
Novel Susceptibility Genes Drive Familial Non-Medullary Thyroid Cancer in a Large Consanguineous Kindred

 , ,
, ,
Abstract
1. Background
2. Results
2.1. Clinical Findings
2.2. Evaluating and Prioritizing Potential Candidate Variants
2.3. Evolutionary Conservation and the Effects of the Amino Acid Changes on Protein Conformation
2.4. Analysis of Protein Interactions
2.5. Evaluating Somatic Candidate Variants
- A heterozygote missense variation in X-Ray Repair Cross Complementing 1 gene (XRCC1, Gene ID: 12828) on chromosome 19:44,047,825 (GRCh37/hg19), c.1727A > C (NM_006297.2), p.Asn576Thr. This gene was reported to have mutations causing FNMTC [38]. Patient IVA5 was the only family member to present heterozygosity of the variation in his gDNA sequence. This variant was present in approximately 4% of our collection of Bedouin exomes. No clinical evidence was found for this variant, and it is classified as a benign variant (BS1 and BS2) according to ACMG [48].
- A heterozygote missense variation in HRAS proto-oncogene (Gene ID: 5173) on chromosome 11:533,875 (GRCh37/hg19), c.181C > A (NM_005343.4), p.Gln61Lys. It is one of the 13 genes with genetic alterations in excised malignant thyroid nodules [57]. None of the family patients presented the variation in their gDNA sequence. Moreover, the variation was absent in the public databases (gnomAD browser, 1000 Genomes, ExAC, and EVS), our collection of Bedouin exomes, the database of 77 Bedouins exomes [49], and the Qatari genome of more than 1000 exomes, with a majority of the Bedouin population [50]. According to ACMG, HRAS gene variation c.181C > A is classified as a pathogenic variant (PM1, PP2, PM2, PM5, and PP5). The variation is linked to DTC, specifically to FTC, and the follicular variant of PTC, the prominent histopathological variant among the PTC patients in the described family [60,61]. In addition, it was classified as a pathogenic variant in ClinVar and UniProt classification.
3. Discussion
4. Patients and Methods
4.1. Family Member Evaluation
4.2. Exome Capture and Sequencing
4.3. DNA Purification from the FFPE Tissue
4.4. Genetic Analysis
4.5. Verification of the Variations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seib, C.D.; Sosa, J.A. Evolving Understanding of the Epidemiology of Thyroid Cancer. Endocrinol. Metab. Clin. North Am. 2018, 48, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Shaha, A.R.; Migliacci, J.C.; Nixon, I.; Wang, L.Y.; Wong, R.J.; Morris, L.G.; Patel, S.G.; Shah, J.P.; Tuttle, R.M.; Ganly, I. Stage migration with the new American Joint Committee on Cancer (AJCC) staging system (8th edition) for differentiated thyroid cancer. Surgery 2019, 165, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Lamartina, L.; Grani, G.; Durante, C.; Filetti, S.; Cooper, D.S. Screening for differentiated thyroid cancer in selected populations. Lancet Diabetes Endocrinol. 2020, 8, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Eng, C.; Chen, B. Familial risks for nonmedullary thyroid cancer. J. Clin. Endocrinol. Metab. 2005, 90, 5747–5753. [Google Scholar] [CrossRef]
