
Targeting Protein–Protein Interfaces with Peptides: The Contribution of Chemical Combinatorial Peptide Library Approaches


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Identification of Protein Partnerships and Characterization of PPI
3. Molecules Targeting PPI
4. Peptides as Effective PPI Modulators
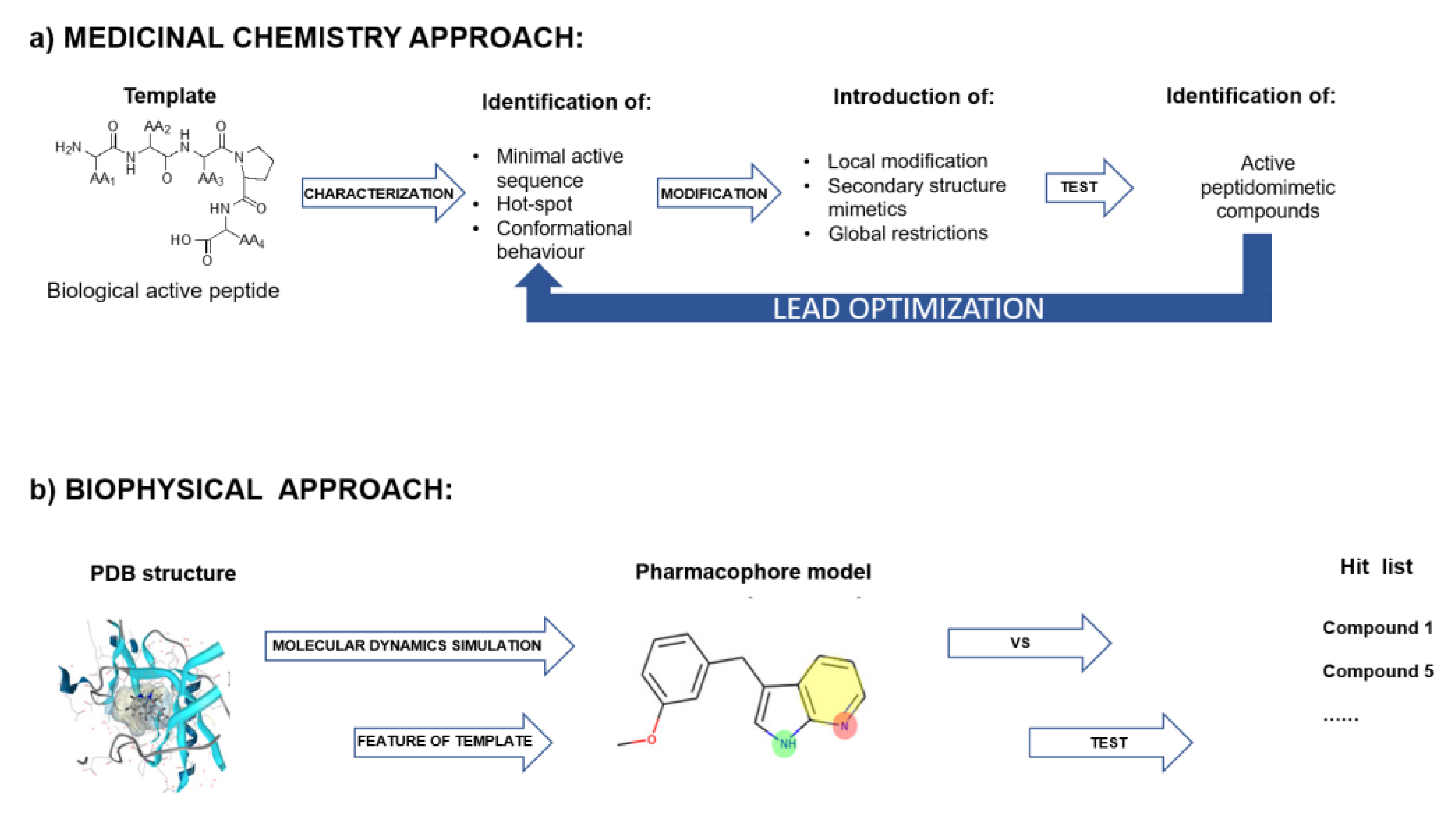
5. Experimental Approaches Used for the Identification and Optimization of Peptides Able to Modulate PPI
5.1. Protein Dissection Approach: Limited Proteolysis and MS Analysis
5.2. Combinatorial Approaches
5.2.1. Phage Display
5.2.2. Chemical Synthesis of Peptide Libraries
6. Successful Applications of Synthetic Combinatorial Approaches in Human Diseases
6.1. Neurological Diseases
6.2. Cancer
6.3. Infectious Diseases
7. Conclusions and Perspectives
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Corbi-Verge, C.; Kim, P.M. Motif mediated protein-protein interactions as drug targets. Cell Commun. Signal. 2016, 14, 8. [Google Scholar] [CrossRef]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef]
- Stein, A.; Aloy, P. Novel peptide-mediated interactions derived from high-resolution 3-dimensional structures. PLoS Comput. Biol. 2010, 6, e1000789. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.R.; Diboun, I.; Dessailly, B.H.; Lees, J.G.; Orengo, C. Transient Protein-Protein Interactions: Structural, Functional, and Network Properties. Structure 2010, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.M.; Oleinikovas, V.; Shammas, S.L.; Wong, C.T.; De Sancho, D.; Baker, C.M.; Clarke, J. Interplay between partner and ligand facilitates the folding and binding of an intrinsically disordered protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15420–15425. [Google Scholar]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Azzarito, V.; Long, K.; Murphy, N.S.; Wilson, A.J. Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Lugovskoy, A.A.; McCarty, J.S.; Li, P.; Wagner, G. Solution structure of DFF40 and DFF45 N-terminal domain complex and mutual chaperone activity of DFF40 and DFF45. Proc. Natl. Acad. Sci. USA 2001, 98, 6051–6055. [Google Scholar] [CrossRef]
- Wright, P.; Dyson, H. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Ishiyama, N.; Lee, S.H.; Liu, S.; Li, G.Y.; Smith, M.J.; Reichardt, L.F.; Ikura, M. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell 2010, 141, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8375. [Google Scholar] [CrossRef]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed]
- Thanasomboon, R.; Kalapanulak, S.; Netrphan, S.; Saithong, T. Exploring dynamic protein-protein interactions in cassava through the integrative interactome network. Sci. Rep. 2020, 10, 6510. [Google Scholar] [PubMed]
- Venkatesan, K.; Rual, J.F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.; Goh, K.I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.D.; Qvit, N.; Mochly-Rosen, D. Peptides and peptidomimetics as regulators of protein-protein interactions. Curr. Opin. Struct. Biol. 2017, 44, 59–66. [Google Scholar] [PubMed]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [PubMed]
- Aramburu, J.; Yaffe, M.B.; Lopez-Rodriguez, C.; Cantley, L.C.; Hogan, P.G.; Rao, A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 1999, 285, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt RM, C.; Putavet, D.A.; Klein JD, D.; Derks KW, J.; Bourgeois BR, M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Poleszak, K.; Kaminska, B. Short peptides interfering with signaling pathways as new therapeutic tools for cancer treatment. Future Med. Chem. 2017, 9, 199–221. [Google Scholar] [CrossRef]
- Hetian, L.; Ping, A.; Shumei, S.; Xiaoying, L.; Luowen, H.; Jian, W.; Lin, M.; Meisheng, L.; Junshan, Y.; Chengchao, S. A novel peptide isolated from a phage display library inhibits tumor growth and metastasis by blocking the binding of vascular endothelial growth factor to its kinase domain receptor. J. Biol. Chem. 2002, 277, 43137–43142. [Google Scholar] [CrossRef]
- Shi, X.D.; Sun, K.; Hu, R.; Liu, X.Y.; Hu, Q.M.; Sun, X.Y.; Yao, B.; Sun, N.; Hao, J.R.; Wei, P.; et al. Blocking the Interaction between EphB2 and ADDLs by a Small Peptide Rescues Impaired Synaptic Plasticity and Memory Deficits in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2016, 36, 11959–11973. [Google Scholar] [CrossRef] [PubMed]
- Bruzzoni-Giovanelli, H.; Alezra, V.; Wolff, N.; Dong, C.Z.; Tuffery, P.; Rebollo, A. Interfering peptides targeting protein-protein interactions: The next generation of drugs? Drug Discov. Today 2018, 23, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N. Protein-protein interaction detection: Methods and analysis. Int. J. Proteomics 2014, 2014, 147648. [Google Scholar] [CrossRef] [PubMed]
- Bian, W.; Jiang, H.; Feng, S.; Chen, J.; Wang, W.; Li, X. Protocol for establishing a protein-protein interaction network using tandem affinity purification followed by mass spectrometry in mammalian cells. STAR Protoc. 2022, 3, 101569. [Google Scholar] [CrossRef]
- Struk, S.; Braem, L.; Walton, A.; De Keyser, A.; Boyer, F.D.; Persiau, G.; De Jaeger, G.; Gevaert, K.; Goormachtig, S. Quantitative Tandem Affinity Purification, an Effective Tool to Investigate Protein Complex Composition in Plant Hormone Signaling: Strigolactones in the Spotlight. Front. Plant Sci. 2018, 9, 528. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Adelmant, G.; Garg, B.K.; Tavares, M.; Card, J.D.; Marto, J.A. Tandem Affinity Purification and Mass Spectrometry (TAP-MS) for the Analysis of Protein Complexes. Curr. Protoc. Protein Sci. 2019, 96, e84. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Chen, T. Using TR-FRET to Investigate Protein-Protein Interactions: A Case Study of PXR-Coregulator Interaction. Adv. Protein Chem. Struct. Biol. 2018, 110, 31–63. [Google Scholar] [PubMed]
