC Cancer 2019, 19, 738. [Google Scholar] [CrossRef]
- Sartorelli, V.; Lauberth, S.M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 2020, 27, 521–528. [Google Scholar] [CrossRef]
- Milde-Langosch, K.; Janke, S.; Wagner, I.; Schröder, C.; Streichert, T.; Bamberger, A.-M.; Jänicke, F.; Löning, T. Role of Fra-2 in breast cancer: Influence on tumor cell invasion and motility. Breast Cancer Res. Treat. 2008, 107, 337–347. [Google Scholar] [CrossRef]
- He, H.; Song, D.; Sinha, I.; Hessling, B.; Li, X.; Haldosen, L.-A.; Zhao, C. Endogenous interaction profiling identifies DDX5 as an oncogenic coactivator of transcription factor Fra-1. Oncogene 2019, 38, 5725–5738. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Liu, Z.-R. P68 RNA Helicase Mediates PDGF-Induced Epithelial Mesenchymal Transition by Displacing Axin from β-Catenin. Cell 2006, 127, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Panchbhai, N.; Turaga, C.R.; Sharma, M.; Satyanarayana, G.; Liu, Z.-R. P68 RNA Helicase facilitates Breast Cancer progression by promoting Proliferation and Migration via PDGFR-β/AR axis. J. Cancer 2021, 12, 6543–6552. [Google Scholar] [CrossRef]
- Song, D.; He, H.; Sinha, I.; Hases, L.; Yan, F.; Archer, A.; Haldosen, L.-A.; Zhao, C.; Williams, C. Blocking Fra-1 sensitizes triple-negative breast cancer to PARP inhibitor. Cancer Lett. 2021, 506, 23–34. [Google Scholar] [CrossRef]
- Ordonez, L.D.; Hay, T.; McEwen, R.; Polanska, U.M.; Hughes, A.; Delpuech, O.; Cadogan, E.; Powell, S.; Dry, J.; Tornillo, G.; et al. Rapid activation of epithelial-mesenchymal transition drives PARP inhibitor resistance in Brca2 -mutant mammary tumours. Oncotarget 2019, 10, 2586–2606. [Google Scholar] [CrossRef]
- Bamberger, A.-M.; Methner, C.; Lisboa, B.W.; Städtler, C.; Schulte, H.M.; Löning, T.; Milde-Langosch, K. Expression pattern of the AP-1 family in breast cancer: Association of fosB expression with a well-differentiated, receptor-positive tumor phenotype. Int. J. Cancer 1999, 84, 533–538. [Google Scholar] [CrossRef]
- Kharman-Biz, A.; Gao, H.; Ghiasvand, R.; Zhao, C.; Zendehdel, K.; Dahlman-Wright, K. Expression of activator protein-1 (AP-1) family members in breast cancer. BMC Cancer 2013, 13, 441. [Google Scholar] [CrossRef]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; de Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The Snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 2017, 36, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liao, W.; Wu, Q.; Huang, X.; Pan, Z.; Chen, W.; Gu, S.; Huang, Z.; Wang, Y.; Tang, X.; et al. LINC00152 upregulates ZEB1 expression and enhances epithelial-mesenchymal transition and oxaliplatin resistance in esophageal cancer by interacting with EZH2. Cancer Cell Int. 2020, 20, 569. [Google Scholar] [CrossRef] [PubMed]
- Gallenne, T.; Ross, K.N.; Visser, N.L.; Salony; Desmet, C.J.; Wittner, B.S.; Wessels, L.F.A.; Ramaswamy, S.; Peeper, D.S. Systematic functional perturbations uncover a prognostic genetic network driving human breast cancer. Oncotarget 2017, 8, 20572–20587. [Google Scholar] [CrossRef]
