
Telmisartan Is a Promising Agent for Managing Neuropathic Pain and Delaying Opioid Analgesic Tolerance in Rats

 ,
, 
 , , , , ,
, , , , ,  , , and
, , and 
Abstract
:
1. Introduction
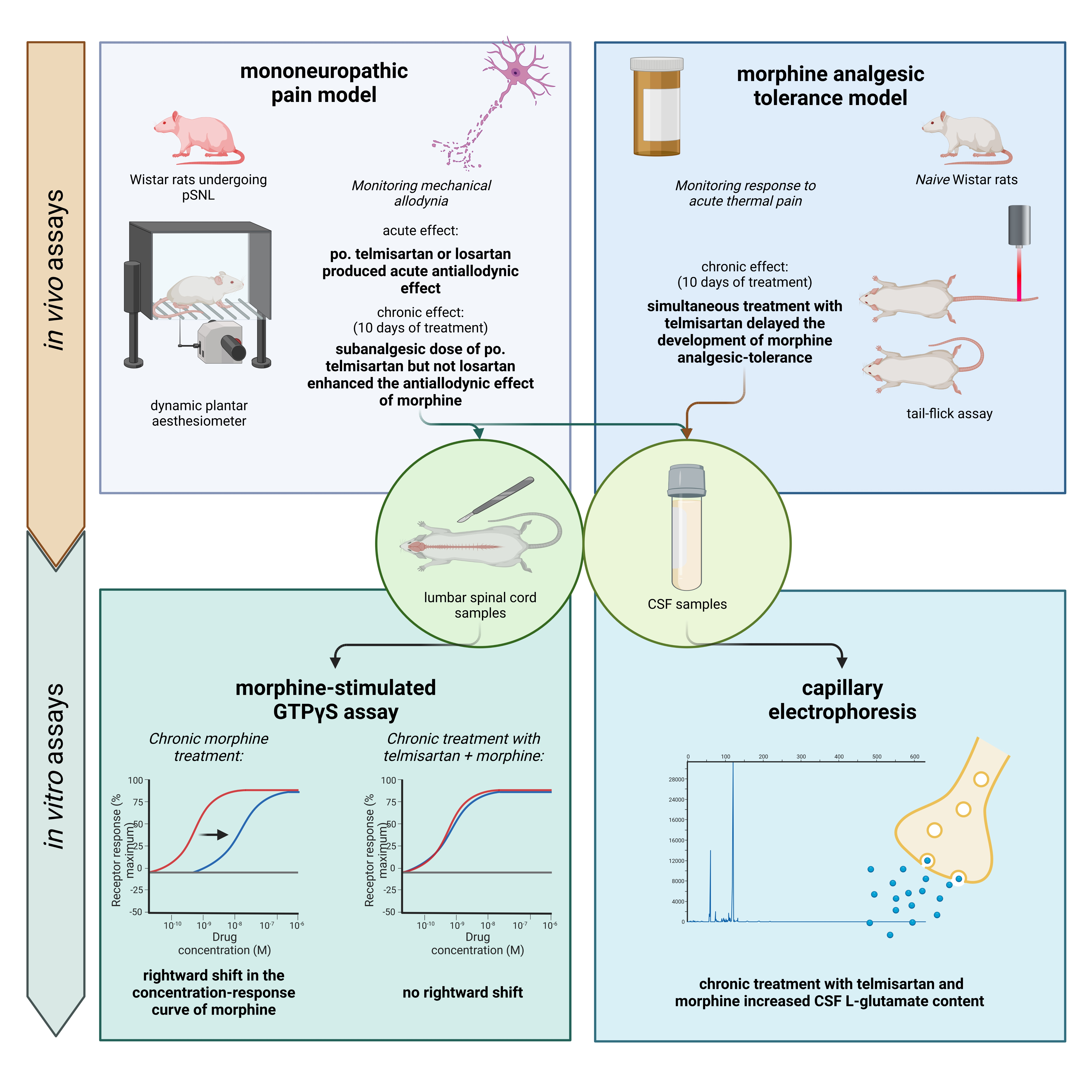
2. Results
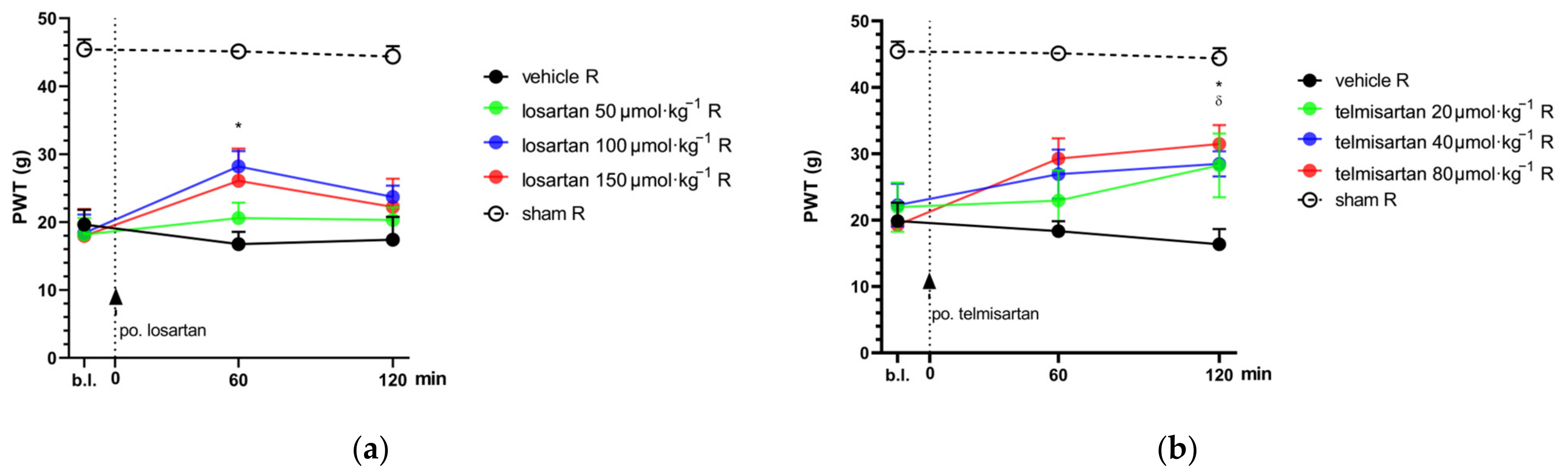
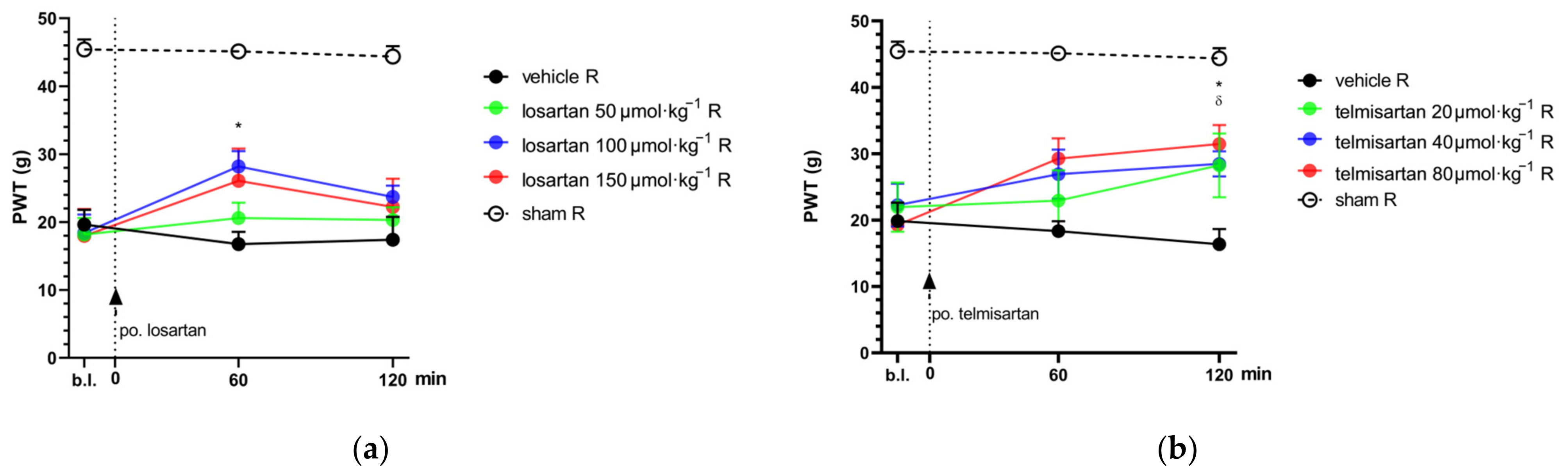
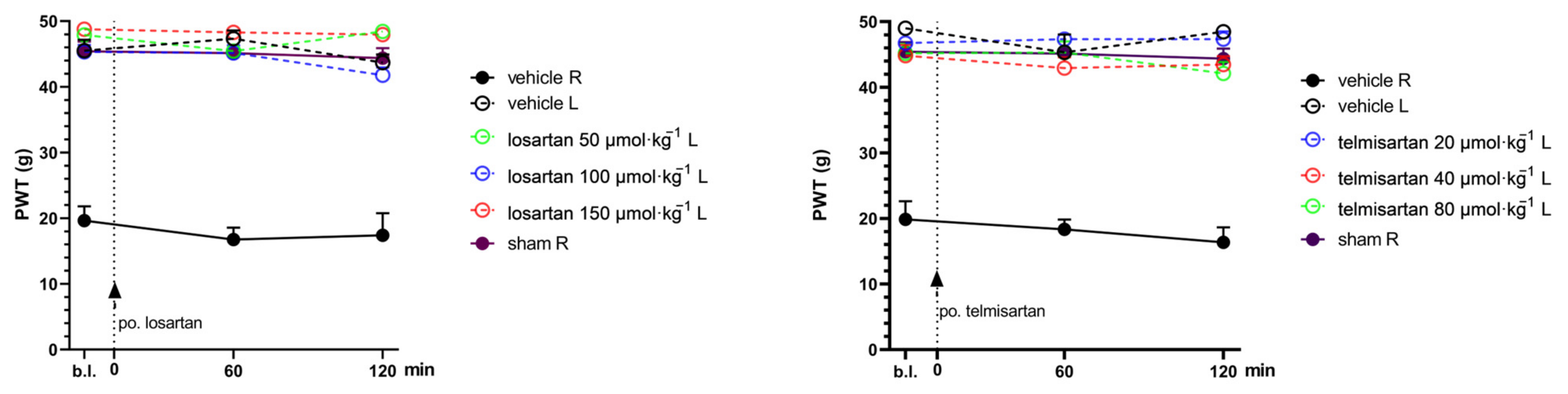
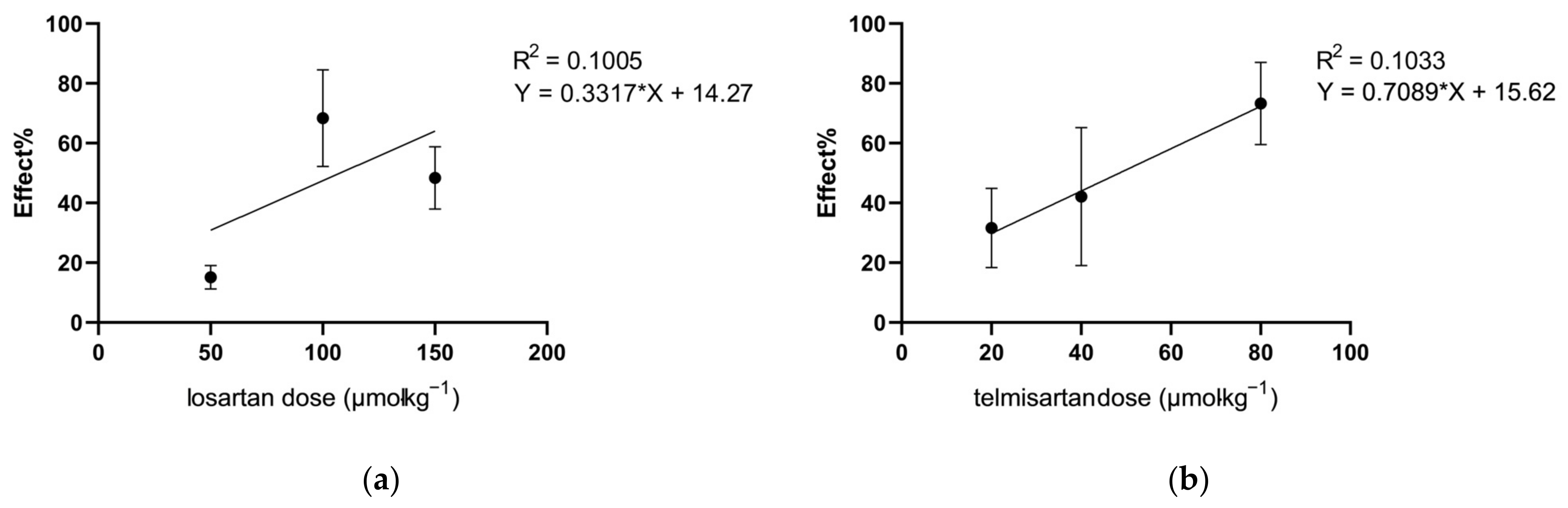
2.1. Telmisartan or Losartan Produces Acute Antiallodynic Effect in Neuropathic Pain Evoked by Sciatic Nerve Injury
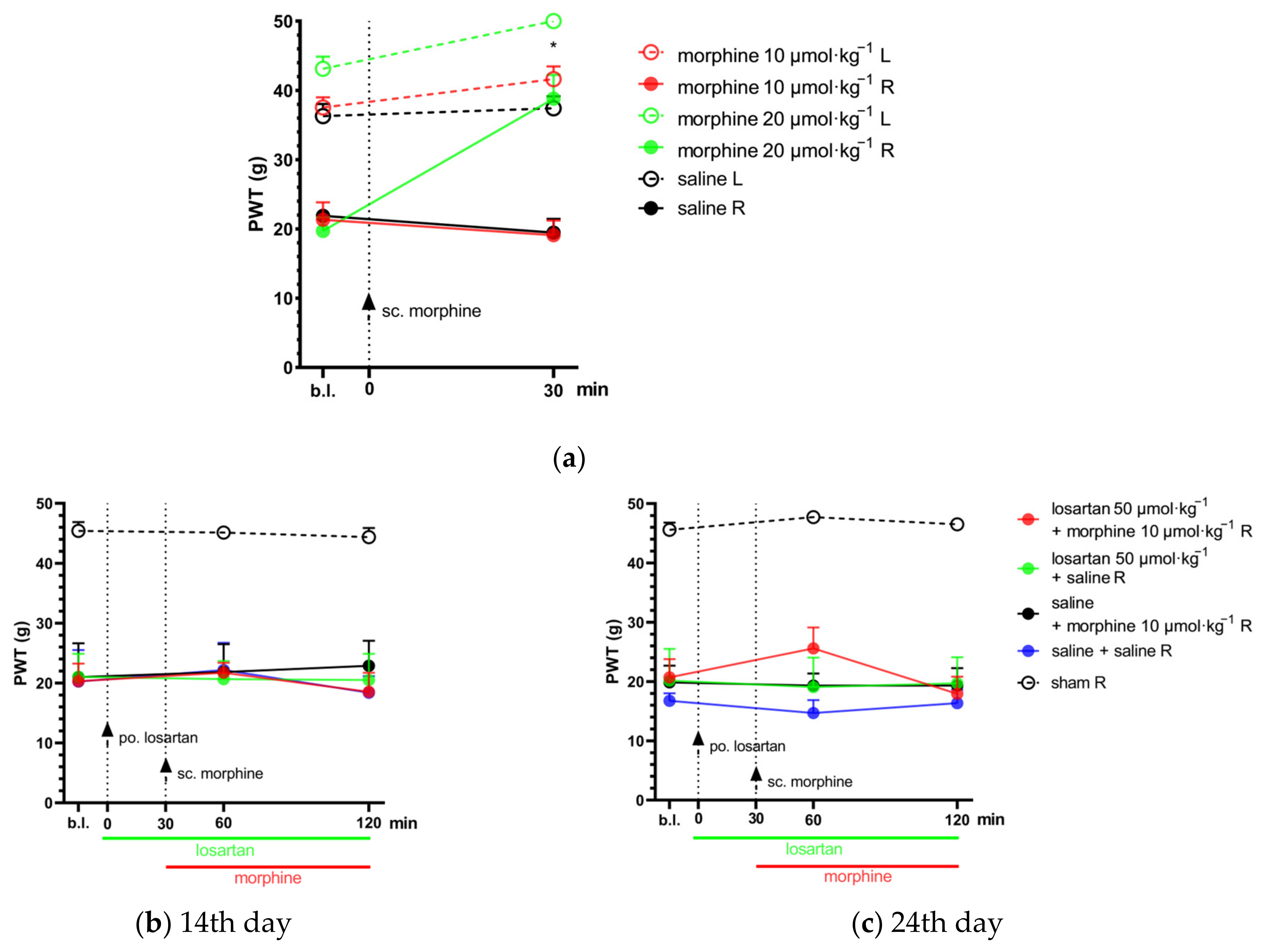
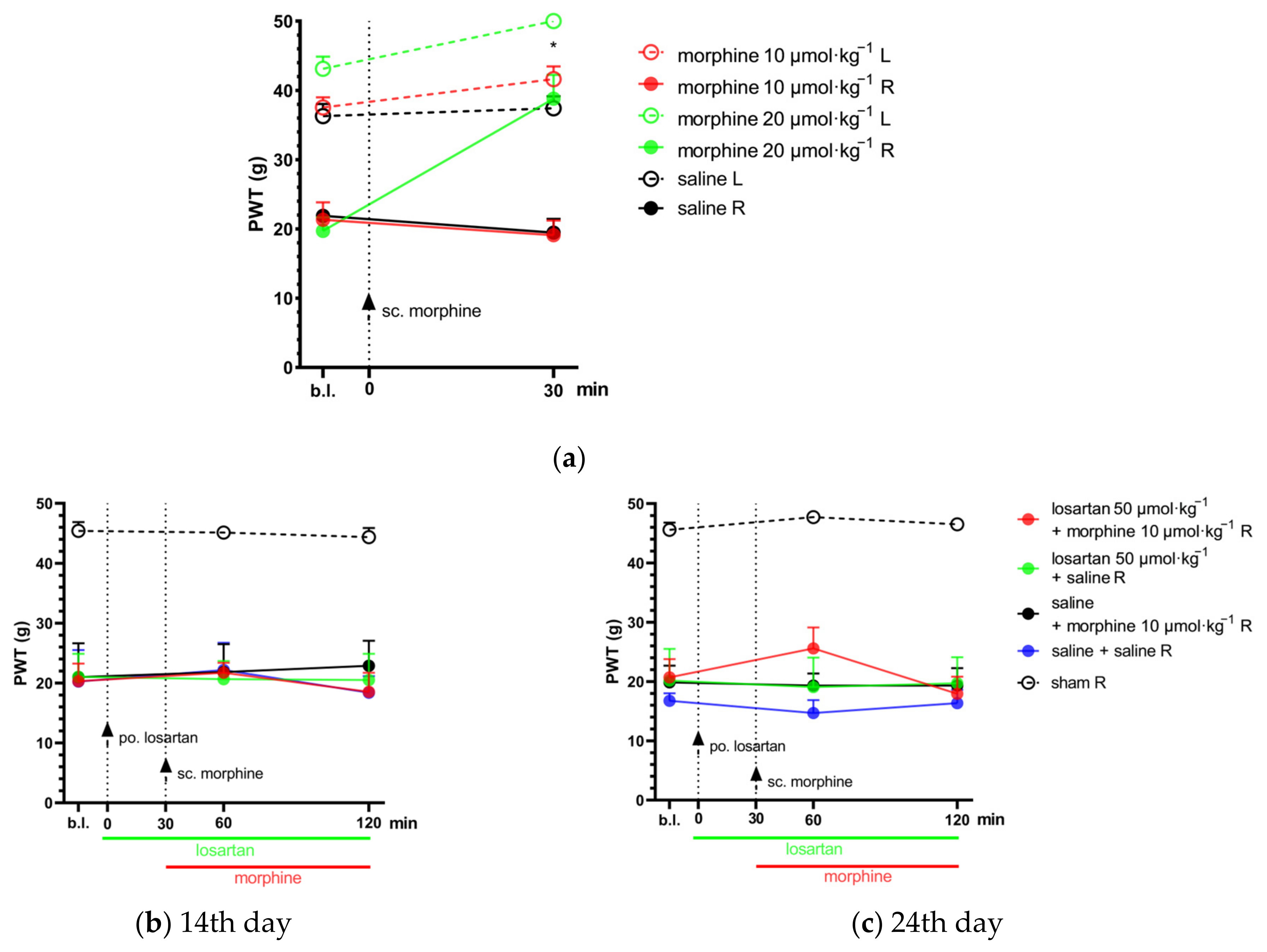
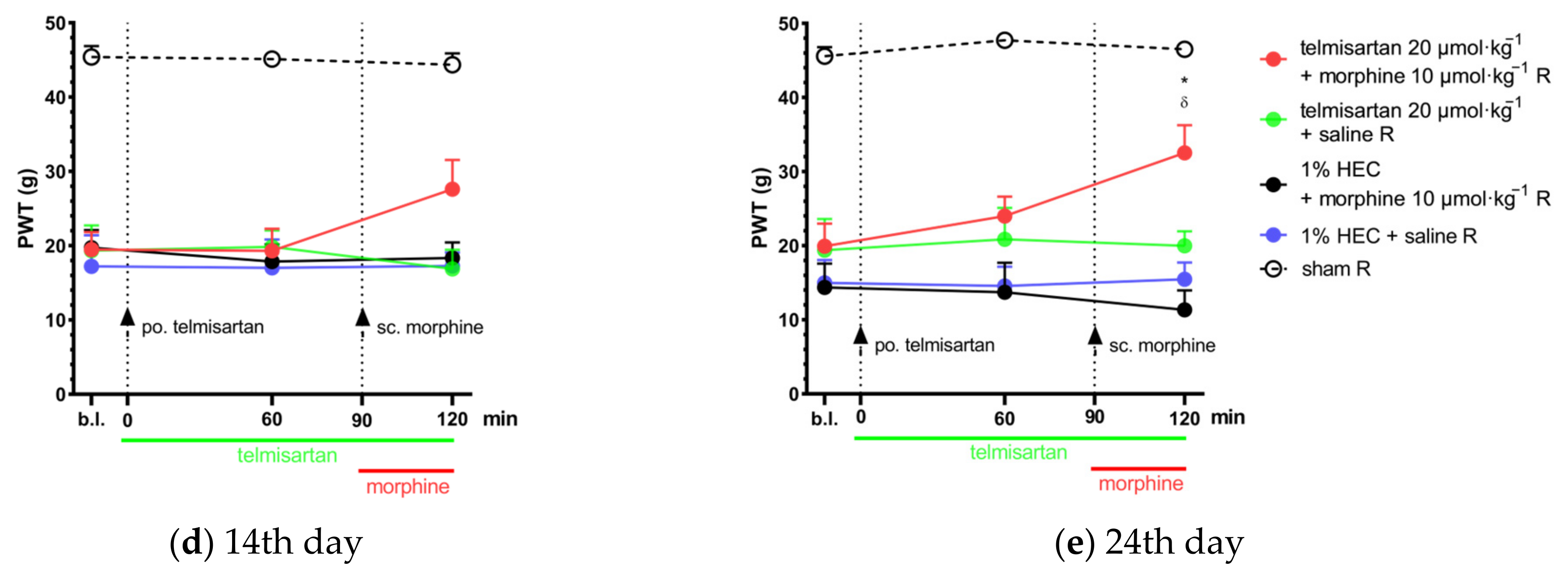
2.2. Combination of Telmisartan and Morphine in Subanalgesic Doses Alleviates Neuropathic Pain Evoked by Sciatic Nerve Injury
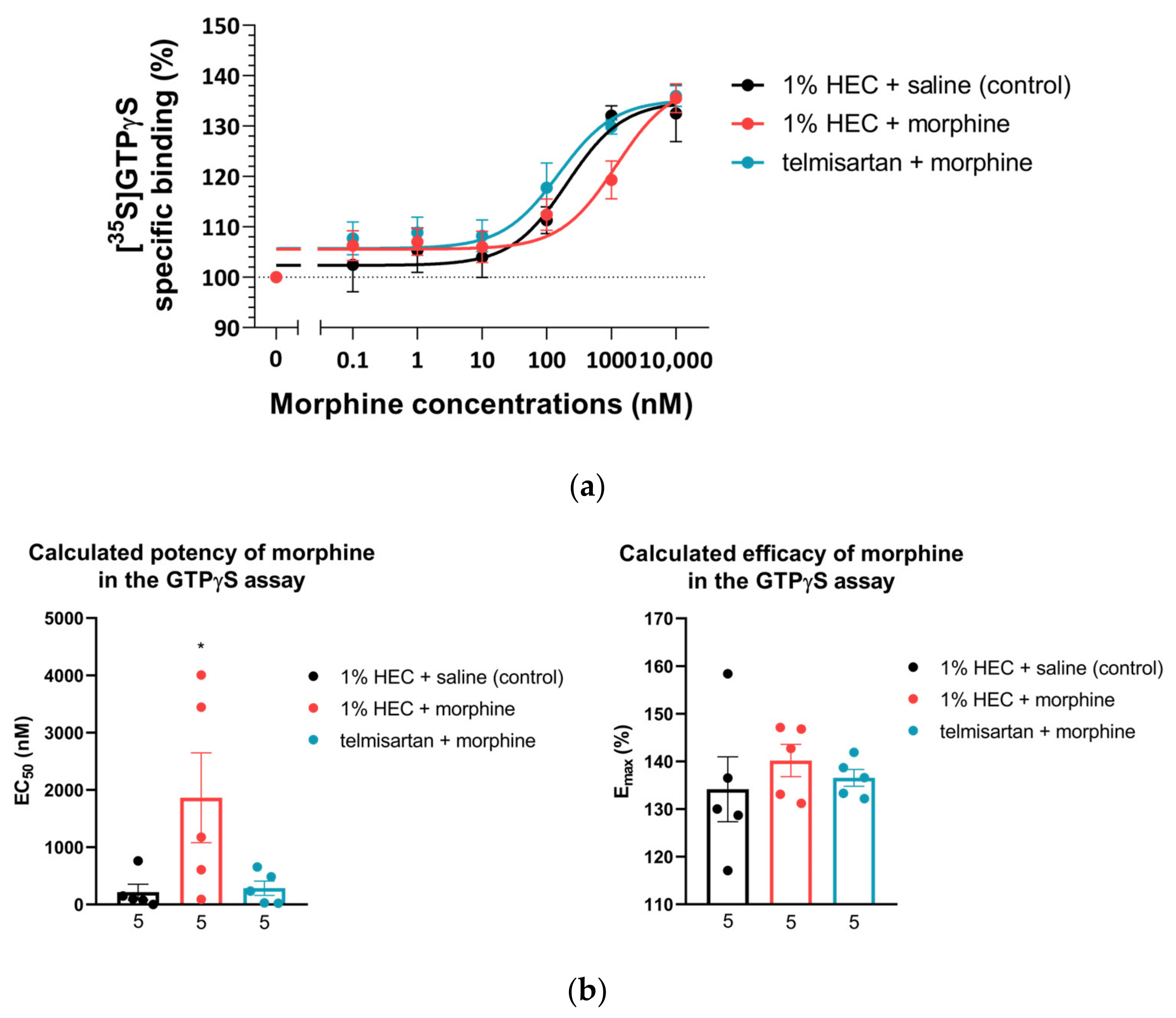
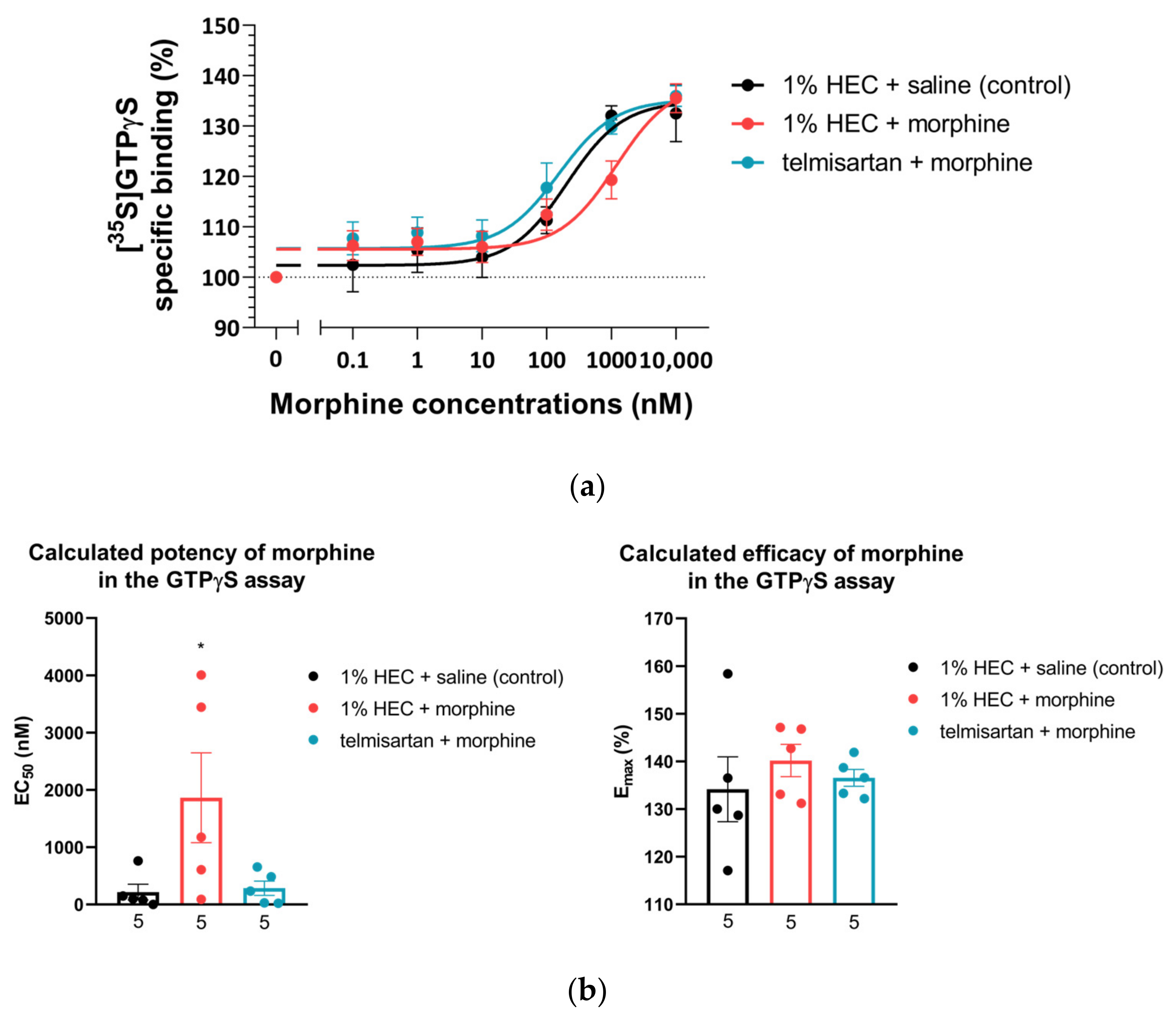
2.3. Impact of Chronic Telmisartan and Morphine Combination Treatment on Morphine-Stimulated [35S]GTPγS Binding in Spinal Cord Membranes of Neuropathic Rats
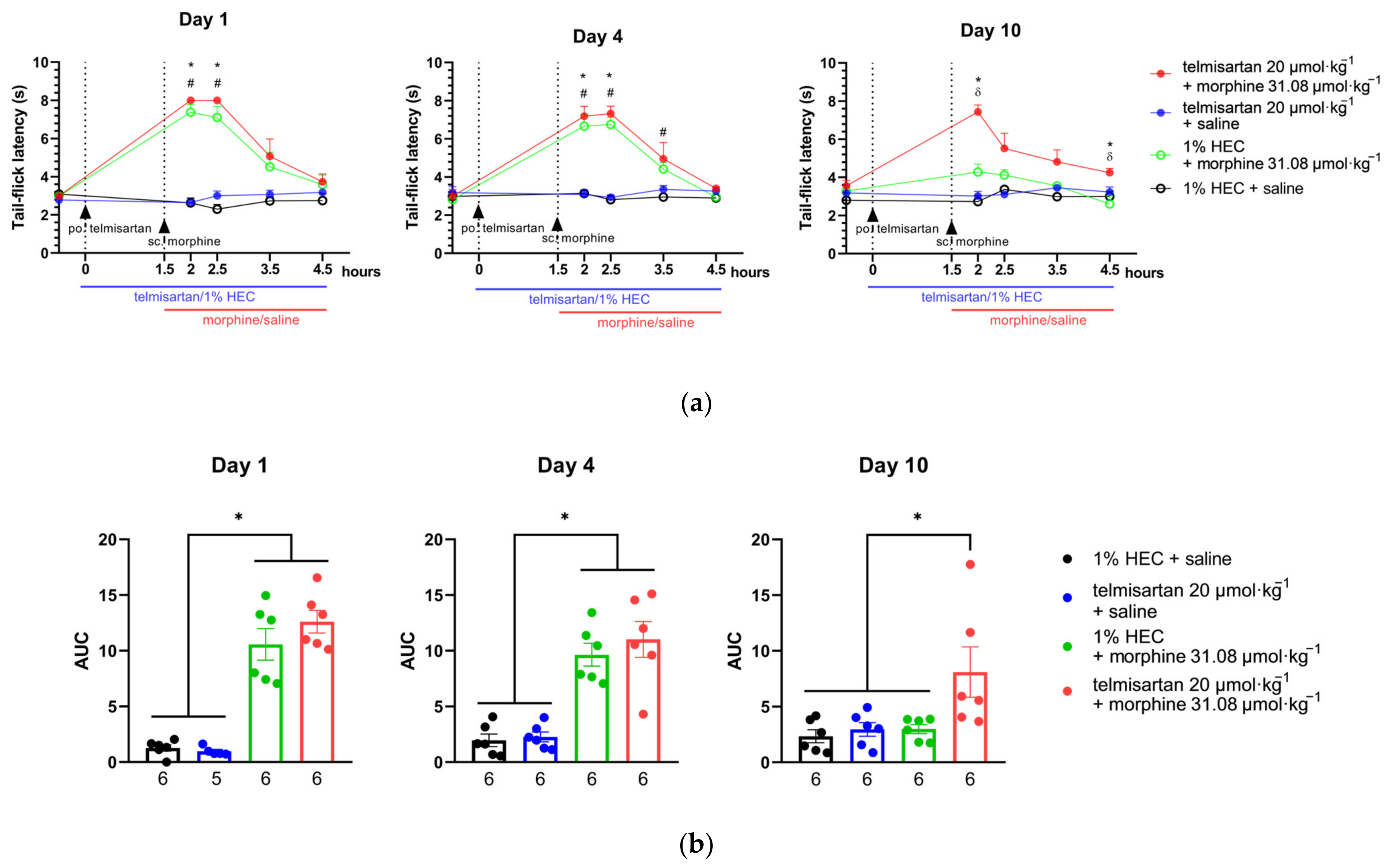
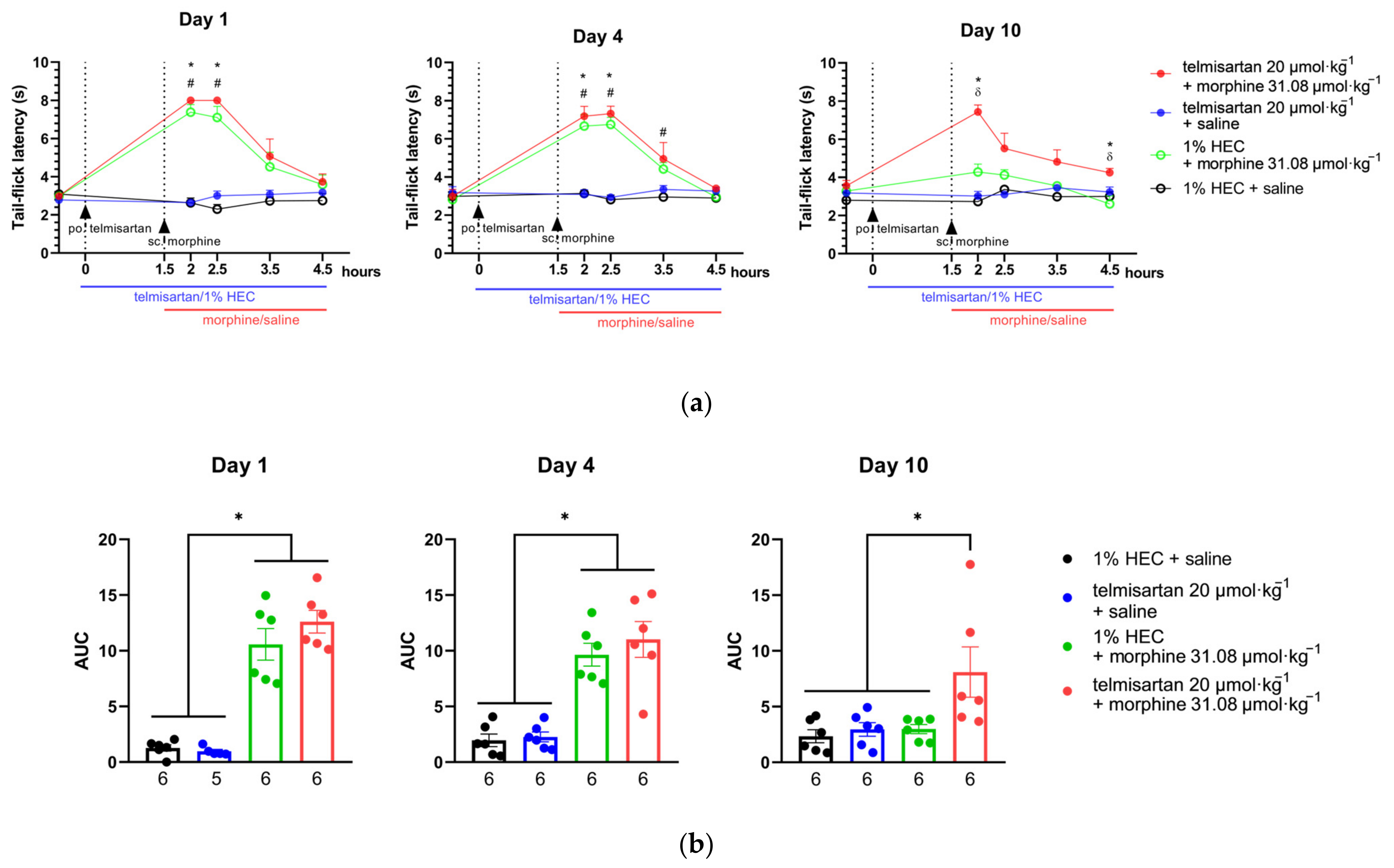
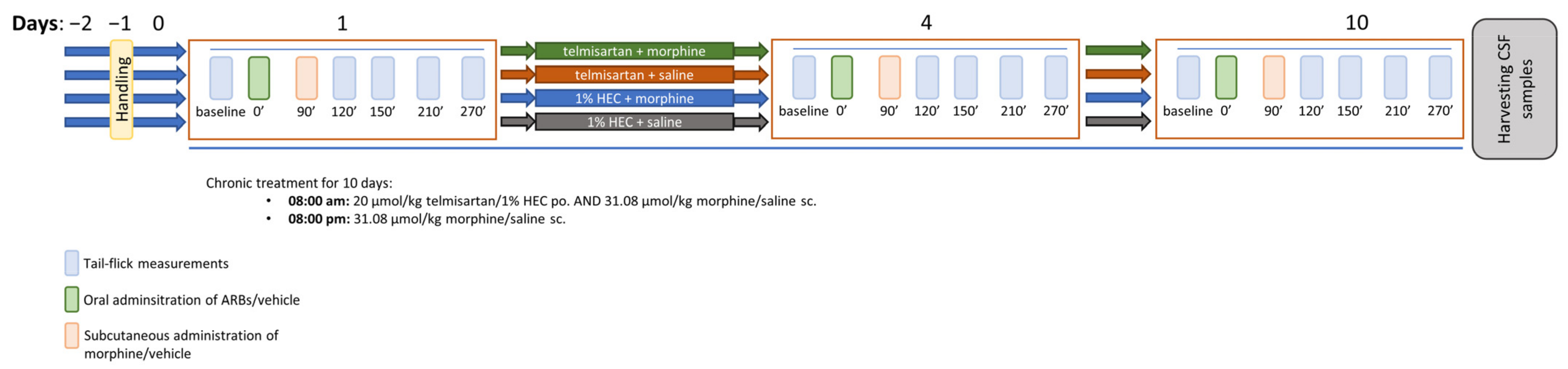
2.4. Telmisartan Delays the Development of Morphine Analgesic Tolerance in Rat Tail-Flick Assay
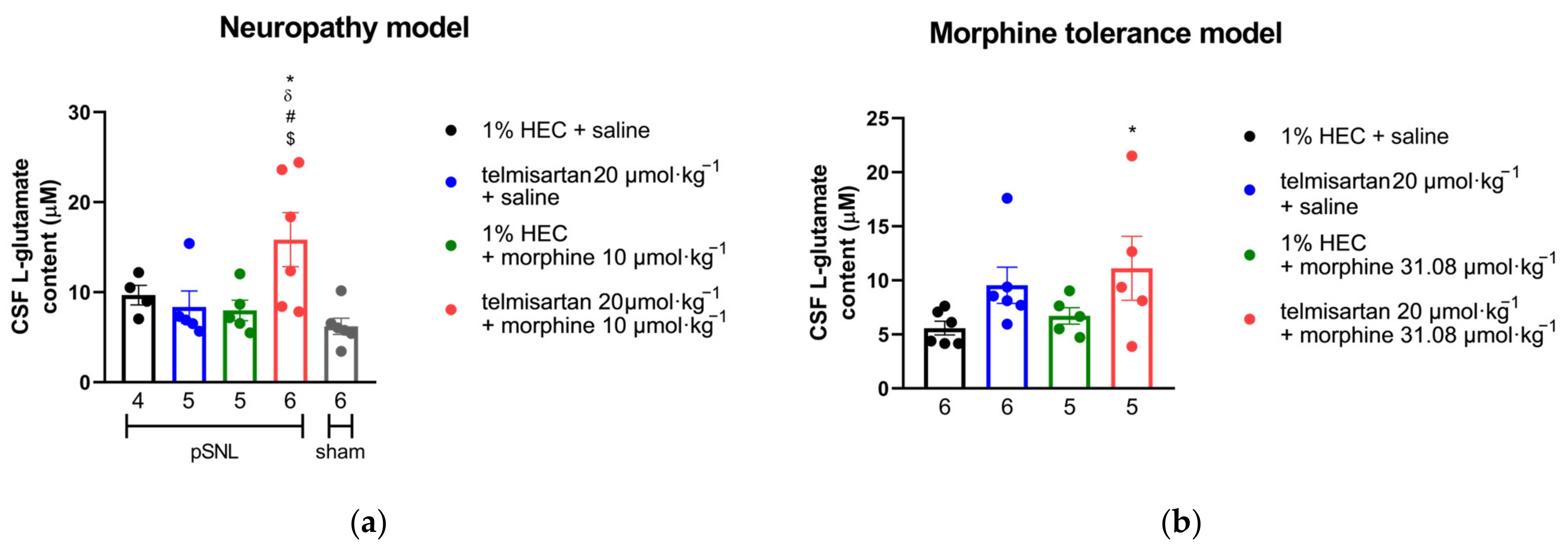
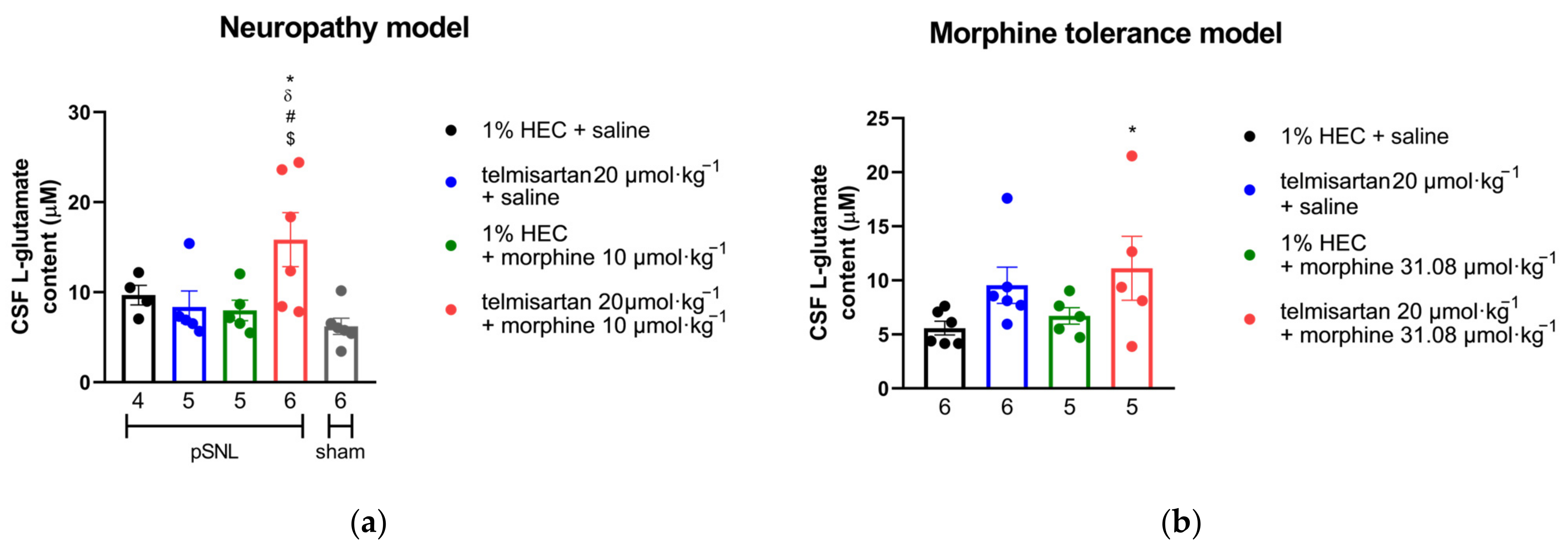
2.5. Impact of Angiotensin Receptor Blockers and Morphine on the L-Glutamate Content of the CSF
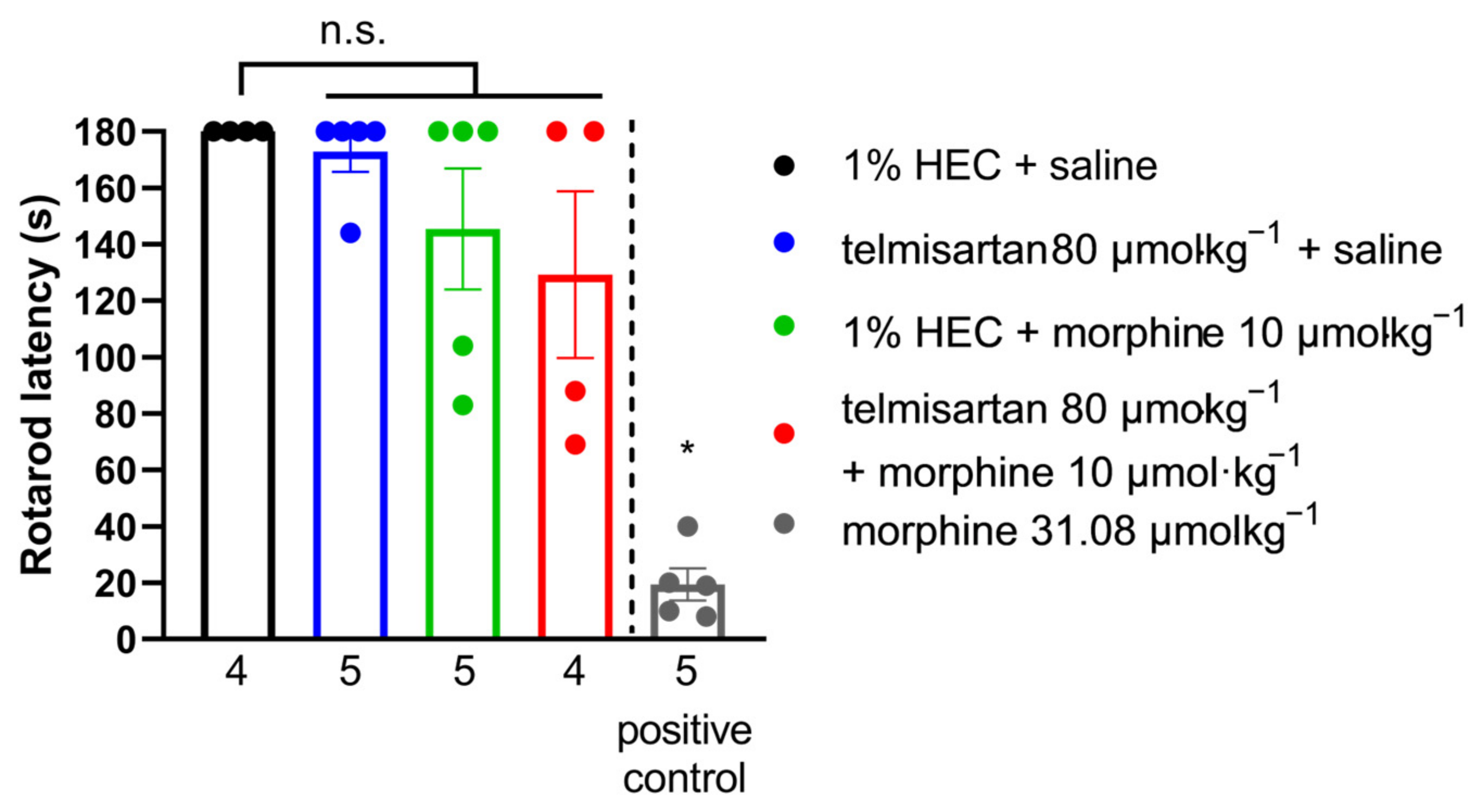
2.6. Impact of Efficient Test Compounds on the Motor Coordination of Rats
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Materials
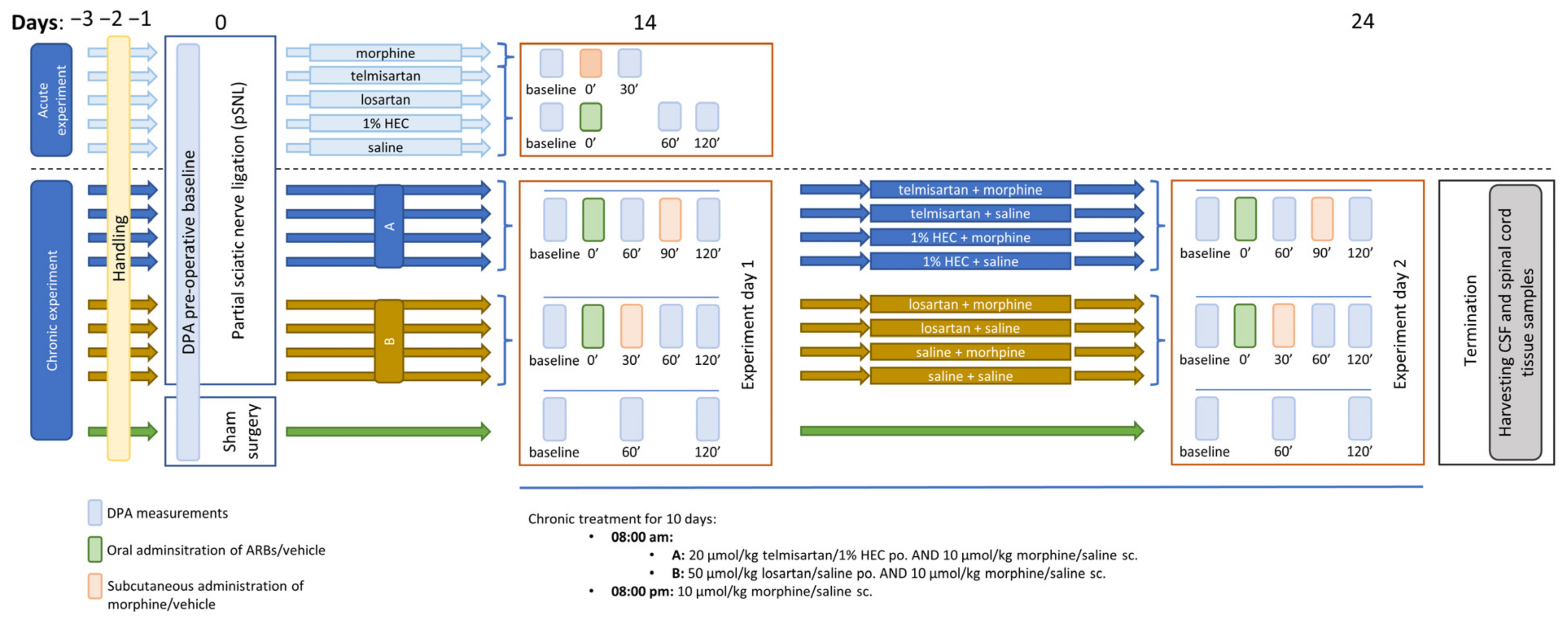
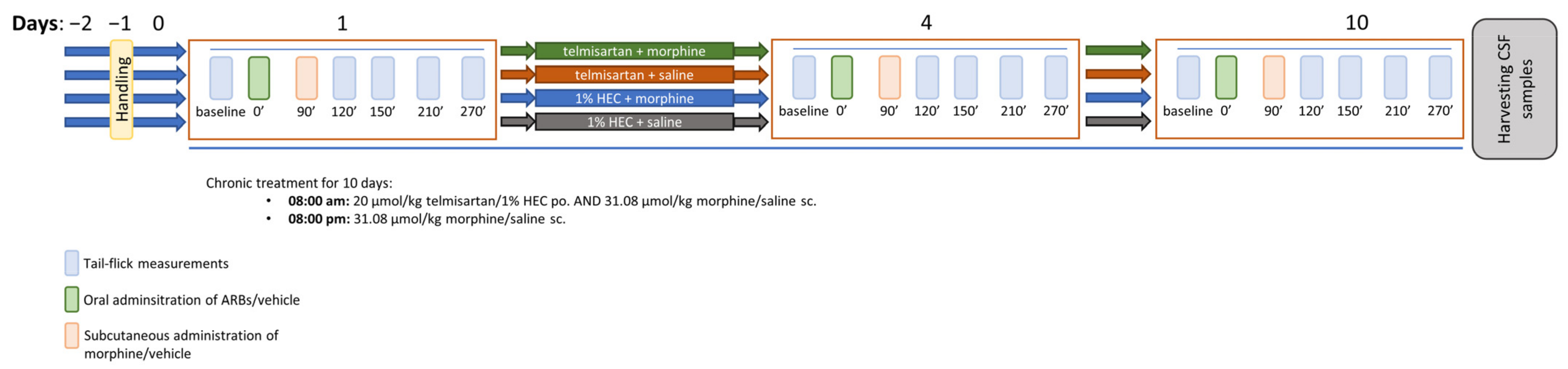
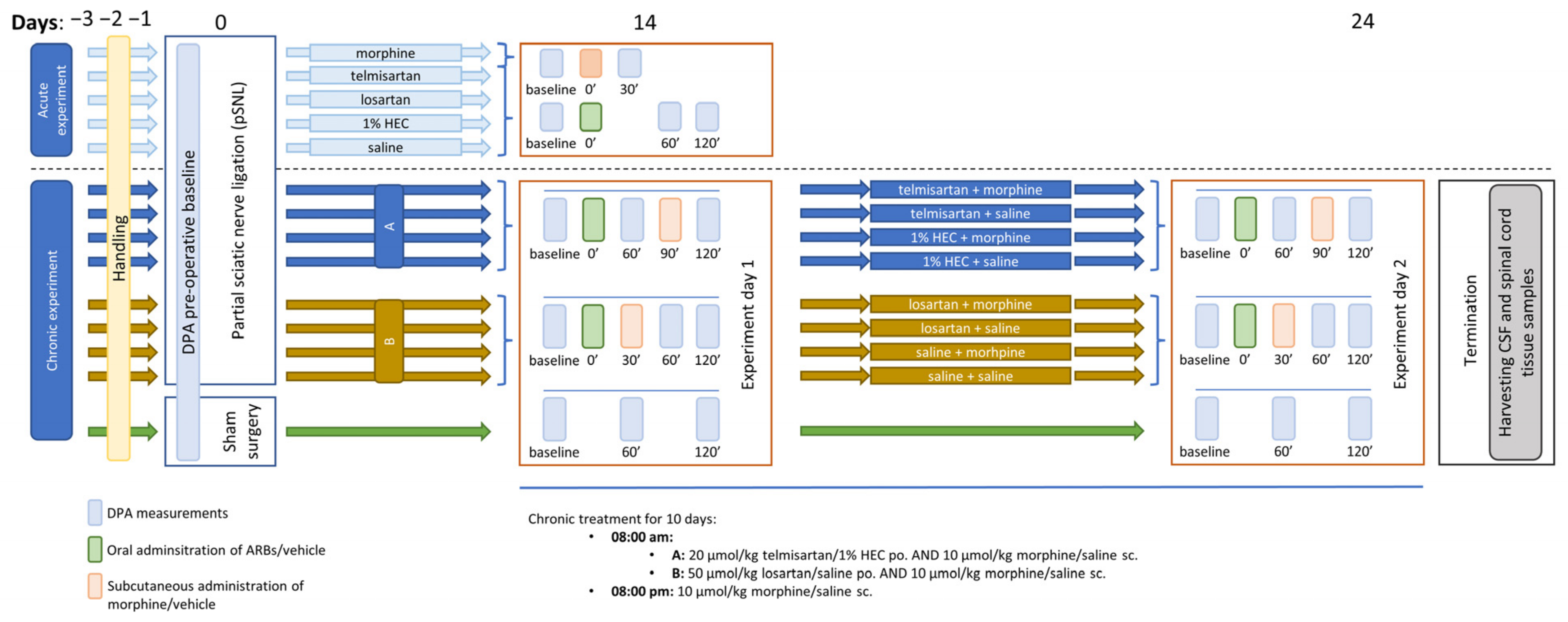
4.3. Experimental Protocols
4.3.1. Mononeuropathic Pain Model
4.3.2. Morphine Analgesic-Tolerance Model
4.3.3. Morphine-Stimulated [35S]GTPγS Binding Assay
4.3.4. Capillary Electrophoresis Analysis of Glutamate Content
4.3.5. Motor Function Testing
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


References
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Torrance, N.; Smith, B.H.; Bennett, M.I.; Lee, A.J. The Epidemiology of Chronic Pain of Predominantly Neuropathic Origin. Results from a General Population Survey. J. Pain 2006, 7, 281–289. [Google Scholar] [CrossRef]
- van Hecke, O.; Torrance, N.; Smith, B.H. Chronic pain epidemiology and its clinical relevance. Br. J. Anaesth. 2013, 111, 13–18. [Google Scholar] [CrossRef]
