
Mitochondrial Quality Control and Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
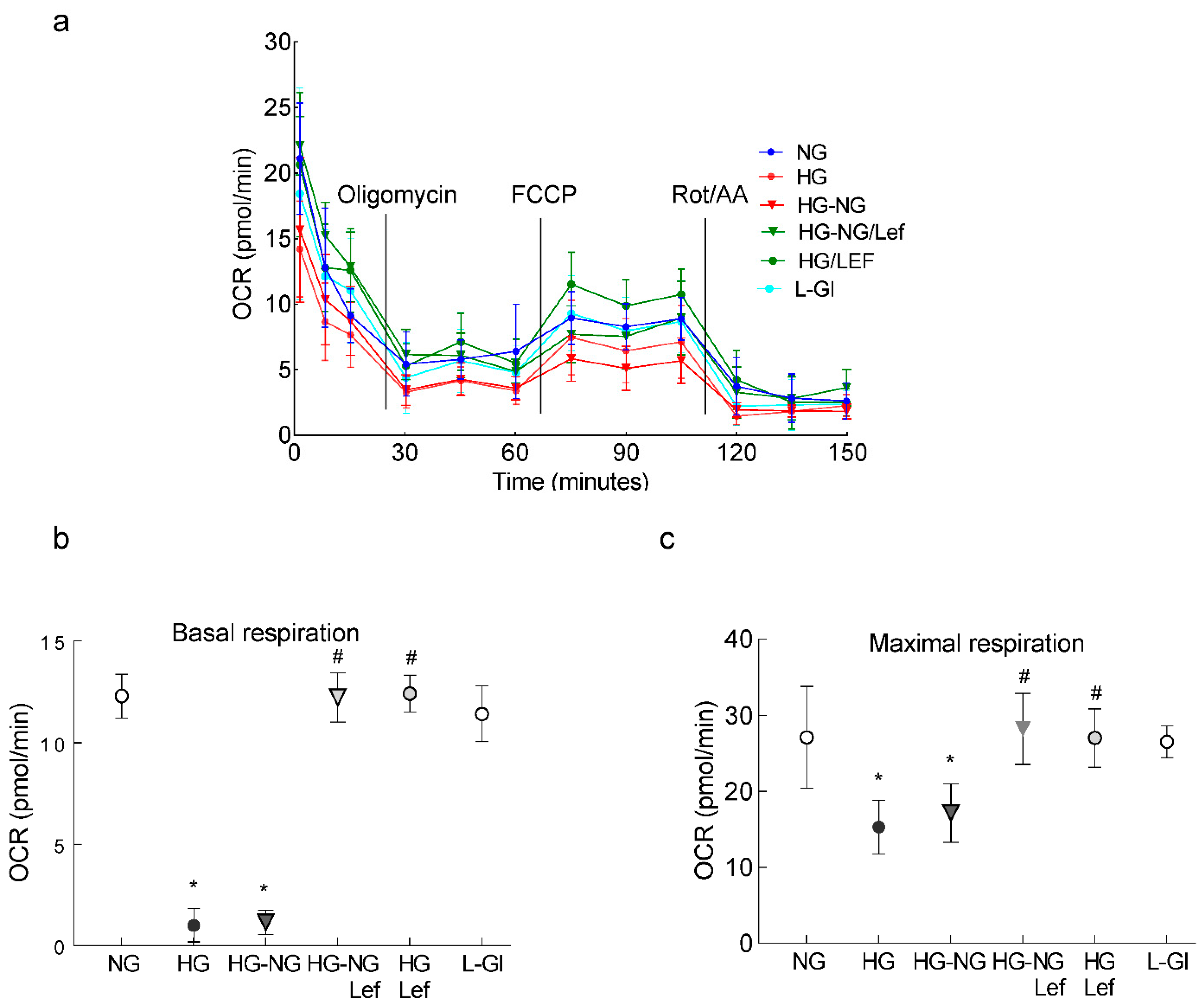
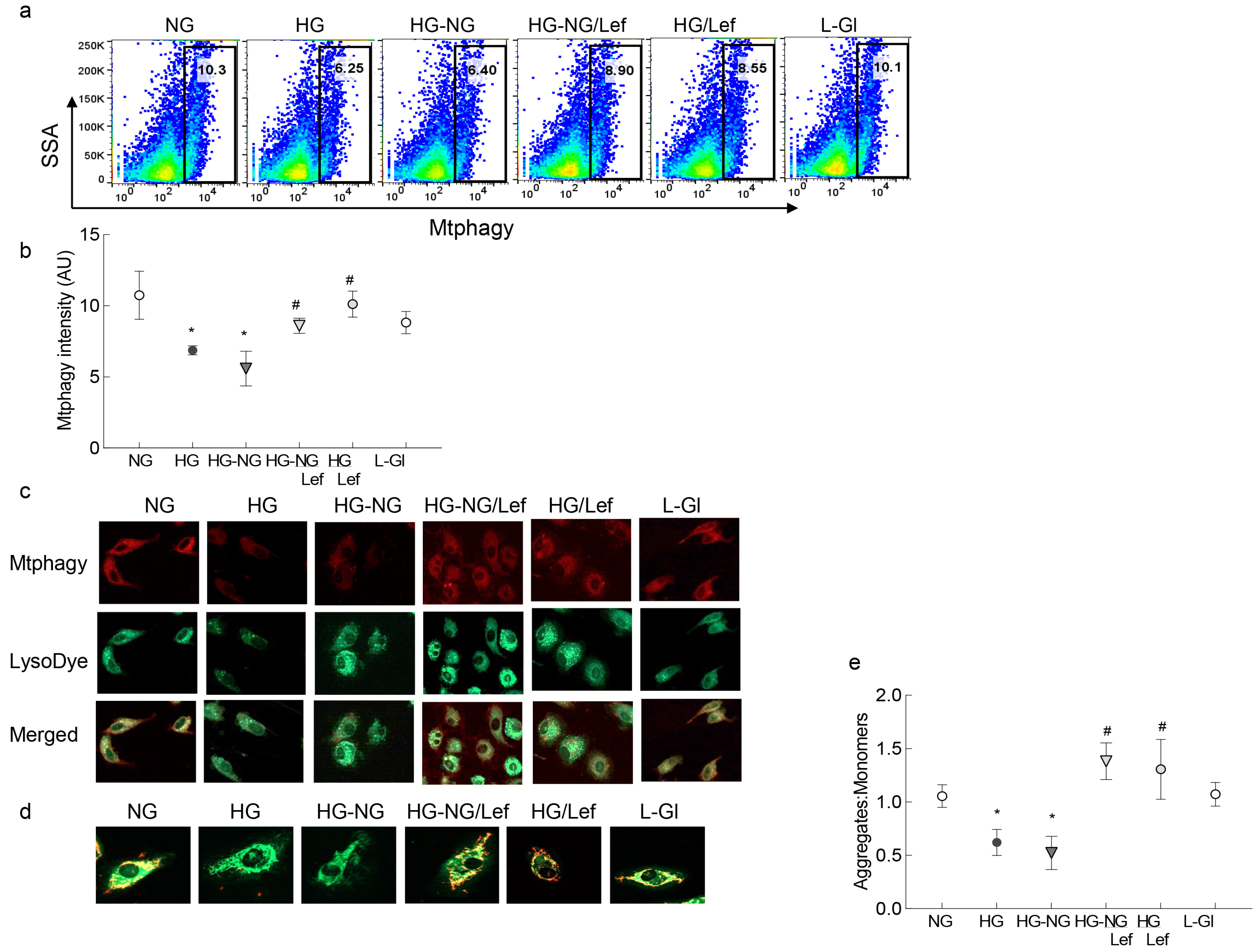
2. Results
3. Discussion
4. Methods and Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) Research Group; Lachin, J.M.; White, N.H.; Hainsworth, D.P.; Sun, W.; Cleary, P.A.; Nathan, D.M. Effect of intensive diabetes therapy on the progression of diabetic retinopathy in patients with type 1 diabetes: 18 years of follow-up in the DCCT/EDIC. Diabetes 2015, 64, 631–642. [Google Scholar] [CrossRef]
- Hainsworth, D.P.; Bebu, I.; Aiello, L.P.; Sivitz, W.; Gubitosi-Klug, R.; Malone, J.; White, N.H.; Danis, R.; Wallia, A.; Gao, X.; et al. Risk Factors for Retinopathy in Type 1 Diabetes: The DCCT/EDIC Study. Diabetes Care 2019, 42, 875–882. [Google Scholar] [CrossRef] [PubMed]
- White, N.H.; Sun, W.; Cleary, P.A.; Tamborlane, W.V.; Danis, R.P.; Hainsworth, D.P.; Davis, M.D.; DCCT-EDIC Research Team. Effect of prior intensive therapy on 10-year progression of retinopathy in DCCT/EDIC: Comparison of adults and adolescents. Diabetes 2010, 59, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P. Diabetic retinopathy and other ocular findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care 2014, 37, 17–23. [Google Scholar] [CrossRef]
- Writing Team for the DCCT/EDIC Research Group; Gubitosi-Klug, R.A.; Sun, W.; Cleary, P.A.; Braffett, B.H.; Aiello, L.P.; Das, A.; Tamborlane, W.; Klein, R. Effects of Prior Intensive Insulin Therapy and Risk Factors on Patient-Reported Visual Function Outcomes in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Cohort. JAMA Ophthalmol. 2016, 134, 137–145. [Google Scholar] [CrossRef]
- Engerman, R.L.; Kern, T.S. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 1987, 36, 808–812. [Google Scholar] [CrossRef]
- Kowluru, R.A. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes 2003, 52, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Zhong, Q.; Kanwar, M. Metabolic memory and diabetic retinopathy: Role of inflammatory mediators in retinal pericytes. Exp. Eye Res. 2010, 90, 617–623. [Google Scholar] [CrossRef]