- Zhou, W.; Ye, X.; Xu, J.; Cao, M.-G.; Fang, Z.-Y.; Li, L.-Y.; Guan, G.-H.; Liu, Q.; Qian, Y.-H.; Xie, D. The lncRNA H19 mediates breast cancer cell plasticity during EMT and MET plasticity by differentially sponging miR-200b/c and let-7b. Sci. Signal. 2017, 10, eaak9557. [Google Scholar] [CrossRef] [PubMed]
- Li, R.-H.; Chen, M.; Liu, J.; Shao, C.-C.; Guo, C.-P.; Wei, X.-L.; Li, Y.-C.; Huang, W.-H.; Zhang, G.-J. Long noncoding RNA ATB promotes the epithelial-mesenchymal transition by upregulating the miR-200c/Twist1 axe and predicts poor prognosis in breast cancer. Cell Death Dis. 2018, 9, 1171. [Google Scholar] [CrossRef]
- Kim, J.; Piao, H.-L.; Kim, B.-J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Yin, Y.; Meng, Q.; Lyu, Y. The role of EMT-related lncRNA in the process of triple-negative breast cancer metastasis. Biosci. Rep. 2021, 41, BSR20203121. [Google Scholar] [CrossRef]
- Godde, N.J.; Sheridan, J.M.; Smith, L.K.; Pearson, H.B.; Britt, K.L.; Galea, R.C.; Yates, L.L.; Visvader, J.E.; Humbert, P.O. Scribble Modulates the MAPK/Fra1 Pathway to Disrupt Luminal and Ductal Integrity and Suppress Tumour Formation in the Mammary Gland. PLoS Genet. 2014, 10, e1004323. [Google Scholar] [CrossRef]
- Zhan, L.; Rosenberg, A.; Bergami, K.C.; Yu, M.; Xuan, Z.; Jaffe, A.B.; Allred, C.; Muthuswamy, S.K. Deregulation of Scribble Promotes Mammary Tumorigenesis and Reveals a Role for Cell Polarity in Carcinoma. Cell 2008, 135, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; Elsum, I.A.; King, C.L.; Kinross, K.M.; Richardson, H.E.; Humbert, P.O. Loss of human Scribble cooperates with H-Ras to promote cell invasion through deregulation of MAPK signalling. Oncogene 2008, 27, 5988–6001. [Google Scholar] [CrossRef]
- Morel, A.-P.; Hinkal, G.W.; Thomas, C.; Fauvet, F.; Courtois-Cox, S.; Wierinckx, A.; Devouassoux-Shisheboran, M.; Treilleux, I.; Tissier, A.; Gras, B.; et al. EMT Inducers Catalyze Malignant Transformation of Mammary Epithelial Cells and Drive Tumorigenesis towards Claudin-Low Tumors in Transgenic Mice. PLoS Genet. 2012, 8, e1002723. [Google Scholar] [CrossRef]
- Knight, J.F.; Lesurf, R.; Zhao, H.; Pinnaduwage, D.; Davis, R.R.; Saleh, S.M.I.; Zuo, D.; Naujokas, M.A.; Chughtai, N.; Herschkowitz, J.I.; et al. Met synergizes with p53 loss to induce mammary tumors that possess features of claudin-low breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E1301–E1310. [Google Scholar] [CrossRef]
- O’Leary, K.A.; Rugowski, D.E.; Sullivan, R.; Schuler, L.A. Prolactin cooperates with loss of p53 to promote claudin-low mammary carcinomas. Oncogene 2014, 33, 3075–3082. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, A.; Perurena, N.; Guruceaga, E.; Mazur, P.K.; Martinez-Canarias, S.; Zandueta, C.; Valencia, K.; Arricibita, A.; Gwinn, D.; Sayles, L.C.; et al. An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer. Nat. Commun. 2017, 8, 14294. [Google Scholar] [CrossRef]