- Lin, H.-T.; Liu, F.-C.; Lin, S.-F.; Kuo, C.-F.; Chen, Y.-Y.; Yu, H.-P. Familial Aggregation and Heritability of Nonmedullary Thyroid Cancer in an Asian Population: A Nationwide Cohort Study. J. Clin. Endocrinol. Metab. 2020, 105, e2521–e2530. [Google Scholar] [CrossRef]
- Ammar, S.A.; Alobuia, W.M.; Kebebew, E. An update on familial nonmedullary thyroid cancer. Endocrine 2020, 68, 502–507. [Google Scholar] [CrossRef]
- Moses, W.; Weng, J.; Kebebew, E. Prevalence, Clinicopathologic features, and somatic genetic mutation profile in familial versus sporadic nonmedullary thyroid cancer. Thyroid 2011, 21, 367–371. [Google Scholar] [CrossRef]
- Kamani, T.; Charkhchi, P.; Zahedi, A.; Akbari, M.R. Genetic susceptibility to hereditary non-medullary thyroid cancer. Hered. Cancer Clin. Pract. 2022, 20, 9. [Google Scholar] [CrossRef]
- Mazeh, H.; Sippel, R.S. Familial nonmedullary thyroid carcinoma. Thyroid 2013, 23, 1049–1056. [Google Scholar] [CrossRef]
- Capezzone, M.; Sagnella, A.; Cantara, S.; Fralassi, N.; Maino, F.; Forleo, R.; Brilli, L.; Pilli, T.; Cartocci, A.; Castagna, M.G. Risk of Second Malignant Neoplasm in Familial Non-Medullary Thyroid Cancer Patients. Front. Endocrinol. 2022, 13, 845954. [Google Scholar] [CrossRef]
- Park, Y.J.; Ahn, H.Y.; Choi, H.S.; Kim, K.W.; Park, D.J.; Cho, B.Y. The long-term outcomes of the second generation of familial nonmedullary thyroid carcinoma are more aggressive than sporadic cases. Thyroid 2012, 22, 356–362. [Google Scholar] [CrossRef]
- Robenshtok, E.; Tzvetov, G.; Grozinsky-Glasberg, S.; Shraga-Slutzky, I.; Weinstein, R.; Lazar, L.; Serov, S.; Singer, J.; Hirsch, D.; Shimon, I.; et al. Clinical characteristics and outcome of familial nonmedullary thyroid cancer: A retrospective controlled study. Thyroid 2011, 21, 43–48. [Google Scholar] [CrossRef]
- Cirello, V. Familial non-medullary thyroid carcinoma: Clinico-pathological features, current knowledge and novelty regarding genetic risk factors. Minerva Endocrinol. 2020, 46, 5–20. [Google Scholar] [CrossRef]
- Charkes, N.D. On the Prevalence of Familial Nonmedullary Thyroid Cancer in Multiply Affected Kindreds. Thyroid 2006, 16, 181–186. [Google Scholar] [CrossRef]
- Cavaco, B.M.; Batista, P.F.; Sobrinho, L.G.; Leite, V. Mapping a New Familial Thyroid Epithelial Neoplasia Susceptibility Locus to Chromosome 8p23.1-p22 by High-Density Single-Nucleotide Polymorphism Genome-Wide Linkage Analysis. J. Clin. Endocrinol. Metab. 2008, 93, 4426–4430. [Google Scholar] [CrossRef]
- Yang, S.P.; Ngeow, J. Familial non-medullary thyroid cancer: Unraveling the genetic maze. Endocr. Relat. Cancer 2016, 23, R577–R595. [Google Scholar] [CrossRef]
- McKay, J.D.; Lesueur, F.; Jonard, L.; Pastore, A.; Williamson, J.; Hoffman, L.; Burgess, J.; Duffield, A.; Papotti, M.; Stark, M.; et al. Localization of a Susceptibility Gene for Familial Nonmedullary Thyroid Carcinoma to Chromosome 2q. Am. J. Hum. Genet. 2001, 69, 440–446. [Google Scholar] [CrossRef]