- Okamoto, K.; Sako, Y. Recent advances in FRET for the study of protein interactions and dynamics. Curr. Opin. Struct Biol. 2017, 46, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Ko, W.; Lee, H.S.; Shin, I. Analysis of Protein-Protein Interaction in a Single Live Cell by Using a FRET System Based on Genetic Code Expansion Technology. J. Am. Chem. Soc. 2019, 141, 4273–4281. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Niu, Q.; Cicka, D.; Du, Y.; Mo, X.; Fu, H. A time-resolved fluorescence resonance energy transfer screening assay for discovery of protein-protein interaction modulators. STAR Protoc. 2021, 2, 100804. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Ventura, S.; Aviles, F.X. Protein complementation assays: Approaches for the in vivo analysis of protein interactions. FEBS Lett. 2009, 583, 1684–1691. [Google Scholar] [CrossRef]
- Li, P.; Wang, L.; Di, L.J. Applications of Protein Fragment Complementation Assays for Analyzing Biomolecular Interactions and Biochemical Networks in Living Cells. J. Proteome Res. 2019, 18, 2987–2998. [Google Scholar] [CrossRef]
- Valtonen, S.; Vuorinen, E.; Kariniemi, T.; Eskonen, V.; Le Quesne, J.; Bushell, M.; Harma, H.; Kopra, K. Nanomolar Protein-Protein Interaction Monitoring with a Label-Free Protein-Probe Technique. Anal. Chem. 2020, 92, 15781–15788. [Google Scholar] [CrossRef]
- Soltermann, F.; Struwe, W.B.; Kukura, P. Label-free methods for optical in vitro characterization of protein-protein interactions. Phys. Chem. Chem. Phys. 2021, 23, 16488–16500. [Google Scholar] [CrossRef]
- Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Characterization of protein-protein interactions by isothermal titration calorimetry. Methods Mol. Biol. 2004, 261, 35–54. [Google Scholar]
- Chen, X.; Liu, Y.; Huang, J.; Liu, W.; Huang, J.; Zhang, Y.; Fu, W. Label-free techniques for laboratory medicine applications. Front. Lab. Med. 2017, 1, 82–85. [Google Scholar] [CrossRef]
- Speight, R.E.; Cooper, M.A. A survey of the 2010 quartz crystal microbalance literature. J. Mol. Recognit. 2012, 25, 451–473. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.T.; Mercer-Smith, A.R.; Johal, M.S. Quartz microbalance technology for probing biomolecular interactions. Methods Mol. Biol. 2015, 1278, 153–164. [Google Scholar] [PubMed]
- Meyerkord, C.L.; Fu, H. Protein-Protein Interactions; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1278. [Google Scholar]
- Birchenough, H.L.; Jowitt, T.A. Quartz Crystal Microbalance with Dissipation Monitoring (QCM-D): Preparing Functionalized Lipid Layers for the Study of Complex Protein-Ligand Interactions. Methods Mol. Biol. 2021, 2263, 183–197. [Google Scholar]
- Jia, J.; Jin, J.; Chen, Q.; Yuan, Z.; Li, H.; Bian, J.; Gui, L. Eukaryotic expression, Co-IP and MS identify BMPR-1B protein-protein interaction network. Biol. Res. 2020, 53, 24. [Google Scholar] [CrossRef]
- Galletta, B.J.; Rusan, N.M. A yeast two-hybrid approach for probing protein-protein interactions at the centrosome. Methods Cell Biol. 2015, 129, 251–277. [Google Scholar] [PubMed]
- Macalino, S.J.Y.; Basith, S.; Clavio, N.A.B.; Chang, H.; Kang, S.; Choi, S. Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery. Molecules 2018, 23, 1963. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Tuncbag, N.; Gursoy, A. Predicting Protein-Protein Interactions from the Molecular to the Proteome Level. Chem. Rev. 2016, 116, 4884–4909. [Google Scholar] [CrossRef]
- Walhout, A.J.; Sordella, R.; Lu, X.; Hartley, J.L.; Temple, G.F.; Brasch, M.A.; Thierry-Mieg, N.; Vidal, M. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 2000, 287, 116–122. [Google Scholar]
- Singh, R.; Devkota, K.; Sledzieski, S.; Berger, B.; Cowen, L. Topsy-Turvy: Integrating a global view into sequence-based PPI prediction. Bioinformatics 2022, 38 (Suppl. 1), i264–i272. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, J.; Luo, X.; Zhu, W.; Yu, K.; Chen, K.; Li, Y.; Jiang, H. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar] [CrossRef]
- Li, S.; Wu, S.; Wang, L.; Li, F.; Jiang, H.; Bai, F. Recent advances in predicting protein-protein interactions with the aid of artificial intelligence algorithms. Curr. Opin. Struct. Biol. 2022, 73, 102344. [Google Scholar] [CrossRef]
- Callaway, E. The revolution will not be crystallized: A new method sweeps through structural biology. Nature 2015, 525, 172–174. [Google Scholar] [CrossRef]
- Kumar, V.; Mahato, S.; Munshi, A.; Kulharia, M. PPInS: A repository of protein-protein interaction sitesbase. Sci. Rep. 2018, 8, 12453. [Google Scholar] [CrossRef]
- Binder, J.L.; Berendzen, J.; Stevens, A.O.; He, Y.; Wang, J.; Dokholyan, N.V.; Oprea, T.I. AlphaFold illuminates half of the dark human proteins. Curr. Opin. Struct. Biol. 2022, 74, 102372. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Islam, S.; Tankhilevich, E.; Sternberg, M.J.E. The AlphaFold Database of Protein Structures: A Biologist’s Guide. J. Mol. Biol. 2022, 434, 167336. [Google Scholar] [CrossRef]
- Humphreys, I.R.; Pei, J.; Baek, M.; Krishnakumar, A.; Anishchenko, I.; Ovchinnikov, S.; Zhang, J.; Ness, T.J.; Banjade, S.; Bagde, S.R.; et al. Computed structures of core eukaryotic protein complexes. Science 2021, 374, eabm4805. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Xiong, D.; Wierbowski, S.; Li, L.; Liang, S.; Yu, H. Deep learning methods for 3D structural proteome and interactome modeling. Curr. Opin. Struct. Biol. 2022, 73, 102329. [Google Scholar] [CrossRef]
- Xiao, S.; Tian, H.; Tao, P. PASSer2.0: Accurate Prediction of Protein Allosteric Sites Through Automated Machine Learning. Front. Mol. Biosci. 2022, 9, 879251. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Xue, L.; Liu, F.; Li, Y.; Jing, R.; Luo, J. The applications of deep learning algorithms on in silico druggable proteins identification. J. Adv. Res. 2022, 41, 219–231. [Google Scholar] [CrossRef]
- Huang, W.; Lu, S.; Huang, Z.; Liu, X.; Mou, L.; Luo, Y.; Zhao, Y.; Liu, Y.; Chen, Z.; Hou, T.; et al. Allosite: A method for predicting allosteric sites. Bioinformatics 2013, 29, 2357–2359. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, N.M.; Bandyopadhyay, A. High throughput virtual screening (HTVS) of peptide library: Technological advancement in ligand discovery. Eur. J. Med. Chem. 2022, 243, 114766. [Google Scholar] [CrossRef]
- Jin, L.; Wang, W.; Fang, G. Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, L.; Chothia, C.; Janin, J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999, 285, 2177–2198. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.alliedmarketresearch.com/on-demand-updates/A11789 (accessed on 20 June 2021).