- Oldrini, B.; Curiel-García, Á.; Marques, C.; Matia, V.; Uluçkan, Ö.; Graña-Castro, O.; Torres-Ruiz, R.; Rodriguez-Perales, S.; Huse, J.T.; Squatrito, M. Somatic genome editing with the RCAS-TVA-CRISPR-Cas9 system for precision tumor modeling. Nat. Commun. 2018, 9, 1466. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Podsypanina, K.; Huang, S.; McGrath, A.; Toneff, M.J.; Bogoslovskaia, E.; Zhang, X.; Moraes, R.C.; Fluck, M.; Allred, D.C.; et al. Introduction of oncogenes into mammary glands in vivo with an avian retroviral vector initiates and promotes carcinogenesis in mouse models. Proc. Natl. Acad. Sci. USA 2006, 103, 17396–17401. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Unterkircher, T.; Kroon, P.; Oldrini, B.; Izzo, A.; Dramaretska, Y.; Ferrarese, R.; Kling, E.; Schnell, O.; Nelander, S.; et al. NF1 regulates mesenchymal glioblastoma plasticity and aggressiveness through the AP-1 transcription factor FOSL1. eLife 2021, 10, e64846. [Google Scholar] [CrossRef]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef]
- Cao, S.; Schnelzer, A.; Hannemann, N.; Schett, G.; Soulat, D.; Bozec, A. The Transcription Factor FRA-1/AP-1 Controls Lipocalin-2 Expression and Inflammation in Sepsis Model. Front. Immunol. 2021, 12, 701675. [Google Scholar] [CrossRef]
- Schreiber, M.; Wang, Z.-Q.; Jochum, W.; Fetka, I.; Elliott, C.; Wagner, E.F. Placental vascularisation requires the AP-1 component Fra1. Development 2000, 127, 4937–4948. [Google Scholar] [CrossRef] [PubMed]
- Galvagni, F.; Orlandini, M.; Oliviero, S. Role of the AP-1 transcription factor FOSL1 in endothelial cells adhesion and migration. Cell Adhes. Migr. 2013, 7, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-Mediated Angiogenesis Links EMT-Induced Cancer Stemness to Tumor Initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.; De Val, S.; Neal, A. Endothelial-Specific Cre Mouse Models: Is Your Cre CREdibile? Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2550–2561. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Lu, S.; Simin, K.; Khan, A.; Mercurio, A.M. Analysis of Integrin β4 Expression in Human Breast Cancer: Association with Basal-like Tumors and Prognostic Significance. Clin. Cancer Res. 2008, 14, 1050–1058. [Google Scholar] [CrossRef]
- Li, M.; Jiang, X.; Wang, G.; Zhai, C.; Liu, Y.; Li, H.; Zhang, Y.; Yu, W.; Zhao, Z. ITGB4 is a novel prognostic factor in colon cancer. J. Cancer 2019, 10, 5223–5233. [Google Scholar] [CrossRef]
- Oldak, M.; Maksym, R.B.; Sperling, T.; Yaniv, M.; Smola, H.; Pfister, H.J.; Malejczyk, J.; Smola, S. Human Papillomavirus Type 8 E2 Protein Unravels JunB/Fra-1 as an Activator of the β4-Integrin Gene in Human Keratinocytes. J. Virol. 2010, 84, 1376–1386. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lv, Z.; Zhang, S.; Wang, Z.; He, L.; Tang, M.; Pu, W.; Zhao, H.; Zhang, Z.; Shi, Q.; et al. Genetic Fate Mapping of Transient Cell Fate Reveals N-Cadherin Activity and Function in Tumor Metastasis. Dev. Cell 2020, 54, 593–607.e5. [Google Scholar] [CrossRef] [PubMed]
- Lüönd, F.; Sugiyama, N.; Bill, R.; Bornes, L.; Hager, C.; Tang, F.; Santacroce, N.; Beisel, C.; Ivanek, R.; Bürglin, T.; et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 2021, 56, 3203–3221.e11. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Colaprico, A.; Silva, T.C.; Chen, J.; An, H.; Ban, Y.; Huang, H.; Wang, L.; James, J.L.; Balko, J.M.; et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat. Commun. 2021, 12, 6276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Donaher, J.L.; Das, S.; Li, X.; Reinhardt, F.; Krall, J.A.; Lambert, A.W.; Thiru, P.; Keys, H.R.; Khan, M.; et al. Genome-wide CRISPR screen identifies PRC2 and KMT2D-COMPASS as regulators of distinct EMT trajectories that contribute differentially to metastasis. Nat. Cell Biol. 2022, 24, 554–564. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Jolly, M.K.; Mani, S.A.; Levine, H. Hybrid epithelial/mesenchymal phenotype(s): The ‘fittest’ for metastasis? Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 151–157. [Google Scholar] [CrossRef]