- Mendlik, M.T.; Uritsky, T.J. Treatment of Neuropathic Pain. Curr. Treat. Options Neurol. 2015, 17, 50. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Sindrup, S.H.; Jensen, T.S. The evidence for pharmacological treatment of neuropathic pain. Pain 2010, 150, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Fornasari, D. Pharmacotherapy for Neuropathic Pain: A Review. Pain Ther. 2017, 6, 25–33. [Google Scholar] [CrossRef]
- Attal, N.; Cruccu, G.; Baron, R.; Haanpää, M.; Hansson, P.; Jensen, T.S.; Nurmikko, T. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur. J. Neurol. 2010, 17, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, R.H.; O’Connor, A.B.; Backonja, M.; Farrar, J.T.; Finnerup, N.B.; Jensen, T.S.; Kalso, E.A.; Loeser, J.D.; Miaskowski, C.; Nurmikko, T.J.; et al. Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain 2007, 132, 237–251. [Google Scholar] [CrossRef]
- Jefferies, K. Treatment of Neuropathic Pain. Semin. Neurol. 2010, 30, 425–432. [Google Scholar] [CrossRef]
- Caraceni, A.; Hanks, G.; Kaasa, S.; Bennett, M.I.; Brunelli, C.; Cherny, N.; Dale, O.; De Conno, F.; Fallon, M.; Hanna, M.; et al. Use of opioid analgesics in the treatment of cancer pain: Evidence-based recommendations from the EAPC. Lancet Oncol. 2012, 13, e58–e68. [Google Scholar] [CrossRef]
- Cherny, N.I. Opioid Analgesics: Comparative Features and Prescribing Guidelines. Drugs 1996, 51, 713–737. [Google Scholar] [CrossRef] [PubMed]
- Schug, S.A.; Palmer, G.M.; Scott, D.A.; Halliwell, R.; Trinca, J. Acute pain management: Scientific evidence, fourth edition, 2015. Med. J. Aust. 2016, 204, 315–317.e1. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Navarro, M.; Maldonado, R.; Baños, J.E. Why mu-opioid agonists have less analgesic efficacy in neuropathic pain? Eur. J. Pain 2019, 23, 435–454. [Google Scholar] [CrossRef]
- Balogh, M.; Zádor, F.; Zádori, Z.S.; Shaqura, M.; Király, K.; Mohammadzadeh, A.; Varga, B.; Lázár, B.; Mousa, S.A.; Hosztafi, S.; et al. Efficacy-based perspective to overcome reduced opioid analgesia of advanced painful diabetic neuropathy in rats. Front. Pharmacol. 2019, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.R.; Sweigart, K.L.; Lakoski, J.M.; Pan, H.L. Functional μ opioid receptors are reduced in the spinal cord dorsal horn of diabetic rats. Anesthesiology 2002, 97, 1602–1608. [Google Scholar] [CrossRef]