- Available online: https://www.globenewswire.com/en/news-release/2023/03/15/2628105/0/en/Monoclonal-Antibodies-Market-Size-is-projected-to-reach-USD-390-58-Billion-by-2030-growing-at-a-CAGR-of-10-2-Straits-Research.html#:~:text=It%20is%20projected%20to%20reach,the%20global%20monoclonal%20antibodies%20market (accessed on 15 March 2023).
- Brekke, O.H.; Loset, G.A. New technologies in therapeutic antibody development. Curr. Opin. Pharmacol. 2003, 3, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Gieselmann, L.; Kreer, C.; Ercanoglu, M.S.; Lehnen, N.; Zehner, M.; Schommers, P.; Potthoff, J.; Gruell, H.; Klein, F. Effective high-throughput isolation of fully human antibodies targeting infectious pathogens. Nat. Protoc. 2021, 16, 3639–3671. [Google Scholar] [CrossRef] [PubMed]
- Descotes, J. Immunotoxicity of monoclonal antibodies. MAbs 2009, 1, 104–111. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use. Guideline on Immunogenicity Assessment of Monoclonal Antibodies Intended for In Vivo Clinical Use; European Medicines Agency: London, UK, 2010.
- Zhu, G.; Ye, M.; Donovan, M.J.; Song, E.; Zhao, Z.; Tan, W. Nucleic acid aptamers: An emerging frontier in cancer therapy. Chem. Commun. 2012, 48, 10472–10480. [Google Scholar] [CrossRef]
- Silwal, A.P.; Thennakoon SK, S.; Arya, S.P.; Postema, R.M.; Jahan, R.; Phuoc CM, T.; Tan, X. DNA aptamers inhibit SARS-CoV-2 spike-protein binding to hACE2 by an RBD- independent or dependent approach. Theranostics 2022, 12, 5522–5536. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. Drugs targeting protein-protein interactions. ChemMedChem Chem. Enabling Drug Discov. 2006, 1, 400–411. [Google Scholar]
- Shin, W.H.; Kumazawa, K.; Imai, K.; Hirokawa, T.; Kihara, D. Current Challenges and Opportunities in Designing Protein-Protein Interaction Targeted Drugs. Adv. Appl. Bioinform. Chem. 2020, 13, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Whitty, A.; Kumaravel, G. Between a rock and a hard place? Nat. Chem. Biol. 2006, 2, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Nevola, L.; Giralt, E. Modulating protein-protein interactions: The potential of peptides. Chem. Commun. 2015, 51, 3302–3315. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Wells, J.A. A hot spot of binding energy in a hormone-receptor interface. Science 1995, 267, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Mabonga, L.; Kappo, A.P. Protein-protein interaction modulators: Advances, successes and remaining challenges. Biophys. Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef] [PubMed]
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef] [PubMed]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Hayes, H.C.; Luk LY, P.; Tsai, Y.H. Approaches for peptide and protein cyclisation. Org. Biomol. Chem. 2021, 19, 3983–4001. [Google Scholar] [CrossRef]
- Qvit, N.; Rubin SJ, S.; Urban, T.J.; Mochly-Rosen, D.; Gross, E.R. Peptidomimetic therapeutics: Scientific approaches and opportunities. Drug Discov. Today 2017, 22, 454–462. [Google Scholar] [CrossRef]
- Takayama, K.; Hitachi, K.; Okamoto, H.; Saitoh, M.; Odagiri, M.; Ohfusa, R.; Shimada, T.; Taguchi, A.; Taniguchi, A.; Tsuchida, K.; et al. Development of Myostatin Inhibitory d-Peptides to Enhance the Potency, Increasing Skeletal Muscle Mass in Mice. ACS Med. Chem. Lett. 2022, 13, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J.; Van Gaal, L.F. Liraglutide (Victoza): Human glucagon-like peptide-1 used in once daily injection for the treatment of type 2 diabetes. Rev. Med. Liege 2010, 65, 464–470. [Google Scholar]
- Ahmadi, S.; Shahsavani, M.B.; Tavaf, Z.; Albaghlany, R.M.; Kumar, A.; Moosavi-Movahedi, A.A.; Yousefi, R. A novel strategy for production of liraglutide precursor peptide and development of a new long-acting incretin mimic. PLoS ONE 2022, 17, e0266833. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.M.; Juhl, C.R.; Torekov, S.S. Benefit-Risk Assessment of Obesity Drugs: Focus on Glucagon-like Peptide-1 Receptor Agonists. Drug Saf. 2019, 42, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Gu, J.; Shao, W.; Pang, J.; Qian, X.; Jin, T. Comparison of Beneficial Metabolic Effects of Liraglutide and Semaglutide in Male C57BL/6J Mice. Can J. Diabetes 2022, 46, 216–224.e2. [Google Scholar] [CrossRef] [PubMed]
- Page, K.; McCool, W.F.; Guidera, M. Examination of the Pharmacology of Oxytocin and Clinical Guidelines for Use in Labor. J. Midwifery Womens Health 2017, 62, 425–433. [Google Scholar] [CrossRef]
- Chen, T.; Wang, Y.; Hao, Z.; Hu, Y.; Li, J. Parathyroid hormone and its related peptides in bone metabolism. Biochem. Pharmacol. 2021, 192, 114669. [Google Scholar] [CrossRef] [PubMed]
- Rettori, V.; Canteros, G.; Renoso, R.; Gimeno, M.; McCann, S.M. Oxytocin stimulates the release of luteinizing hormone-releasing hormone from medial basal hypothalamic explants by releasing nitric oxide. Proc. Natl. Acad. Sci. USA 1997, 94, 2741–2744. [Google Scholar] [CrossRef]
- Bork, K.; Yasothan, U.; Kirkpatrick, P. Icatibant. Nat. Rev. Drug Discov. 2008, 7, 801–802. [Google Scholar] [CrossRef]
- Case, D.B.; Wallace, J.M.; Keim, H.J.; Sealey, J.E.; Laragh, J.H. Usefulness and limitations of saralasin, a partial competitive agonist of angioten II, for evaluating the renin and sodium factors in hypertensive patients. Am. J. Med. 1976, 60, 825–836. [Google Scholar] [CrossRef]
- Thiagarajan, P.; Wu, K.K. Mechanisms of antithrombotic drugs. Adv. Pharmacol. 1999, 46, 297–324. [Google Scholar] [PubMed]
- Erdmann, F.; Weiwad, M. Calcineurin inhibitors: Status quo and perspectives. Biomol. Concepts 2011, 2, 65–78. [Google Scholar] [CrossRef]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998, 4, 1302–1307. [Google Scholar] [CrossRef]
- Dal Corso, A.; Pignataro, L.; Belvisi, L.; Gennari, C. alphavbeta3 Integrin-Targeted Peptide/Peptidomimetic-Drug Conjugates: In-Depth Analysis of the Linker Technology. Curr. Top Med. Chem. 2016, 16, 314–329. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled alpha-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Lee, A.C.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.transparencymarketresearch.com/peptide-therapeutics-market.html (accessed on 27 October 2022).