- Malchoff, C.D.; Sarfarazi, M.; Tendler, B.; Forouhar, F.; Whalen, G.; Joshi, V.; Malchoff, D.M. Papillary Thyroid Carcinoma Associated with Papillary Renal Neoplasia: Genetic Linkage Analysis of a Distinct Heritable Tumor Syndrome. J. Clin. Endocrinol. Metab. 2000, 85, 1758–1764. [Google Scholar]
- Gara, S.K.; Jia, L.; Merino, M.J.; Agarwal, S.K.; Zhang, L.; Cam, M.; Patel, D.; Kebebew, E. Germline HABP2 Mutation Causing Familial Nonmedullary Thyroid Cancer. New Engl. J. Med. 2015, 373, 448–455. [Google Scholar] [CrossRef]
- Pereira, J.S.; Da Silva, J.G.; Tomaz, R.A.; Pinto, A.E.; Bugalho, M.J.; Leite, V.; Cavaco, B.M. Identification of a novel germline FOXE1 variant in patients with familial non-medullary thyroid carcinoma (FNMTC). Endocrine 2014, 49, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.S.W.; Lang, B.H.; Liu, T.; Shum, C.K.; So, M.T.; Lau, D.K.; Garcia-Barceló, M. A Germline Mutation (A339V) in Thyroid Transcription Factor-1 (TITF-1/NKX2.1) in Patients with Multinodular Goiter and Papillary Thyroid Carcinoma. JNCI J. Natl. Cancer Inst. 2009, 101, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, Y.; Yin, G.; Zhang, J.; Wen, W.; Ruan, X.; Li, D.; Zhang, S.; Cai, W.; Gao, M.; et al. C14orf93 ( RTFC ) is identified as a novel susceptibility gene for familial nonmedullary thyroid cancer. Biochem. Biophys. Res. Commun. 2017, 482, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yu, T.; Chen, L.; Xie, D.; Wang, F.; Fu, L.; Cheng, C.; Li, Y.; Zhu, X.; Miao, G. A Germline CHEK2 Mutation in a Family with Papillary Thyroid Cancer. Thyroid 2020, 30, 924–930. [Google Scholar] [CrossRef]
- Hińcza, K.; Kowalik, A.; Kowalska, A. Current Knowledge of Germline Genetic Risk Factors for the Development of Non-Medullary Thyroid Cancer. Genes 2019, 10, 482. [Google Scholar] [CrossRef]
- Diquigiovanni, C.; Bergamini, C.; Evangelisti, C.; Isidori, F.; Vettori, A.; Tiso, N.; Argenton, F.; Costanzini, A.; Iommarini, L.; Anbunathan, H.; et al. Mutant MYO1F alters the mitochondrial network and induces tumor proliferation in thyroid cancer. Int. J. Cancer 2018, 143, 1706–1719. [Google Scholar] [CrossRef]
- Frio, T.R.; Bahubeshi, A.; Kanellopoulou, C.; Hamel, N.; Niedziela, M.; Sabbaghian, N.; Pouchet, C.; Gilbert, L.; O’Brien, P.K.; Serfas, K.; et al. DICER1 Mutations in Familial Multinodular Goiter With and Without Ovarian Sertoli-Leydig Cell Tumors. JAMA 2011, 305, 68–77. [Google Scholar] [CrossRef]
- Sauer, M.; Barletta, J.A. Proceedings of the North American Society of Head and Neck Pathology, Los Angeles, CA, March 20, 2022: DICER1-Related Thyroid Tumors. Head Neck Pathol. 2022, 16, 190–199. [Google Scholar] [CrossRef]
- Onder, S.; Mete, O.; Yilmaz, I.; Bayram, A.; Bagbudar, S.; Altay, A.Y.; Issin, G.; Terzi, N.K.; Iscan, Y.; Sormaz, I.C.; et al. DICER1 Mutations Occur in More Than One-Third of Follicular-Patterned Pediatric Papillary Thyroid Carcinomas and Correlate with a Low-Risk Disease and Female Gender Predilection. Endocr. Pathol. 2022, 33, 437–445. [Google Scholar] [CrossRef]