- Shaqura, M.; Khalefa, B.I.; Shakibaei, M.; Winkler, J.; Al-Khrasani, M.; Fürst, S.; Mousa, S.A.; Schäfer, M. Reduced number, G protein coupling, and antinociceptive efficacy of spinal mu-opioid receptors in diabetic rats are reversed by nerve growth factor. J. Pain 2013, 14, 720–730. [Google Scholar] [CrossRef]
- Zurek, J.R.; Nadeson, R.; Goodchild, C.S. Spinal and supraspinal components of opioid antinociception in streptozotocin induced diabetic neuropathy in rats. Pain 2001, 90, 57–63. [Google Scholar] [CrossRef]
- Ueda, H.; Ueda, M. Mechanisms underlying morphine analgesic tolerance and dependence. Front. Biosci. 2009, 14, 5260–5272. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for themanagement of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Kaur, S.; Bali, A.; Singh, N.; Jaggi, A.S. Demystifying the dual role of the angiotensin system in neuropathic pain. Neuropeptides 2022, 94, 102260. [Google Scholar] [CrossRef] [PubMed]
- Balogh, M.; Aguilar, C.; Nguyen, N.T.; Shepherd, A.J. Angiotensin receptors and neuropathic pain. Pain Rep. 2021, 6, e869. [Google Scholar] [CrossRef] [PubMed]
- Király, K.; Karádi, D.; Zádor, F.; Mohammadzadeh, A.; Galambos, A.R.; Balogh, M.; Riba, P.; Tábi, T.; Zádori, Z.S.; Szökő, É.; et al. Shedding light on the pharmacological interactions between µ-opioid analgesics and angiotensin receptor modulators: A new option for treating chronic pain. Molecules 2021, 26, 6168. [Google Scholar] [CrossRef]
- Balogh, M.; Zádori, Z.S.; Lázár, B.; Karádi, D.; László, S.; Mousa, S.A.; Hosztafi, S.; Zádor, F.; Riba, P.; Schäfer, M.; et al. The Peripheral versus Central Antinociception of a Novel Opioid Agonist: Acute Inflammatory Pain in Rats. Neurochem. Res. 2018, 43, 1250–1257. [Google Scholar] [CrossRef]
- Jensen, T.S.; Finnerup, N.B. Allodynia and hyperalgesia in neuropathic pain: Clinical manifestations and mechanisms. Lancet Neurol. 2014, 13, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Al-Rejaie, S.S.; Abuohashish, H.M.; Ahmed, M.M.; Arrejaie, A.S.; Aleisa, A.M.; AlSharari, S.D. Telmisartan inhibits hyperalgesia and inflammatory progression in a diabetic neuropathic pain model of Wistar rats. Neurosciences 2015, 20, 115–123. [Google Scholar] [CrossRef]
- Ogata, Y.; Nemoto, W.; Nakagawasai, O.; Yamagata, R.; Tadano, T.; Tan-No, K. Involvement of spinal angiotensin II system in streptozotocin-induced diabetic neuropathic pain in mice. Mol. Pharmacol. 2016, 90, 205–213. [Google Scholar] [CrossRef]