- Mizutani, M.; Kern, T.S.; Lorenzi, M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J. Clin. Investig. 1996, 97, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Et Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef]
- Duraisamy, A.J.; Mohammad, G.; Kowluru, R.A. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1617–1626. [Google Scholar] [CrossRef]
- Mohammad, G.; Kowluru, R.A. Mitochondrial Dynamics in the Metabolic Memory of Diabetic Retinopathy. J. Diabetes Res. 2022, 2022, 3555889. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Chan, P.S. Metabolic memory in diabetes-from in vitro oddity to in vivo problem: Role of apoptosis. Brain Res. Bull. 2010, 81, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mohammad, G. Epigenetics and Mitochondrial Stability in the Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy. Sci. Rep. 2020, 10, 6655. [Google Scholar] [CrossRef] [PubMed]
- Miret-Casals, L.; Sebastián, D.; Brea, J.; Rico-Leo, E.M.; Palacín, M.; Fernández-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278.e264. [Google Scholar] [CrossRef] [PubMed]
- Madsen-Bouterse, S.A.; Mohammad, G.; Kanwar, M.; Kowluru, R.A. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid. Redox Signal. 2010, 13, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924-1931. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Galloway, C.A.; Lee, H.; Yoon, Y. Mitochondrial morphology-emerging role in bioenergetics. Free Radic. Biol. Med. 2012, 53, 2218–2228. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef]
- Cortassa, S.; Aon, M.A.; Winslow, R.L.; O’Rourke, B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys. J. 2004, 87, 2060–2073. [Google Scholar] [CrossRef]
- Fragoso, Y.D.; Brooks, J.B. Leflunomide and teriflunomide: Altering the metabolism of pyrimidines for the treatment of autoimmune diseases. Expert. Rev. Clin. Pharm. 2015, 8, 315–320. [Google Scholar] [CrossRef]
- Maremanda, K.P.; Sundar, I.K.; Rahman, I. Role of inner mitochondrial protein OPA1 in mitochondrial dysfunction by tobacco smoking and in the pathogenesis of COPD. Redox Biol. 2021, 45, 102055. [Google Scholar] [CrossRef]