- Santiago, K.; Chen Wongworawat, Y.; Khan, S. Differential MicroRNA-Signatures in Thyroid Cancer Subtypes. J. Oncol. 2020, 2020, 2052396. [Google Scholar] [CrossRef]
- Swierniak, M.; Wojcicka, A.; Czetwertynska, M.; Stachlewska, E.; Maciag, M.; Wiechno, W.; Gornicka, B.; Bogdanska, M.; Koperski, L.; De La Chapelle, A.; et al. In-Depth Characterization of the MicroRNA Transcriptome in Normal Thyroid and Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, E1401–E1409. [Google Scholar] [CrossRef]
- Lin, R.-X.; Yang, S.-L.; Jia, Y.; Wu, J.-C.; Xu, Z.; Zhang, H. Epigenetic regulation of papillary thyroid carcinoma by long non-coding RNAs. Semin. Cancer Biol. 2022, 83, 253–260. [Google Scholar] [CrossRef]
- Tomsic, J.; He, H.; Akagi, K.; Liyanarachchi, S.; Pan, Q.; Bertani, B.; Nagy, R.; Symer, D.E.; Blencowe, B.J.; de la Chapelle, A. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci. Rep. 2015, 5, 10566. [Google Scholar] [CrossRef]
- Ye, F.; Gao, H.; Xiao, L.; Zuo, Z.; Liu, Y.; Zhao, Q.; Chen, H.; Feng, W.; Fu, B.; Sun, L.; et al. Whole exome and target sequencing identifies MAP2K5 as novel susceptibility gene for familial non-medullary thyroid carcinoma. Int. J. Cancer 2018, 144, 1321–1330. [Google Scholar] [CrossRef]
- Marques, I.J.; Gomes, M.I.; Pojo, M.; Pires, M.C.; Moura, M.M.; Cabrera, R.; Santos, C.; van Ijcken, W.F.J.; Teixeira, M.R.; Ramalho, J.S.; et al. Identification of SPRY4 as a Novel Candidate Susceptibility Gene for Familial Nonmedullary Thyroid Cancer. Thyroid 2021, 31, 1366–1375. [Google Scholar] [CrossRef]
- He, H.; Bronisz, A.; Liyanarachchi, S.; Nagy, R.; Li, W.; Huang, Y.; Akagi, K.; Saji, M.; Kula, D.; Wojcicka, A.; et al. SRGAP1Is a Candidate Gene for Papillary Thyroid Carcinoma Susceptibility. J. Clin. Endocrinol. Metab. 2013, 98, E973–E980. [Google Scholar] [CrossRef]
- Liu, D.; Yang, C.; Bojdani, E.; Murugan, A.K.; Xing, M. Identification of RASAL1 as a Major Tumor Suppressor Gene in Thyroid Cancer. Gynecol. Oncol. 2013, 105, 1617–1627. [Google Scholar] [CrossRef]
- A Ryu, R.; Tae, K.; Min, H.J.; Jeong, J.H.; Cho, S.H.; Lee, S.H.; Ahn, Y.H. XRCC1 Polymorphisms and Risk of Papillary Thyroid Carcinoma in a Korean Sample. J. Korean Med. Sci. 2011, 26, 991–995. [Google Scholar] [CrossRef]
- Yu, Y.; Dong, L.; Li, D.; Chuai, S.; Wu, Z.; Zheng, X.; Cheng, Y.; Han, L.; Yu, J.; Gao, M. Targeted DNA Sequencing Detects Mutations Related to Susceptibility among Familial Non-medullary Thyroid Cancer. Sci. Rep. 2015, 5, 16129. [Google Scholar] [CrossRef]
- Srivastava, A.; Kumar, A.; Giangiobbe, S.; Bonora, E.; Hemminki, K.; Försti, A.; Bandapalli, O. Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways. Biomolecules 2019, 9, 605. [Google Scholar] [CrossRef]