- Kim, E.; Hwang, S.H.; Kim, H.K.; Abdi, S.; Kim, H.K. Losartan, an Angiotensin II Type 1 Receptor Antagonist, Alleviates Mechanical Hyperalgesia in a Rat Model of Chemotherapy-Induced Neuropathic Pain by Inhibiting Inflammatory Cytokines in the Dorsal Root Ganglia. Mol. Neurobiol. 2019, 56, 7408–7419. [Google Scholar] [CrossRef]
- Kalynovska, N.; Diallo, M.; Sotakova-Kasparova, D.; Palecek, J. Losartan attenuates neuroinflammation and neuropathic pain in paclitaxel-induced peripheral neuropathy. J. Cell. Mol. Med. 2020, 24, 7949–7958. [Google Scholar] [CrossRef]
- Hegazy, N.; Rezq, S.; Fahmy, A. Mechanisms Involved in Superiority of Angiotensin Receptor Blockade over ACE Inhibition in Attenuating Neuropathic Pain Induced in Rats. Neurotherapeutics 2020, 17, 1031–1047. [Google Scholar] [CrossRef]
- Jaggi, A.S.; Singh, N. Exploring the potential of telmisartan in chronic constriction injury-induced neuropathic pain in rats. Eur. J. Pharmacol. 2011, 667, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Yoon, Y.W.; Chung, J.M. Comparison of three rodent neuropathic pain models. Exp. Brain Res. 1997, 113, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.C.O.; Romero, T.R.; Pacheco, D.F.; Perez, A.C.; Savernini, A.; Santos, R.R.; Duarte, I.D. Participation of AT1 and Mas receptors in the modulation of inflammatory pain. Peptides 2014, 61, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.J.; Fan, X. The Possible Role of the Angiotensin System in the Pathophysiology of Schizophrenia: Implications for Pharmacotherapy. CNS Drugs 2019, 33, 539–547. [Google Scholar] [CrossRef]
- Wincewicz, D.; Braszko, J.J. Telmisartan attenuates cognitive impairment caused by chronic stress in rats. Pharmacol. Rep. 2014, 66, 436–441. [Google Scholar] [CrossRef]
- Gohlke, P.; Weiss, S.; Jansen, A.; Wienen, W.; Stangier, J.; Rascher, W.; Culman, J.; Unger, T. AT1 receptor antagonist telmisartan administered peripherally inhibits central responses to angiotensin II in conscious rats. J. Pharmacol. Exp. Ther. 2001, 298, 62–70. [Google Scholar]
- Konno, S.; Hirooka, Y.; Kishi, T.; Sunagawa, K. Sympathoinhibitory effects of telmisartan through the reduction of oxidative stress in the rostral ventrolateral medulla of obesity-induced hypertensive rats. J. Hypertens. 2012, 30, 1992–1999. [Google Scholar] [CrossRef]