- Brogi, S. Computational Approaches for Drug Discovery. Molecules 2019, 24, 3061. [Google Scholar] [CrossRef]
- Chang, L.; Mondal, A.; Perez, A. Towards rational computational peptide design. Front. Bioinform. 2022, 2, 1046493. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Srivastava, D.; Sahu, M.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Artificial intelligence to deep learning: Machine intelligence approach for drug discovery. Mol. Divers 2021, 25, 1315–1360. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Luna, J.; Grisoni, F.; Weskamp, N.; Schneider, G. Artificial intelligence in drug discovery: Recent advances and future perspectives. Expert. Opin. Drug Discov. 2021, 16, 949–959. [Google Scholar] [CrossRef]
- Pasrija, P.; Jha, P.; Upadhyaya, P.; Khan, M.S.; Chopra, M. Machine Learning and Artificial Intelligence: A Paradigm Shift in Big Data-Driven Drug Design and Discovery. Curr. Top Med. Chem. 2022, 22, 1692–1727. [Google Scholar] [PubMed]
- Stanzione, F.; Giangreco, I.; Cole, J.C. Use of molecular docking computational tools in drug discovery. Prog. Med. Chem. 2021, 60, 273–343. [Google Scholar] [PubMed]
- Andreani, J.; Guerois, R. Evolution of protein interactions: From interactomes to interfaces. Arch. Biochem. Biophys. 2014, 554, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Petrotchenko, E.V.; Borchers, C.H. Protein Chemistry Combined with Mass Spectrometry for Protein Structure Determination. Chem. Rev. 2022, 122, 7488–7499. [Google Scholar] [CrossRef] [PubMed]
- Viparelli, F.; Cassese, A.; Doti, N.; Paturzo, F.; Marasco, D.; Dathan, N.A.; Monti, S.M.; Basile, G.; Ungaro, P.; Sabatella, M.; et al. Targeting of PED/PEA-15 molecular interaction with phospholipase D1 enhances insulin sensitivity in skeletal muscle cells. J. Biol. Chem. 2008, 283, 21769–21778. [Google Scholar] [CrossRef]
- Sandomenico, A.; Monti, S.M.; Sabatella, M.; De Capua, A.; Tornatore, L.; Doti, N.; Viparelli, F.; Dathan, N.A.; Pedone, C.; Ruvo, M.; et al. Protein-protein interactions: A simple strategy to identify binding sites and peptide antagonists. Chem. Biol. Drug Des. 2009, 73, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Irwin, A.; Hartson, S.D.; Matts, R.L. Functional dissection of cdc37: Characterization of domain structure and amino acid residues critical for protein kinase binding. Biochemistry 2003, 42, 12577–12588. [Google Scholar] [CrossRef]
- Rega, C.; Russo, R.; Foca, A.; Sandomenico, A.; Iaccarino, E.; Raimondo, D.; Milanetti, E.; Tornatore, L.; Franzoso, G.; Pedone, P.V.; et al. Probing the interaction interface of the GADD45beta/MKK7 and MKK7/DTP3 complexes by chemical cross-linking mass spectrometry. Int. J. Biol. Macromol. 2018, 114, 114–123. [Google Scholar] [CrossRef]
- Bruckmann, C.; Tamburri, S.; De Lorenzi, V.; Doti, N.; Monti, A.; Mathiasen, L.; Cattaneo, A.; Ruvo, M.; Bachi, A.; Blasi, F. Mapping the native interaction surfaces of PREP1 with PBX1 by cross-linking mass-spectrometry and mutagenesis. Sci. Rep. 2020, 10, 16809. [Google Scholar] [CrossRef] [PubMed]
- Sztacho, M.; Šalovská, B.; Červenka, J.; Balaban, C.; Hoboth, P.; Hozák, P. Limited Proteolysis-Coupled Mass Spectrometry Identifies Phosphatidylinositol 4,5-Bisphosphate Effectors in Human Nuclear Proteome. Cells 2021, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.K.; Mollapour, M.; Smith, J.R.; Truman, A.; Hu, B.; Good, V.M.; Panaretou, B.; Neckers, L.; Clarke, P.A.; Workman, P.; et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol. Cell 2008, 31, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.J., Jr.; Prince, T.; Cheng, J.; Stevenson, M.A.; Calderwood, S.K. Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer 2008, 8, 491–495. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Fu, W.T.; Jiang, Z.Y.; You, Q.D.; Xu, X.L. Optimization and bioevaluation of Cdc37-derived peptides: An insight into Hsp90-Cdc37 protein-protein interaction modulators. Bioorg. Med. Chem. 2017, 25, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bao, Q.C.; Xu, X.L.; Jiang, F.; Gu, K.; Jiang, Z.Y.; Zhang, X.J.; Guo, X.K.; You, Q.D.; Sun, H.P. Discovery and identification of Cdc37-derived peptides targeting the Hsp90–Cdc37 protein–protein interaction. RSC Adv. 2015, 5, 96138–96145. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, X.; Zhou, J.; Zhang, L.; Xu, X.; Zhang, L.; You, Q.; Wang, L. Design, synthesis and bioevaluation of inhibitors targeting HSP90-CDC37 protein-protein interaction based on a hydrophobic core. Eur. J. Med. Chem. 2021, 210, 112959. [Google Scholar] [CrossRef] [PubMed]
- Admassu, H.; Gasmalla MA, A.; Yang, R.; Zhao, W. Bioactive Peptides Derived from Seaweed Protein and Their Health Benefits: Antihypertensive, Antioxidant, and Antidiabetic Properties. J. Food Sci. 2018, 83, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vaquero, M.; Mora, L.; Hayes, M. In Vitro and In Silico Approaches to Generating and Identifying Angiotensin-Converting Enzyme I Inhibitory Peptides from Green Macroalga Ulva lactuca. Mar. Drugs 2019, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, C.; Mora-Soler, L.; Gallagher, E.; O’Connor, P.; Prieto, J.; Soler-Vila, A.; Hayes, M. Isolation and characterization of bioactive pro-peptides with in vitro renin inhibitory activities from the macroalga Palmaria palmata. J. Agric. Food Chem. 2012, 60, 7421–7427. [Google Scholar] [CrossRef] [PubMed]
- Harnedy, P.A.; FitzGerald, R.J. In vitro assessment of the cardioprotective, anti-diabetic and antioxidant potential of Palmaria palmata protein hydrolysates. J. Appl. Phycol. 2013, 25, 1793–1803. [Google Scholar] [CrossRef]
- Sun, S.; Xu, X.; Sun, X.; Zhang, X.; Chen, X.; Xu, N. Preparation and Identification of ACE Inhibitory Peptides from the Marine Macroalga Ulva intestinalis. Mar. Drugs 2019, 17, 179. [Google Scholar] [CrossRef]
- Farina, B.; Di Sorbo, G.; Chambery, A.; Caporale, A.; Leoni, G.; Russo, R.; Mascanzoni, F.; Raimondo, D.; Fattorusso, R.; Ruvo, M.; et al. Structural and biochemical insights of CypA and AIF interaction. Sci. Rep. 2017, 7, 1138. [Google Scholar] [CrossRef]
- Chelko, S.P.; Keceli, G.; Carpi, A.; Doti, N.; Agrimi, J.; Asimaki, A.; Beti, C.B.; Miyamoto, M.; Amat-Codina, N.; Bedja, D.; et al. Exercise triggers CAPN1-mediated AIF truncation, inducing myocyte cell death in arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2021, 13, eabf0891. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Palumbo, R.; Monti, A.; Fontana, E.; Nebbioso, A.; Ruvo, M.; Altucci, L.; Doti, N. Relevance of AIF/CypA Lethal Pathway in SH-SY5Y Cells Treated with Staurosporine. Int. J. Mol. Sci. 2021, 23, 265. [Google Scholar] [CrossRef] [PubMed]
- Doti, N.; Reuther, C.; Scognamiglio, P.L.; Dolga, A.M.; Plesnila, N.; Ruvo, M.; Culmsee, C. Inhibition of the AIF/CypA complex protects against intrinsic death pathways induced by oxidative stress. Cell Death Dis. 2014, 5, e993. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Guterman, S.; Kent, R.D.; Ladner, R.; Ley, A.; Markland, W.; Roberts, B.L. Direct evolution of novel binding proteins. US Patent 5,223,409, 1 March 1991. [Google Scholar]