- Fagin, J.A.; Wells, S.A., Jr. Biologic and Clinical Perspectives on Thyroid Cancer. New Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhou, M.; Li, X.; Gu, S.; Cao, Y.; Xing, T.; Chen, W.; Chu, C.; Gu, F.; Zhou, J.; et al. SLC34A2 simultaneously promotes papillary thyroid carcinoma growth and invasion through distinct mechanisms. Oncogene 2020, 39, 2658–2675. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, Y.; Lee, E.K.; Son, H.-Y.; Im, S.-W.; Kwak, S.-J.; Yoon, J.W.; Kim, M.J.; Kim, J.; Choi, H.S.; Ryu, C.H.; et al. Genome-Wide Association Study Reveals Distinct Genetic Susceptibility of Thyroid Nodules From Thyroid Cancer. J. Clin. Endocrinol. Metab. 2018, 103, 4384–4394. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Levine, D.A. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Sarquis, M.; Moraes, D.C.; Bastos-Rodrigues, L.; Azevedo, P.G.; Ramos, A.V.; Reis, F.V.; Dande, P.V.; Paim, I.; Friedman, E.; De Marco, L. Germline Mutations in Familial Papillary Thyroid Cancer. Endocr. Pathol. 2020, 31, 14–20. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, T.; Sun, J.; Wang, F.; Cheng, C.; He, S.; Chen, L.; Xie, D.; Fu, L.; Guan, X.; et al. Germ-line mutations in WDR77 predispose to familial papillary thyroid cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2026327118. [Google Scholar] [CrossRef]
- Vriens, M.R.; Suh, I.; Moses, W.; Kebebew, E. Clinical Features and Genetic Predisposition to Hereditary Nonmedullary Thyroid Cancer. Thyroid 2009, 19, 1343–1349. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef]
- AlSalem, A.B.; Halees, A.S.; Anazi, S.; Alshamekh, S.; Alkuraya, F.S. Autozygome Sequencing Expands the Horizon of Human Knockout Research and Provides Novel Insights into Human Phenotypic Variation. PLOS Genet. 2013, 9, e1004030. [Google Scholar] [CrossRef]
- A Fakhro, K.; Staudt, M.R.; Ramstetter, M.D.; Robay, A.; A Malek, J.; Badii, R.; Al-Marri, A.A.-N.; Khalil, C.A.; Al-Shakaki, A.; Chidiac, O.; et al. The Qatar genome: A population-specific tool for precision medicine in the Middle East. Hum. Genome Var. 2016, 3, 16016. [Google Scholar] [CrossRef]
- Keller, B.A.; Volkening, K.; Droppelmann, C.A.; Ang, L.C.; Rademakers, R.; Strong, M.J. Co-aggregation of RNA binding proteins in ALS spinal motor neurons: Evidence of a common pathogenic mechanism. Acta Neuropathol. 2012, 124, 733–747. [Google Scholar] [CrossRef]
- Horikawa, I.; Fujita, K.; Jenkins, L.M.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.P.; Appella, E.; Harris, C.C. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53-mediated senescence. Nat. Commun. 2014, 5, 4706. [Google Scholar] [CrossRef]
- Di Lello, P.; Jenkins LM, M.; Jones, T.N.; Nguyen, B.D.; Hara, T.; Yamaguchi, H.; Omichinski, J.G. Structure of the Tfb1/p53 Complex: Insights into the Interaction between the p62/Tfb1 Subunit of TFIIH and the Activation Domain of p53. Mol. Cell 2006, 22, 731–740. [Google Scholar] [CrossRef]
- Yuan, F.; Sun, Q.; Zhang, S.; Ye, L.; Xu, Y.; Deng, G.; Xu, Z.; Zhang, S.; Liu, B.; Chen, Q. The dual role of p62 in ferroptosis of glioblastoma according to p53 status. Cell Biosci. 2022, 12, 20. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, J.-S.; Hong, D.; Lee, S.-H.; Kim, N.; Lee, W.-K.; Sung, T.-W.; Gong, Y.-D.; Kim, S.-Y. Renal cell carcinoma escapes death by p53 depletion through transglutaminase 2-chaperoned autophagy. Cell Death Dis. 2016, 7, e2163. [Google Scholar] [CrossRef]