- Wang, J.M.; Tan, J.; Leenen, F.H.H. Central nervous system blockade by peripheral administration of AT1 receptor blockers. J. Cardiovasc. Pharmacol. 2003, 41, 593–599. [Google Scholar] [CrossRef]
- Wang, J.; Pang, T.; Hafko, R.; Benicky, J.; Sanchez-Lemus, E.; Saavedra, J.M. Telmisartan ameliorates glutamate-induced neurotoxicity: Roles of AT 1 receptor blockade and PPARγ activation. Neuropharmacology 2014, 79, 249–261. [Google Scholar] [CrossRef]
- Kurtz, T.W. Treating the metabolic syndrome: Telmisartan as a peroxisome proliferator-activated receptor-gamma activator. Acta Diabetol. 2005, 42, s9–s16. [Google Scholar] [CrossRef]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ding, Z.; Ma, S.; Zou, Y.; Yang, X.; Ding, Z.; Zhang, Y.; Zhu, X.; Schäfer, M.K.E.; Guo, Q.; et al. Targeting aurora kinase B alleviates spinal microgliosis and neuropathic pain in a rat model of peripheral nerve injury. J. Neurochem. 2020, 152, 72–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, L.-J.; Wang, J.; Li, D.; Ren, W.-J.; Peng, J.; Wei, X.; Xu, T.; Xin, W.-J.; Pang, R.-P.; et al. TNF-α differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J. Neurosci. 2017, 37, 871–881. [Google Scholar] [CrossRef]
- Pottorf, T.S.; Rotterman, T.M.; McCallum, W.M.; Haley-Johnson, Z.A.; Alvarez, F.J. The Role of Microglia in Neuroinflammation of the Spinal Cord after Peripheral Nerve Injury. Cells 2022, 11, 2083. [Google Scholar] [CrossRef]
- Li, X.; Guo, Q.; Ye, Z.; Wang, E.; Zou, W.; Sun, Z.; He, Z.; Zhong, T.; Weng, Y.; Pan, Y. PPAR γ Prevents Neuropathic Pain by Down-Regulating CX3CR1 and Attenuating M1 Activation of Microglia in the Spinal Cord of Rats Using a Sciatic Chronic Constriction Injury Model. Front. Neurosci. 2021, 15, 620525. [Google Scholar] [CrossRef] [PubMed]
- Schupp, M.; Lee, L.D.; Frost, N.; Umbreen, S.; Schmidt, B.; Unger, T.; Kintscher, U. Regulation of peroxisome proliferator—Activated receptor γ activity by losartan metabolites. Hypertension 2006, 47, 586–589. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Coats, B.D.; Lewis, S.S.; Zhang, Y.; Sprunger, D.B.; Rezvani, N.; Baker, E.M.; Jekich, B.M.; Wieseler, J.L.; Somogyi, A.A.; et al. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav. Immun. 2008, 22, 1178–1189. [Google Scholar] [CrossRef]