- Moon, S.J.; Kim, E.K.; Jhun, J.Y.; Lee, H.J.; Lee, W.S.; Park, S.H.; Cho, M.L.; Min, J.K. The active metabolite of leflunomide, A77 1726, attenuates inflammatory arthritis in mice with spontaneous arthritis via induction of heme oxygenase-1. J. Transl. Med. 2017, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Pellattiero, A.; Scorrano, L. Flaming Mitochondria: The Anti-inflammatory Drug Leflunomide Boosts Mitofusins. Cell Chem. Biol. 2018, 25, 231–233. [Google Scholar] [CrossRef]
- Abdel-Hamid, N.M.; Abass, S.A.; Eldomany, R.A.; Abdel-Kareem, M.A.; Zakaria, S. Dual regulating of mitochondrial fusion and Timp-3 by leflunomide and diallyl disulfide combination suppresses diethylnitrosamine-induced hepatocellular tumorigenesis in rats. Life Sci. 2022, 294, 120369. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019, 5, e126915. [Google Scholar] [CrossRef] [PubMed]
- Anzell, A.R.; Maizy, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury. Mol. Neurobiol. 2018, 55, 2547–2564. [Google Scholar] [CrossRef]
- Mohammad, G.; Kowluru, R.A. Nuclear genome-encoded long noncoding RNAs and mitochondrial damage in diabetic retinopathy. Cells 2021, 10, 3271. [Google Scholar] [CrossRef]
- Kumar, J.; Mohammad, G.; Alka, K.; Kowluru, R.A. Mitochondrial Genome-Encoded Long Noncoding RNA and Mitochondrial Stability in Diabetic Retinopathy. Diabetes 2022, 72, 520–531. [Google Scholar] [CrossRef]
- Navneet, S.; Zhao, J.; Wang, J.; Mysona, B.; Barwick, S.; Ammal Kaidery, N.; Saul, A.; Kaddour-Djebbar, I.; Bollag, W.B.; Thomas, B.; et al. Hyperhomocysteinemia-induced death of retinal ganglion cells: The role of Muller glial cells and NRF2. Redox Biol. 2019, 24, 101199. [Google Scholar] [CrossRef]