- Luo, X.-L.; Lin, L.; Hu, H.; Hu, F.-L.; Lin, Y.; Luo, M.-L.; Wang, L.; He, Y.-Q. Development and characterization of mammary intraductal (MIND) spontaneous metastasis models for triple-negative breast cancer in syngeneic mice. Sci. Rep. 2020, 10, 4681. [Google Scholar] [CrossRef]
- Dobrolecki, L.E.; Airhart, S.D.; Alferez, D.G.; Aparicio, S.; Behbod, F.; Bentires-Alj, M.; Brisken, C.; Bult, C.J.; Cai, S.; Clarke, R.B.; et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016, 35, 547–573. [Google Scholar] [CrossRef]
- Guillen, K.P.; Fujita, M.; Butterfield, A.J.; Scherer, S.D.; Bailey, M.H.; Chu, Z.; DeRose, Y.S.; Zhao, L.; Cortes-Sanchez, E.; Yang, C.-H.; et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat. Cancer 2022, 3, 232–250. [Google Scholar] [CrossRef]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-Modulated Pulses of ERK Activity Transmit Quantitative Proliferation Signals. Mol. Cell 2013, 49, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.S.; Ng, S.; Hostetter, G.; Azam, S.H.; Der, C.J. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018, 34, 807–822.e7. [Google Scholar] [CrossRef]
- Guo, P.; Yang, J.; Huang, J.; Auguste, D.T.; Moses, M.A. Therapeutic genome editing of triple-negative breast tumors using a noncationic and deformable nanolipogel. Proc. Natl. Acad. Sci. USA 2019, 116, 18295–18303. [Google Scholar] [CrossRef]
- Yu, M.; Ghamsari, L.; Rotolo, J.A.; Kappel, B.J.; Mason, J.M. Combined computational and intracellular peptide library screening: Towards a potent and selective Fra1 inhibitor. RSC Chem. Biol. 2021, 2, 656–668. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Humphreys, I.R.; Pei, J.; Baek, M.; Krishnakumar, A.; Anishchenko, I.; Ovchinnikov, S.; Zhang, J.; Ness, T.J.; Banjade, S.; Bagde, S.R.; et al. Computed structures of core eukaryotic protein complexes. Science 2021, 374, eabm4805. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casalino, L.; Talotta, F.; Matino, I.; Verde, P. FRA-1 as a Regulator of EMT and Metastasis in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 8307. https://doi.org/10.3390/ijms24098307
Casalino L, Talotta F, Matino I, Verde P. FRA-1 as a Regulator of EMT and Metastasis in Breast Cancer. International Journal of Molecular Sciences. 2023; 24(9):8307. https://doi.org/10.3390/ijms24098307
Chicago/Turabian StyleCasalino, Laura, Francesco Talotta, Ilenia Matino, and Pasquale Verde. 2023. "FRA-1 as a Regulator of EMT and Metastasis in Breast Cancer" International Journal of Molecular Sciences 24, no. 9: 8307. https://doi.org/10.3390/ijms24098307
APA StyleCasalino, L., Talotta, F., Matino, I., & Verde, P. (2023). FRA-1 as a Regulator of EMT and Metastasis in Breast Cancer. International Journal of Molecular Sciences, 24(9), 8307. https://doi.org/10.3390/ijms24098307