- Madia, P.A.; Navani, D.M.; Yoburn, B.C. [35S]GTPγS binding and opioid tolerance and efficacy in mouse spinal cord. Pharmacol. Biochem. Behav. 2012, 101, 155–165. [Google Scholar] [CrossRef]
- Kohno, T.; Ji, R.-R.; Ito, N.; Allchorne, A.J.; Befort, K.; Karchewski, L.A.; Woolf, C.J. Peripheral axonal injury results in reduced μ opioid receptor pre- and post-synaptic action in the spinal cord. Pain 2005, 117, 77–87. [Google Scholar] [CrossRef]
- De Guglielmo, G.; Kallupi, M.; Scuppa, G.; Stopponi, S.; Demopulos, G.; Gaitanaris, G.; Ciccocioppo, R. Analgesic tolerance to morphine is regulated by PPARγ. Br. J. Pharmacol. 2014, 171, 5407–5416. [Google Scholar] [CrossRef]
- Morgenweck, J.; Griggs, R.B.; Donahue, R.R.; Zadina, J.E.; Taylor, B.K. PPARγ activation blocks development and reduces established neuropathic pain in rats. Neuropharmacology 2013, 70, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Rivat, C.; Sebaihi, S.; Van Steenwinckel, J.; Fouquet, S.; Kitabgi, P.; Pohl, M.; Parsadaniantz, S.M.; Goazigo, A.R.-L. Src family kinases involved in CXCL12-induced loss of acute morphine analgesia. Brain Behav. Immun. 2014, 38, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.M.; Jung, H.; Ripsch, M.S.; Miller, R.J.; White, F.A. CXCR4 signaling mediates morphine-induced tactile hyperalgesia. Brain Behav. Immun. 2011, 25, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of μ-opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 2013, 65, 223–254. [Google Scholar] [CrossRef] [PubMed]
- Al-Khrasani, M.; Mohammadzadeh, A.; Balogh, M.; Király, K.; Barsi, S.; Hajnal, B.; Köles, L.; Zádori, Z.S.; Harsing, L.G. Glycine transporter inhibitors: A new avenue for managing neuropathic pain. Brain Res. Bull. 2019, 152, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, A.; Lakatos, P.P.; Balogh, M.; Zádor, F.; Karádi, D.; Zádori, Z.S.; Király, K.; Galambos, A.R.; Barsi, S.; Riba, P.; et al. Pharmacological evidence on augmented antiallodynia following systemic co-treatment with glyt-1 and glyt-2 inhibitors in rat neuropathic pain model. Int. J. Mol. Sci. 2021, 22, 2479. [Google Scholar] [CrossRef] [PubMed]
- Rotman, N.; Wahli, W. PPAR modulation of kinase-linked receptor signaling in physiology and disease. Physiology 2010, 25, 176–185. [Google Scholar] [CrossRef]
- Pang, T.; Wang, J.; Benicky, J.; Sánchez-Lemus, E.; Saavedra, J.M. Telmisartan directly ameliorates the neuronal inflammatory response to IL-1β partly through the JNK/c-Jun and NADPH oxidase pathways. J. Neuroinflamm. 2012, 9, 102. [Google Scholar] [CrossRef]