- Saw, P.E.; Song, E.W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef] [PubMed]
- Pero, S.C.; Oligino, L.; Daly, R.J.; Soden, A.L.; Liu, C.; Roller, P.P.; Li, P.; Krag, D.N. Identification of novel non-phosphorylated ligands, which bind selectively to the SH2 domain of Grb7. J. Biol. Chem. 2002, 277, 11918–11926. [Google Scholar] [CrossRef] [PubMed]
- Pero, S.C.; Shukla, G.S.; Armstrong, A.L.; Peterson, D.; Fuller, S.P.; Godin, K.; Kingsley-Richards, S.L.; Weaver, D.L.; Bond, J.; Krag, D.N. Identification of a small peptide that inhibits the phosphorylation of ErbB2 and proliferation of ErbB2 overexpressing breast cancer cells. Int. J. Cancer 2004, 111, 951–960. [Google Scholar] [CrossRef]
- Agarwal, G.; Gabrani, R. Identification of Peptide Binders to Truncated Recombinant Chikungunya Virus Envelope Protein 2 Using Phage Display Technology and Their In Silico Characterization. Protein Pept. Lett. 2021, 28, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Perlas, C.; Varese, M.; Guardiola, S.; Sanchez-Navarro, M.; Garcia, J.; Teixido, M.; Giralt, E. Protein Chemical Synthesis Combined with Mirror-Image Phage Display Yields d-Peptide EGF Ligands that Block the EGF-EGFR Interaction. Chembiochem 2019, 20, 2079–2084. [Google Scholar] [CrossRef] [PubMed]
- Lipok, M.; Szlachcic, A.; Kindela, K.; Czyrek, A.; Otlewski, J. Identification of a peptide antagonist of the FGF1-FGFR1 signaling axis by phage display selection. FEBS Open Bio 2019, 9, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Maruta, F.; Parker, A.L.; Fisher, K.D.; Hallissey, M.T.; Ismail, T.; Rowlands, D.C.; Chandler, L.A.; Kerr, D.J.; Seymour, L.W. Identification of FGF receptor-binding peptides for cancer gene therapy. Cancer Gene Ther. 2002, 9, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araujo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol./Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Nixon, A.E.; Sexton, D.J.; Ladner, R.C. Drugs derived from phage display: From candidate identification to clinical practice. MAbs 2014, 6, 73–85. [Google Scholar] [CrossRef]
- Deyle, K.; Kong, X.D.; Heinis, C. Phage Selection of Cyclic Peptides for Application in Research and Drug Development. Acc. Chem. Res. 2017, 50, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Xu, B. Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol. Concepts 2016, 7, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Shukla, G.S.; Krag, D.N. Phage-displayed combinatorial peptide libraries in fusion to beta-lactamase as reporter for an accelerated clone screening: Potential uses of selected enzyme-linked affinity reagents in downstream applications. Comb. Chem. High Throughput Screen. 2010, 13, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.S.; Chen, P.C.; Hampton, J.T.; Tharp, J.M.; Reed, C.A.; Das, S.K.; Wang, D.S.; Hayatshahi, H.S.; Shen, Y.; Liu, J.; et al. A Genetically Encoded, Phage-Displayed Cyclic-Peptide Library. Angew. Chem. 2019, 58, 15904–15909. [Google Scholar] [CrossRef] [PubMed]
- Ledsgaard, L.; Ljungars, A.; Rimbault, C.; Sorensen, C.V.; Tulika, T.; Wade, J.; Wouters, Y.; McCafferty, J.; Laustsen, A.H. Advances in antibody phage display technology. Drug Discov. Today 2022, 27, 2151–2169. [Google Scholar] [CrossRef] [PubMed]
- Jaroszewicz, W.; Morcinek-Orlowska, J.; Pierzynowska, K.; Gaffke, L.; Wegrzyn, G. Phage display and other peptide display technologies. FEMS Microbiol. Rev. 2022, 46, fuab052. [Google Scholar] [CrossRef]
- Aillaud, I.; Kaniyappan, S.; Chandupatla, R.R.; Ramirez, L.M.; Alkhashrom, S.; Eichler, J.; Horn AH, C.; Zweckstetter, M.; Mandelkow, E.; Sticht, H.; et al. A novel D-amino acid peptide with therapeutic potential (ISAD1) inhibits aggregation of neurotoxic disease-relevant mutant Tau and prevents Tau toxicity in vitro. Alzheimer’s Res. Ther. 2022, 14, 15. [Google Scholar] [CrossRef]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierski, W.M.; Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Geysen, H.M.; Meloen, R.H.; Barteling, S.J. Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc. Natl. Acad. Sci. USA 1984, 81, 3998–4002. [Google Scholar] [CrossRef]
- Houghten, R.A. General method for the rapid solid-phase synthesis of large numbers of peptides: Specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. USA 1985, 82, 5131–5135. [Google Scholar] [CrossRef] [PubMed]
- Bozovicar, K.; Bratkovic, T. Evolving a Peptide: Library Platforms and Diversification Strategies. Int. J. Mol. Sci. 2019, 21, 215. [Google Scholar] [CrossRef] [PubMed]
- Furka, A. Forty years of combinatorial technology. Drug Discov. Today 2022, 27, 103308. [Google Scholar] [CrossRef] [PubMed]
- Sandomenico, A.; Caporale, A.; Doti, N.; Cross, S.; Cruciani, G.; Chambery, A.; De Falco, S.; Ruvo, M. Synthetic Peptide Libraries: From Random Mixtures to In Vivo Testing. Curr. Med. Chem. 2020, 27, 997–1016. [Google Scholar] [CrossRef]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM Database of Bioactive Peptides: Current Opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Bo, W.; Zheng, X.; Hao, Y.; Li, B.; Zheng, J.; Liang, G. DFBP: A comprehensive database of food-derived bioactive peptides for peptidomics research. Bioinformatics 2022, 38, 3275–3280. [Google Scholar] [CrossRef]
- Fiore-Gartland, A.; Manso, B.A.; Friedrich, D.P.; Gabriel, E.E.; Finak, G.; Moodie, Z.; Hertz, T.; De Rosa, S.C.; Frahm, N.; Gilbert, P.B.; et al. Pooled-Peptide Epitope Mapping Strategies Are Efficient and Highly Sensitive: An Evaluation of Methods for Identifying Human T Cell Epitope Specificities in Large-Scale HIV Vaccine Efficacy Trials. PLoS ONE 2016, 11, e0147812. [Google Scholar] [CrossRef] [PubMed]
- Heuzenroeder, M.W.; Barton, M.D.; Vanniasinkam, T.; Phumoonna, T. Linear B-cell epitope mapping using enzyme-linked immunosorbent assay for libraries of overlapping synthetic peptides. Methods Mol. Biol. 2009, 524, 137–144. [Google Scholar] [PubMed]
- Martínez-Botas, J.; de la Hoz, B. IgE and IgG4 Epitope Mapping of Food Allergens with a Peptide Microarray Immunoassay. Methods Mol Biol 2023, 2578, 219–236. [Google Scholar]
- Recke, A.; Regensburger, A.K.; Weigold, F.; Muller, A.; Heidecke, H.; Marschner, G.; Hammers, C.M.; Ludwig, R.J.; Riemekasten, G. Autoantibodies in Serum of Systemic Scleroderma Patients: Peptide-Based Epitope Mapping Indicates Increased Binding to Cytoplasmic Domains of CXCR3. Front. Immunol. 2018, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- San Bartolome, C.; Oeo-Santos, C.; San Segundo-Acosta, P.; Munoz-Cano, R.; Martinez-Botas, J.; Bartra, J.; Pascal, M. Epitope Mapping of Allergenic Lipid Transfer Proteins. Methods Mol. Biol. 2021, 2344, 107–117. [Google Scholar] [PubMed]