- Mukherjee, S.; Maddalena, M.; Lü, Y.; Martinez, S.; Nataraj, N.B.; Noronha, A.; Oren, M. Cross-talk between mutant p53 and p62/SQSTM1 augments cancer cell migration by promoting the degradation of cell adhesion proteins. Proc. Natl. Acad. Sci. USA 2022, 119, e2119644119. [Google Scholar] [CrossRef]
- Desai, D.; Lepe, M.; Baloch, Z.W.; Mandel, S.J. ThyroSeq v3 for Bethesda III and IV: An institutional experience. Cancer Cytopathol. 2021, 129, 164–170. [Google Scholar] [CrossRef]
- Fasci, D.; van Ingen, H.; Scheltema, R.A.; Heck, A.J.R. Histone Interaction Landscapes Visualized by Crosslinking Mass Spectrometry in Intact Cell Nuclei. Mol. Cell Proteom. 2018, 17, 2018–2033. [Google Scholar] [CrossRef]
- Wang, Y.; He, H.; Li, W.; Phay, J.; Shen, R.; Yu, L.; Hancioglu, B.; de la Chapelle, A. MYH9 binds to lncRNA gene PTCSC2 and regulates FOXE1 in the 9q22 thyroid cancer risk locus. Proc. Natl. Acad. Sci. USA 2017, 114, 474–479. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W., 2nd; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS Point Mutations and PAX8-PPARγ Rearrangement in Thyroid Tumors: Evidence for Distinct Molecular Pathways in Thyroid Follicular Carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef]
- Buhrman, G.; Wink, G.; Mattos, C. Transformation Efficiency of RasQ61 Mutants Linked to Structural Features of the Switch Regions in the Presence of Raf. Structure 2007, 15, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Manzella, L.; Stella, S.; Pennisi, M.S.; Tirrò, E.; Massimino, M.; Romano, C.; Puma, A.; Tavarelli, M.; Vigneri, P. New Insights in Thyroid Cancer and p53 Family Proteins. Int. J. Mol. Sci. 2017, 18, 1325. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Huiling, H.; Liyanarachchi, S.; Jendrzejewski, J.; Srinivas, M.; Davuluri, R.V.; Nagy, R.; De La Chapelle, A. Genetic Predisposition to Papillary Thyroid Carcinoma: Involvement of FOXE1, TSHR, and a Novel lincRNA Gene, PTCSC. J. Clin. Endocrinol. Metab. 2015, 100, E164–E172. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, Y.; Park, Y.J. Genome-Wide Association Studies of Autoimmune Thyroid Diseases, Thyroid Function, and Thyroid Cancer. Endocrinol. Metab. 2018, 33, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Gueven, N.; Becherel, O.J.; Kijas, A.W.; Chen, P.; Howe, O.; Rudolph, J.H.; Gatti, R.; Date, H.; Onodera, O.; Taucher-Scholz, G.; et al. Aprataxin, a novel protein that protects against genotoxic stress. Hum. Mol. Genet. 2004, 13, 1081–1093. [Google Scholar] [CrossRef]
- Muhammad, E.; Levitas, A.; Singh, S.R.; Braiman, A.; Ofir, R.; Etzion, S.; Sheffield, V.C.; Etzion, Y.; Carrier, L.; Parvari, R. PLEKHM2mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum. Mol. Genet. 2015, 24, 7227–7240. [Google Scholar] [CrossRef]
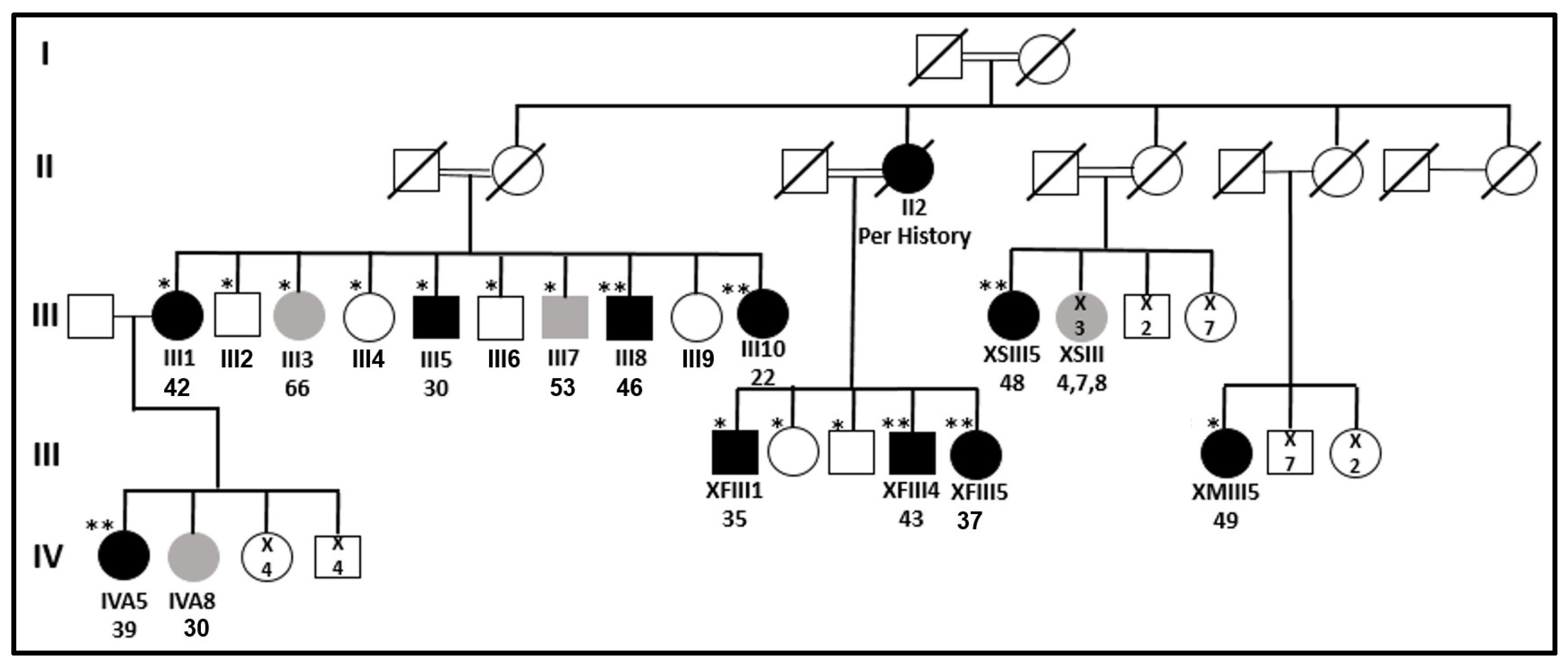
 : top—patient’s code; bottom—patient’s age at diagnosis.