- Christoph, T.; Schiene, K.; Englberger, W.; Parsons, C.G.; Chizh, B.A. The antiallodynic effect of NMDA antagonists in neuropathic pain outlasts the duration of the in vivo NMDA antagonism. Neuropharmacology 2006, 51, 12–17. [Google Scholar] [CrossRef]
- Chen, S.R.; Samoriski, G.; Pan, H.L. Antinociceptive effects of chronic administration of uncompetitive NMDA receptor antagonists in a rat model of diabetic neuropathic pain. Neuropharmacology 2009, 57, 121–126. [Google Scholar] [CrossRef]
- Trujillo, K.A.; Akil, H. Inhibition of opiate tolerance by non-competitive N-d-aspartate receptor antagonists. Brain Res. 1994, 633, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Fürst, S.; Zádori, Z.S.; Zádor, F.; Király, K.; Balogh, M.; László, S.B.; Hutka, B.; Mohammadzadeh, A.; Calabrese, C.; Galambos, A.R.; et al. On the role of peripheral sensory and gut mu opioid receptors: Peripheral analgesia and tolerance. Molecules 2020, 25, 2473. [Google Scholar] [CrossRef] [PubMed]
- Murai, N.; Sekizawa, T.; Gotoh, T.; Watabiki, T.; Takahashi, M.; Kakimoto, S.; Takahashi, Y.; Iino, M.; Nagakura, Y. Spontaneous and evoked pain-associated behaviors in a rat model of neuropathic pain respond differently to drugs with different mechanisms of action. Pharmacol. Biochem. Behav. 2016, 141, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, P.P.; Karádi, D.; Galambos, A.R.; Essmat, N.; Király, K.; Laufer, R.; Geda, O.; Zádori, Z.S.; Tábi, T.; Al-Khrasani, M.; et al. The Acute Antiallodynic Effect of Tolperisone in Rat Neuropathic Pain and Evaluation of Its Mechanism of Action. Int. J. Mol. Sci. 2022, 23, 9564. [Google Scholar] [CrossRef]
- Balogh, M.; Varga, B.K.; Karádi, D.; Riba, P.; Puskár, Z.; Kozsurek, M.; Al-Khrasani, M.; Király, K. Similarity and dissimilarity in antinociceptive effects of dipeptidyl-peptidase 4 inhibitors, Diprotin A and vildagliptin in rat inflammatory pain models following spinal administration. Brain Res. Bull. 2019, 147, 78–85. [Google Scholar] [CrossRef]
- Seltzer, Z.; Dubner, R.; Shir, Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 1990, 43, 205–218. [Google Scholar] [CrossRef]
- Tulunay, F.C.; Takemori, A.E. The increased efficacy of narcotic antagonists induced by various narcotic analgesics. J. Pharmacol. Exp. Ther. 1974, 190, 395–400. [Google Scholar]