- Vanniasinkam, T.; Barton, M.D.; Das, T.P.; Heuzenroeder, M.W. B-Cell Epitope Mapping Using a Library of Overlapping Synthetic Peptides in an Enzyme-Linked Immunosorbent Assay. Methods Mol. Biol. 2018, 1785, 121–128. [Google Scholar] [PubMed]
- Doti, N.; Cassese, A.; Marasco, D.; Paturzo, F.; Sabatella, M.; Viparelli, F.; Dathan, N.; Monti, S.M.; Miele, C.; Formisano, P.; et al. Residues 762-801 of PLD1 mediate the interaction with PED/PEA15. Mol. Biosyst. 2010, 6, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Santa-Coloma, T.A. Overlapping synthetic peptides as a tool to map protein-protein interactions—FSH as a model system of nonadditive interactions. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2022, 1866, 130153. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, H.; Chen, X.; Chen, P.H.; Fischer, D.; Sriraman, V.; Yu, H.N.; Arkinstall, S.; He, X. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 12491–12496. [Google Scholar] [CrossRef]
- Jiang, X.; Fischer, D.; Chen, X.; McKenna, S.D.; Liu, H.; Sriraman, V.; Yu, H.N.; Goutopoulos, A.; Arkinstall, S.; He, X. Evidence for Follicle-stimulating Hormone Receptor as a Functional Trimer. J. Biol. Chem. 2014, 289, 14273–14282. [Google Scholar] [CrossRef]
- Jiang, H.; Deng, R.; Yang, X.; Shang, J.; Lu, S.; Zhao, Y.; Song, K.; Liu, X.; Zhang, Q.; Chen, Y.; et al. Peptidomimetic inhibitors of APC-Asef interaction block colorectal cancer migration. Nat. Chem. Biol. 2017, 13, 994–1001. [Google Scholar] [CrossRef]
- Akbarian, M.; Khani, A.; Eghbalpour, S.; Uversky, V.N. Bioactive Peptides: Synthesis, Sources, Applications, and Proposed Mechanisms of Action. Int. J. Mol. Sci. 2022, 23, 1445. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Teranishi, Y.; Takahashi, H.; Toyosato, M.; Notake, M.; Nakanishi, S.; Numa, S. Isolation and structural organization of the human preproenkephalin gene. Nature 1982, 297, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, M.; Seva, C.; Fourmy, D. Cholecystokinin and gastrin receptors. Physiol. Rev. 2006, 86, 805–847. [Google Scholar] [CrossRef] [PubMed]
- Somsen, B.A.; Craenmehr FW, B.; Liu, W.W.; Koops, A.A.; Pennings MA, M.; Visser, E.J.; Ottmann, C.; Cossar, P.J.; Brunsveld, L. Functional mapping of the 14-3-3 hub protein as a guide to design 14-3-3 molecular glues. Chem. Sci. 2022, 13, 13122–13131. [Google Scholar] [CrossRef] [PubMed]
- Goede, A.; Jaeger, I.S.; Preissner, R. SUPERFICIAL--surface mapping of proteins via structure-based peptide library design. BMC Bioinform. 2005, 6, 223. [Google Scholar] [CrossRef]
- Calvanese, L.; Marasco, D.; Doti, N.; Saporito, A.; D’Auria, G.; Paolillo, L.; Ruvo, M.; Falcigno, L. Structural investigations on the Nodal-Cripto binding: A theoretical and experimental approach. Biopolymers 2010, 93, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Hagan, C.L.; Westwood, D.B.; Kahne, D. bam Lipoproteins Assemble BamA in vitro. Biochemistry 2013, 52, 6108–6113. [Google Scholar] [CrossRef] [PubMed]
- Hagan, C.L.; Wzorek, J.S.; Kahne, D. Inhibition of the beta-barrel assembly machine by a peptide that binds BamD. Proc. Natl. Acad. Sci. USA 2015, 112, 2011–2016. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.L.; Weiss, G.A. Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 2001, 5, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Lee, Y.C.; Gates, Z.P.; Ling, Y.; Mortensen, J.C.; Yang, F.S.; Lin, Y.S.; Pentelute, B.L. Binary combinatorial scanning reveals potent poly-alanine-substituted inhibitors of protein-protein interactions. Commun. Chem. 2022, 5, 128. [Google Scholar] [CrossRef]
- Doti, N.; Scognamiglio, P.L.; Madonna, S.; Scarponi, C.; Ruvo, M.; Perretta, G.; Albanesi, C.; Marasco, D. New mimetic peptides of the kinase-inhibitory region (KIR) of SOCS1 through focused peptide libraries. Biochem. J. 2012, 443, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Marasco, D.; Perretta, G.; Sabatella, M.; Ruvo, M. Past and future perspectives of synthetic peptide libraries. Curr. Protein. Pept. Sci. 2008, 9, 447–467. [Google Scholar] [CrossRef] [PubMed]
- Kupai, A.; Vaughan, R.M.; Dickson, B.M.; Rothbart, S.B. A Degenerate Peptide Library Approach to Reveal Sequence Determinants of Methyllysine-Driven Protein Interactions. Front. Cell Dev. Biol. 2020, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Houghten, R.A.; Pinilla, C.; Blondelle, S.E.; Appel, J.R.; Dooley, C.T.; Cuervo, J.H. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 1991, 354, 84–86. [Google Scholar] [CrossRef]
- Barnash, K.D.; Lamb, K.N.; Stuckey, J.I.; Norris, J.L.; Cholensky, S.H.; Kireev, D.B.; Frye, S.V.; James, L.I. Chromodomain Ligand Optimization via Target-Class Directed Combinatorial Repurposing. ACS Chem. Biol. 2016, 11, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8927. [Google Scholar] [CrossRef]
- Perez, J.J. Designing Peptidomimetics. Curr. Top Med. Chem. 2018, 18, 566–590. [Google Scholar] [CrossRef]
- Li Petri, G.; Di Martino, S.; De Rosa, M. Peptidomimetics: An Overview of Recent Medicinal Chemistry Efforts toward the Discovery of Novel Small Molecule Inhibitors. J. Med. Chem. 2022, 65, 7438–7475. [Google Scholar] [CrossRef]
- Doti, N.; Mardirossian, M.; Sandomenico, A.; Ruvo, M.; Caporale, A. Recent Applications of Retro-Inverso Peptides. Int. J. Mol. Sci. 2021, 22, 8677. [Google Scholar] [CrossRef] [PubMed]
- Rai, J. Peptide and protein mimetics by retro and retroinverso analogs. Chem. Biol. Drug Des. 2019, 93, 724–736. [Google Scholar] [CrossRef]
- Taylor, M.; Moore, S.; Mayes, J.; Parkin, E.; Beeg, M.; Canovi, M.; Gobbi, M.; Mann, D.M.; Allsop, D. Development of a proteolytically stable retro-inverso peptide inhibitor of beta-amyloid oligomerization as a potential novel treatment for Alzheimer’s disease. Biochemistry 2010, 49, 3261–3272. [Google Scholar] [CrossRef]
- Austen, B.M.; Paleologou, K.E.; Ali, S.A.; Qureshi, M.M.; Allsop, D.; El-Agnaf, O.M. Designing peptide inhibitors for oligomerization and toxicity of Alzheimer’s beta-amyloid peptide. Biochemistry 2008, 47, 1984–1992. [Google Scholar] [CrossRef]
- Jana, A.K.; Greenwood, A.B.; Hansmann, U.H.E. Small Peptides for Inhibiting Serum Amyloid A Aggregation. ACS Med. Chem. Lett. 2021, 12, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.; Liu, J.; Burgess, K. Minimalist and universal peptidomimetics. Chem. Soc. Rev. 2011, 40, 4411–4421. [Google Scholar] [CrossRef] [PubMed]
- Whitby, L.R.; Boger, D.L. Comprehensive peptidomimetic libraries targeting protein-protein interactions. Acc. Chem. Res. 2012, 45, 1698–1709. [Google Scholar] [CrossRef]
- Horsley, J.R.; Jovcevski, B.; Wegener, K.L.; Yu, J.; Pukala, T.L.; Abell, A.D. Rationally designed peptide-based inhibitor of Abeta42 fibril formation and toxicity: A potential therapeutic strategy for Alzheimer’s disease. Biochem. J. 2020, 477, 2039–2054. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://news.un.org/en/story/2007/02/210312 (accessed on 29 March 2023).