: top—patient’s code; bottom—patient’s age at diagnosis.  : number of individuals.
: top—patient’s code; bottom—patient’s age at diagnosis. : number of individuals.
: number of individuals.
: top—patient’s code; bottom—patient’s age at diagnosis. : number of individuals.


{kind=link}
{kind=link}
| ID | Gender M/F | Current Age | Age at Diagnosis | Pathology or US Results ¥ | Largest Diameter | Variant # | Multifocality # | LN MTS # | RAI # | Status (Year) § |
|---|---|---|---|---|---|---|---|---|---|---|
| III1 | F | 79 | 42 | PTC | NA | NA | NA | NA | NA | Free of disease (2016) |
| III2 | M | 76 | 69 | NT | NT (2016) | |||||
| III3 | F | 73 | 66 | Benign MNG ˄ | 2 cm | Benign MNG (2016) | ||||
| III4 | F | 70 | 63 | NT | NT (2016) | |||||
| III5 | M | 63 | 30 | PTC | NA | NA | NA | Yes | Yes | Lost to follow-up |
| III6 | M | 61 | 54 | NT | NT (2016) | |||||
| III7 | M | 60 | 53 | Benign MNG ~ | 4 cm | Benign MNG (2016) | ||||
| III8 | M | 57 | 46 | PTC | 1.6 cm | Follicular | Yes, 5 foci | No | 100 | Free of disease (2021) |
| III9 | F | 54 | NA | |||||||
| III10 | F | 50 | 22 | PTC | 2.5 cm | Follicular | Yes, 3 foci | No | Yes- Twice | Free of disease (2021) |
| XFIII1 | M | 58 | 35 | PTC | 1.5 cm | Follicular | Yes, 3 foci | Yes | 250 * | Lost to follow-up |
| XFIII2 | F | NA | ||||||||
| XFIII3 | M | NA | ||||||||
| XFIII4 | M | 51 | 43 | PTC | 2.1 cm | Classic/follicular | Yes, “many foci” | Yes | 100 | Free of disease (2022) |
| XFIII5 | F | 50 | 37 | PTC | NA | NA | NA | Yes | 150 | Free of disease (2022) |
| IVA5 @ | F | 53 | 39 | PTC | 1.4 cm | Classic | Yes, at least 4 foci | Yes | 150 | Free of disease (2011) @ |
| IVA8 | F | 45 | 30 | Benign MNG | 3 cm | Lost to follow-up | ||||
| Number of Variants after Filtration | Filtration Criteria for Homozygous or Heterozygous Variations |
|---|---|
| 644,298 → 12,626 | Shared variations between the WESs of patients III8, III10, IVA5, XFIII4, and XFIII5 |
| 12,626 → 84 | Presence in the general databases—1KG, EVS, ExAC, and gnomAD—at frequencies < 1% |
| 84 → 54 | Presence in our internal laboratory exome database of the Bedouin population at frequencies < 1% |
| 54 → 4 | Familial segregation analysis (detailed in Supplementary Table S2) |
| Gene Symbol and Position | Ref/Alt | III1 A | III2 H | III4 H | III5 A | III6 H | III8 A | III10 A | IVA5 A | XFIII1 A | XFIII4 A | XFIII5 A |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ARHGEF28 chr5:73048875 | A > G | −/+ | +/+ | +/+ | −/+ | −/+ | −/+ | −/+ | −/+ | −/+ | −/+ | −/− |
| FBXW10 chr17:18661699 | delCAT | −/+ | −/+ | +/+ | −/+ | +/+ | −/+ | −/+ | −/+ | −/+ | −/+ | −/+ |
| SLC24A4 chr14:92953131 | A > G | −/+ | +/+ | −/+ | −/+ | +/+ | −/+ | −/+ | −/+ | −/+ | −/+ | −/+ |
| SLC47A1 chr17:19459316 | G > A | −/+ | −/+ | +/+ | −/− | +/+ | −/+ | −/+ | −/+ | −/+ | −/+ | −/+ |