- Thounaojam, M.C.; Jadeja, R.N.; Warren, M.; Powell, F.L.; Raju, R.; Gutsaeva, D.; Khurana, S.; Martin, P.M.; Bartoli, M. MicroRNA-34a (miR-34a) Mediates Retinal Endothelial Cell Premature Senescence through Mitochondrial Dysfunction and Loss of Antioxidant Activities. Antioxidants 2019, 8, 328. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Menon, B.; Gierhart, D.L. Beneficial effect of zeaxanthin on retinal metabolic abnormalities in diabetic rats. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, Y.C.; Tsai, M.L.; Liao, W.T.; Hung, C.H. TSLP regulates mitochondrial ROS-induced mitophagy via histone modification in human monocytes. Cell Biosci. 2022, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, S.; Chen, X.; Wang, Z.; Wang, X.; Zhou, Q.; Fang, W.; Zheng, C. Liraglutide prevents high glucose induced HUVECs dysfunction via inhibition of PINK1/Parkin-dependent mitophagy. Mol. Cell. Endocrinol. 2022, 545, 111560. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Tewari, S.; Goldberg, A.F.X.; Kowluru, R.A. Mitochondria biogenesis and the development of diabetic retinopathy. Free Radic. Biol. Med. 2011, 51, 1849–1860. [Google Scholar] [CrossRef]
- Hegedűs, C.; Boros, G.; Fidrus, E.; Kis, G.N.; Antal, M.; Juhász, T.; Janka, E.A.; Jankó, L.; Paragh, G.; Emri, G.; et al. PARP1 Inhibition Augments UVB-Mediated Mitochondrial Changes-Implications for UV-Induced DNA Repair and Photocarcinogenesis. Cancers 2019, 12, 5. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowluru, R.A.; Alka, K. Mitochondrial Quality Control and Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy. Int. J. Mol. Sci. 2023, 24, 8076. https://doi.org/10.3390/ijms24098076
Kowluru RA, Alka K. Mitochondrial Quality Control and Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy. International Journal of Molecular Sciences. 2023; 24(9):8076. https://doi.org/10.3390/ijms24098076
Chicago/Turabian StyleKowluru, Renu A., and Kumari Alka. 2023. "Mitochondrial Quality Control and Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy" International Journal of Molecular Sciences 24, no. 9: 8076. https://doi.org/10.3390/ijms24098076
APA StyleKowluru, R. A., & Alka, K. (2023). Mitochondrial Quality Control and Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy. International Journal of Molecular Sciences, 24(9), 8076. https://doi.org/10.3390/ijms24098076