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef]
- Broadley, S.A.; Hartl, F.U. The role of molecular chaperones in human misfolding diseases. FEBS Lett. 2009, 583, 2647–2653. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Sharma, R.; Sardana, S.; Goyal, P.K.; Piplani, M.; Pandey, A. A Systematic Review of updated mechanistic insights towards Alzheimer’s disease. CNS Neurol. Disord.-Drug Targets 2022. [Google Scholar] [CrossRef] [PubMed]
- Chandel, T.I.; Zaman, M.; Khan, M.V.; Ali, M.; Rabbani, G.; Ishtikhar, M.; Khan, R.H. A mechanistic insight into protein-ligand interaction, folding, misfolding, aggregation and inhibition of protein aggregates: An overview. Int. J. Biol. Macromol. 2018, 106, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Naslund, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef]
- Chafekar, S.M.; Malda, H.; Merkx, M.; Meijer, E.W.; Viertl, D.; Lashuel, H.A.; Baas, F.; Scheper, W. Branched KLVFF tetramers strongly potentiate inhibition of beta-amyloid aggregation. Chembiochem 2007, 8, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Aoraha, E.; Candreava, J.; Kim, J.R. Engineering of a peptide probe for β-amyloid aggregates. Mol. BioSyst 2015, 11, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Dongjoon, I.; Soohyeong, K.; Gyusub, Y.; Da Gyeong, H.; Yu-Gon, E.; Ye, E.L.; Chang, H.S.; Jeong-Mo, C.; Hugh, I.K. Decoding the Roles of Amyloid-β (1–42)’s Key Oligomerization Domains toward Designing Epitope-Specific Aggregation Inhibitors. JACS Au 2023, 3, 1065–1075. [Google Scholar]
- Cruz, M.; Tusell, J.M.; Grillo-Bosch, D.; Albericio, F.; Serratosa, J.; Rabanal, F.; Giralt, E. Inhibition of beta-amyloid toxicity by short peptides containing N-methyl amino acids. J. Pept. Res. 2004, 63, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, K.; Kumaraswamy, P.; Sethuramana, S.; Krishnan, U.M. Self-assembly characteristics of a structural analogue of Tjernberg peptide. RSC Adv. 2014, 4, 16517–16523. [Google Scholar] [CrossRef]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castaño, E.M.; Frangione, B. Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat. Med. 1998, 4, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Xiong, N.; Dong, X.Y.; Zheng, J.; Liu, F.F.; Sun, Y. Design of LVFFARK and LVFFARK-functionalized nanoparticles for inhibiting amyloid β-protein fibrillation and cytotoxicity. ACS Appl. Mater. Interfaces 2015, 7, 5650–5666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dong, X.; Liu, F.; Zheng, J.; Sun, Y. Ac-LVFFARK-NH2 conjugation to β-cyclodextrin exhibits significantly enhanced performance on inhibiting amyloid β-protein fibrillogenesis and cytotoxicity. Biophys. Chem. 2018, 235, 40–47. [Google Scholar] [CrossRef]
- Zhang, H.; Zang, C.; Dong, X.Y.; Zheng, J.; Sun, Y. Design of nonapeptide LVFFARKHH: A bifunctional agent against Cu2+-mediated amyloid β-protein aggregation and cytotoxicity. J. Mol. Recogn. 2018, 31, e2697. [Google Scholar] [CrossRef]
- Mazzaglia, A.; Di Natale, G.; Tosto, R.; Scala, A.; Sortino, G.; Piperno, A.; Casaletto, M.P.; Riminucci, A.; Giuffrida, M.L.; Mineo, P.G.; et al. KLVFF oligopeptide-decorated amphiphilic cyclodextrin nanomagnets for selective amyloid beta recognition and fishing. J. Colloid Interface Sci. 2022, 613, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Cimini, S.; Sclip, A.; Mancini, S.; Colombo, L.; Messa, M.; Cagnotto, A.; Di Fede, G.; Tagliavini, F.; Salmona, M.; Borsello, T. The cell-permeable Aβ1-6A2VTAT(D) peptide reverts synaptopathy induced by Aβ1-42wt. Neurobiol. Dis. 2016, 89, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Catania, M.; Maderna, E.; Morbin, M.; Moda, F.; Colombo, L.; Rossi, A.; Cagnotto, A.; Virgilio, T.; Palamara, L.; et al. Tackling amyloidogenesis in Alzheimer’s disease with A2V variants of Amyloid-β. Sci. Rep. 2016, 6, 20949. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Vali, S.; Sun, S.; Chen, X.; Liang, X.; Drozhzhina, T.; Popugaeva, E.; Bezprozvanny, I. Aβ42-binding peptoids as amyloid aggregation inhibitors and detection ligands. ACS Chem. Neurosci. 2013, 4, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Richman, M.; Wilk, S.; Chemerovski, M.; Wärmländer, S.K.; Wahlström, A.; Gräslund, A.; Rahimipour, S. In vitro and mechanistic studies of an antiamyloidogenic self-assembled cyclic D,L-α-peptide architecture. J. Am. Chem. Soc. 2013, 135, 3474–3484. [Google Scholar] [CrossRef] [PubMed]
- Bartling, C.R.O.; Jensen, T.M.T.; Henry, S.M.; Colliander, A.L.; Sereikaite, V.; Wenzler, M.; Jain, P.; Maric, H.M.; Harpsoe, K.; Pedersen, S.W.; et al. Targeting the APP-Mint2 Protein-Protein Interaction with a Peptide-Based Inhibitor Reduces Amyloid-beta Formation. J. Am. Chem. Soc. 2021, 143, 891–901. [Google Scholar] [CrossRef]
- Zhang, L.; Lovell, S.; De Vita, E.; Jagtap PK, A.; Lucy, D.; Goya Grocin, A.; Kjaer, S.; Borg, A.; Hennig, J.; Miller, A.K.; et al. A KLK6 Activity-Based Probe Reveals a Role for KLK6 Activity in Pancreatic Cancer Cell Invasion. J. Am. Chem. Soc. 2022, 144, 22493–22504. [Google Scholar] [CrossRef]
- Korbakis, D.; Soosaipillai, A.; Diamandis, E.P. Study of kallikrein-related peptidase 6 (KLK6) and its complex with alpha1-antitrypsin in biological fluids. Clin. Chem. Lab. Med. 2017, 55, 1385–1396. [Google Scholar] [CrossRef]
- Ashraf, G.M.; Greig, N.H.; Khan, T.A.; Hassan, I.; Tabrez, S.; Shakil, S.; Sheikh, I.A.; Zaidi, S.K.; Akram, M.; Jabir, N.R.; et al. Protein misfolding and aggregation in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, I.; Edwards Iii, G.; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and Alzheimer disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef]
- Lutz, T.A.; Meyer, U. Amylin at the interface between metabolic and neurodegenerative disorders. Front. Neurosci. 2015, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Bakou, M.; Hille, K.; Kracklauer, M.; Spanopoulou, A.; Frost, C.V.; Malideli, E.; Yan, L.M.; Caporale, A.; Zacharias, M.; Kapurniotu, A. Key aromatic/hydrophobic amino acids controlling a cross-amyloid peptide interaction versus amyloid self-assembly. J. Biol. Chem. 2017, 292, 14587–14602. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhao, Z.; Gao, H.; Rostami, I.; You, Q.; Jia, X.; Wang, C.; Zhu, L.; Yang, Y. Enhanced blood-brain-barrier penetrability and tumor-targeting efficiency by peptide-functionalized poly(amidoamine) dendrimer for the therapy of gliomas. Nanotheranostics 2019, 3, 311–330. [Google Scholar] [CrossRef]
- Demeule, M.; Currie, J.C.; Bertrand, Y.; Che, C.; Nguyen, T.; Regina, A.; Gabathuler, R.; Castaigne, J.P.; Beliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef]
- Thun, M.J.; DeLancey, J.O.; Center, M.M.; Jemal, A.; Ward, E.M. The global burden of cancer: Priorities for prevention. Carcinogenesis 2010, 31, 100–110. [Google Scholar] [CrossRef]