| Gene Symbol Position Ref/Alt | cDNA Protein | GTEx (TPM) @ | Prevalence in the Population of Origin | Prediction Tools | ACMG | ClinVar | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Internal Lab Frequency * | Saudi’s Frequency & | Qatari’s Allele Number $ | |||||||||||
| Het. | Hom. | Het. | Hom. | Het. | Hom. | SIFT | CADD | PolyPhen | |||||
| ARHGEF28 chr5:73048875 A > G | c.323A > G p.Asn108Ser | 19.44 | 0.0057 | 0.0006 | 0 | 0 | 0.003 | 0 | 0.14 (tolerated) | 10.71 | 0.02 (benign) | BS2, BP4 (likely benign) | VUS |
| FBXW10 chr17:18661699 delCAT | c.1317_ 1319delCAT p.Ile440del | 1.22 | 0.0128 | 0 | 0 | 0 | 0 | 0 | - | - | - | PM2, PM4 (VUS) | - |
| SLC24A4 chr14:92953131 A > G | c.1532A > G - | 0.20 | - | - | 0 | 0 | 0.0065 | 0 | - | 4.68 | - | BS2, BP4, BP6 (benign) | Benign |
| SLC47A1 chr17:19459316 G > A | c.862G > A p.Gly288Ser | 7.45 | 0.0147 | 0.0006 | 0.026 | 0.013 | 0.012 | 0 | 0.83 (tolerated) | 22 | 0.73 (possibly damaging) | BP4, PM2 (VUS) | - |
| Number of Variants after Filtration | Filtration Criteria |
|---|---|
| 217,476 → 6697 | Variations that exist in the WES of the malignant thyroid tissue and are absent in the gDNA of the same patient—III8 |
| 6697 → 1527 | Presence in the general databases—1KG, EVS, ExAC and gnomAD—at frequencies < 5% |
| 1527 → 2 | Applying a case panel that presents only variations in genes associated with PTC—see Supplementary Table S1 and list of 13 somatic genes [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majdalani, P.; Yoel, U.; Nasasra, T.; Fraenkel, M.; Haim, A.; Loewenthal, N.; Zarivach, R.; Hershkovitz, E.; Parvari, R. Novel Susceptibility Genes Drive Familial Non-Medullary Thyroid Cancer in a Large Consanguineous Kindred. Int. J. Mol. Sci. 2023, 24, 8233. https://doi.org/10.3390/ijms24098233
Majdalani P, Yoel U, Nasasra T, Fraenkel M, Haim A, Loewenthal N, Zarivach R, Hershkovitz E, Parvari R. Novel Susceptibility Genes Drive Familial Non-Medullary Thyroid Cancer in a Large Consanguineous Kindred. International Journal of Molecular Sciences. 2023; 24(9):8233. https://doi.org/10.3390/ijms24098233
Chicago/Turabian StyleMajdalani, Pierre, Uri Yoel, Tayseer Nasasra, Merav Fraenkel, Alon Haim, Neta Loewenthal, Raz Zarivach, Eli Hershkovitz, and Ruti Parvari. 2023. "Novel Susceptibility Genes Drive Familial Non-Medullary Thyroid Cancer in a Large Consanguineous Kindred" International Journal of Molecular Sciences 24, no. 9: 8233. https://doi.org/10.3390/ijms24098233
APA StyleMajdalani, P., Yoel, U., Nasasra, T., Fraenkel, M., Haim, A., Loewenthal, N., Zarivach, R., Hershkovitz, E., & Parvari, R. (2023). Novel Susceptibility Genes Drive Familial Non-Medullary Thyroid Cancer in a Large Consanguineous Kindred. International Journal of Molecular Sciences, 24(9), 8233. https://doi.org/10.3390/ijms24098233