- Kiraly, K.; Caputi, F.F.; Hanuska, A.; Kató, E.; Balogh, M.; Köles, L.; Palmisano, M.; Riba, P.; Hosztafi, S.; Romualdi, P.; et al. A new potent analgesic agent with reduced liability to produce morphine tolerance. Brain Res. Bull. 2015, 117, 32–38. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Jakó, T.; Szabó, E.; Tábi, T.; Zachar, G.; Csillag, A.; Szöko, É. Chiral analysis of amino acid neurotransmitters and neuromodulators in mouse brain by CE-LIF. Electrophoresis 2014, 35, 2870–2876. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Group (10 Days of Treatment) | GTPγS Binding | |
|---|---|---|
| EC50 ± S.E.M. (nM) | Emax ± S.E.M. (%) | |
| 1% HEC + saline (n = 5) | 217.2 ± 137.8 | 134.1 ± 6.8 |
| 1% HEC + morphine (n = 5) | 1864.0 ± 783.5 *δ | 140.2 ± 3.4 |
| telmisartan + morphine (n = 5) | 285.0 ± 125.1 | 136.5 ± 1.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karádi, D.Á.; Galambos, A.R.; Lakatos, P.P.; Apenberg, J.; Abbood, S.K.; Balogh, M.; Király, K.; Riba, P.; Essmat, N.; Szűcs, E.; et al. Telmisartan Is a Promising Agent for Managing Neuropathic Pain and Delaying Opioid Analgesic Tolerance in Rats. Int. J. Mol. Sci. 2023, 24, 7970. https://doi.org/10.3390/ijms24097970
Karádi DÁ, Galambos AR, Lakatos PP, Apenberg J, Abbood SK, Balogh M, Király K, Riba P, Essmat N, Szűcs E, et al. Telmisartan Is a Promising Agent for Managing Neuropathic Pain and Delaying Opioid Analgesic Tolerance in Rats. International Journal of Molecular Sciences. 2023; 24(9):7970. https://doi.org/10.3390/ijms24097970
Chicago/Turabian StyleKarádi, David Á., Anna Rita Galambos, Péter P. Lakatos, Joost Apenberg, Sarah K. Abbood, Mihály Balogh, Kornél Király, Pál Riba, Nariman Essmat, Edina Szűcs, and et al. 2023. "Telmisartan Is a Promising Agent for Managing Neuropathic Pain and Delaying Opioid Analgesic Tolerance in Rats" International Journal of Molecular Sciences 24, no. 9: 7970. https://doi.org/10.3390/ijms24097970
APA StyleKarádi, D. Á., Galambos, A. R., Lakatos, P. P., Apenberg, J., Abbood, S. K., Balogh, M., Király, K., Riba, P., Essmat, N., Szűcs, E., Benyhe, S., Varga, Z. V., Szökő, É., Tábi, T., & Al-Khrasani, M. (2023). Telmisartan Is a Promising Agent for Managing Neuropathic Pain and Delaying Opioid Analgesic Tolerance in Rats. International Journal of Molecular Sciences, 24(9), 7970. https://doi.org/10.3390/ijms24097970