- Krzyzanowski, A.; Esser, L.M.; Willaume, A.; Prudent, R.; Peter, C.; Hart, P.; Waldmann, H. Development of Macrocyclic PRMT5-Adaptor Protein Interaction Inhibitors. J. Med. Chem. 2022, 65, 15300–15311. [Google Scholar] [CrossRef]
- Yang, X.; Zhong, J.; Zhang, Q.; Qian, J.; Song, K.; Ruan, C.; Xu, J.; Ding, K.; Zhang, J. Rational Design and Structure Validation of a Novel Peptide Inhibitor of the Adenomatous-Polyposis-Coli (APC)-Rho-Guanine-Nucleotide-Exchange-Factor-4 (Asef) Interaction. J. Med. Chem. 2018, 61, 8017–8028. [Google Scholar] [CrossRef] [PubMed]
- Tkachev, V.O.; Menshchaikova, E.B.; Zenkov, N.K. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry 2011, 76, 407–422. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Moll. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Hong, F.; Sekar, K.R.; Freeman, M.L.; Liebler, D.C. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 2005, 280, 31768–31775. [Google Scholar] [CrossRef]
- Zhang, D.D. The Nrf2-Keap1-ARE signaling pathway: The regulation and dual function of Nrf2 in cancer. Antioxid. Redox Signal. 2010, 13, 1623–1626. [Google Scholar] [CrossRef]
- Magesh, S.; Chen, Y.; Hu, L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef]
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef]
- Georgakopoulos, N.D.; Talapatra, S.K.; Gatliff, J.; Kozielski, F.; Wells, G. Modified Peptide Inhibitors of the Keap1-Nrf2 Protein-Protein Interaction Incorporating Unnatural Amino Acids. Chembiochem 2018, 19, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.; Bertrand, H.C.; Tsujita, T.; Naz, S.; El-Bakry, A.; Laoruchupong, J.; Hayes, J.D.; Wells, G. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction. Free Radic. Biol. Med. 2012, 52, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.; Schaap, M.; Pfistera, H.; Wells, G. Peptide inhibitors of the Keap1–Nrf2 protein–protein interaction with improved binding and cellular activity. Org. Biomol. Chem. 2013, 11, 3553–3557. [Google Scholar] [CrossRef] [PubMed]
- Wells, G. Peptide and small molecule inhibitors of the Keap1-Nrf2 protein-protein interaction. Biochem. Soc. Trans. 2015, 43, 674–679. [Google Scholar] [CrossRef]
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J.; et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043. [Google Scholar] [CrossRef] [PubMed]
- Inoyama, D.; Chen, Y.; Huang, X.; Beamer, L.J.; Kong, A.N.; Hu, L. Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction. J. Biomol. Screen 2012, 17, 435–447. [Google Scholar] [CrossRef]
- Steel, R.; Cowan, J.; Payerne, E.; O’Connell, M.A.; Searcey, M. Anti-inflammatory Effect of a Cell-Penetrating Peptide Targeting the Nrf2/Keap1 Interaction. ACS Med. Chem. Lett. 2012, 3, 407–410. [Google Scholar] [CrossRef]
- Al-Ghabkari, A.; Narendran, A. In Vitro Characterization of a Potent p53-MDM2 Inhibitor, RG7112 in Neuroblastoma Cancer Cell Lines. Cancer Biother. Radiopharm. 2019, 34, 252–257. [Google Scholar] [PubMed]
- Patnaik, A.; Tolcher, A.; Beeram, M.; Nemunaitis, J.; Weiss, G.J.; Bhalla, K.; Agrawal, M.; Nichols, G.; Middleton, S.; Beryozkina, A.; et al. Clinical pharmacology characterization of RG7112, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussiere, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef]
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Bottger, A.; Bottger, V.; Sparks, A.; Liu, W.L.; Howard, S.F.; Lane, D.P. Design of a synthetic Mdm2-binding mini protein that activates the p53 response in vivo. Curr. Biol. 1997, 7, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G.; et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar] [CrossRef]
- Hu, B.; Gilkes, D.M.; Chen, J. Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res. 2007, 67, 8810–8817. [Google Scholar] [CrossRef] [PubMed]
- Bernal, F.; Tyler, A.F.; Korsmeyer, S.J.; Walensky, L.D.; Verdine, G.L. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 2007, 129, 2456–2457. [Google Scholar] [CrossRef] [PubMed]
- Iegre, J.; Ahmed, N.S.; Gaynord, J.S.; Wu, Y.; Herlihy, K.M.; Tan, Y.S.; Lopes-Pires, M.E.; Jha, R.; Lau, Y.H.; Sore, H.F.; et al. Stapled peptides as a new technology to investigate protein-protein interactions in human platelets. Chem. Sci. 2018, 9, 4638–4643. [Google Scholar] [CrossRef]
- Rasafar, N.; Barzegar, A.; Mehdizadeh Aghdam, E. Structure-based designing efficient peptides based on p53 binding site residues to disrupt p53-MDM2/X interaction. Sci. Rep. 2020, 10, 11449. [Google Scholar] [CrossRef]
- Robertson, N.S.; Spring, D.R. Using Peptidomimetics and Constrained Peptides as Valuable Tools for Inhibiting Protein(-)Protein Interactions. Molecules 2018, 23, 959. [Google Scholar] [CrossRef]
- Bernal, F.; Wade, M.; Godes, M.; Davis, T.N.; Whitehead, D.G.; Kung, A.L.; Wahl, G.M.; Walensky, L.D. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 2010, 18, 411–422. [Google Scholar] [CrossRef]
- Danial, N.N.; Walensky, L.D.; Zhang, C.Y.; Choi, C.S.; Fisher, J.K.; Molina, A.J.; Datta, S.R.; Pitter, K.L.; Bird, G.H.; Wikstrom, J.D.; et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat. Med. 2008, 14, 144–153. [Google Scholar] [CrossRef]
- Moellering, R.E.; Cornejo, M.; Davis, T.N.; Del Bianco, C.; Aster, J.C.; Blacklow, S.C.; Kung, A.L.; Gilliland, D.G.; Verdine, G.L.; Bradner, J.E. Direct inhibition of the NOTCH transcription factor complex. Nature 2009, 462, 182–188. [Google Scholar] [CrossRef]
- Patgiri, A.; Yadav, K.K.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 2011, 7, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Boggiano, C.; Reixach, N.; Pinilla, C.; Blondelle, S.E. Successful identification of novel agents to control infectious diseases from screening mixture-based peptide combinatorial libraries in complex cell-based bioassays. Biopolymers 2003, 71, 103–116. [Google Scholar] [CrossRef]
- Chong, H.; Wu, X.; Su, Y.; He, Y. Development of potent and long-acting HIV-1 fusion inhibitors. AIDS 2016, 30, 1187–1196. [Google Scholar] [CrossRef]
- Chong, H.; Xue, J.; Xiong, S.; Cong, Z.; Ding, X.; Zhu, Y.; Liu, Z.; Chen, T.; Feng, Y.; He, L.; et al. A Lipopeptide HIV-1/2 Fusion Inhibitor with Highly Potent In Vitro, Ex Vivo, and In Vivo Antiviral Activity. J. Virol. 2017, 91, e00288-17. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar] [CrossRef] [PubMed]
- Jameson, B.A.; Rao, P.E.; Kong, L.I.; Hahn, B.H.; Shaw, G.M.; Hood, L.E.; Kent, S.B. Location and chemical synthesis of a binding site for HIV-1 on the CD4 protein. Science 1988, 240, 1335–1339. [Google Scholar] [CrossRef]
- Robey, F.A.; Harris-Kelson, T.; Robert-Guroff, M.; Batinic, D.; Ivanov, B.; Lewis, M.S.; Roller, P.P. A synthetic conformational epitope from the C4 domain of HIV Gp120 that binds CD4. J. Biol. Chem. 1996, 271, 17990–17995. [Google Scholar] [CrossRef]
- Wild, C.; Oas, T.; McDanal, C.; Bolognesi, D.; Matthews, T. A synthetic peptide inhibitor of human immunodeficiency virus replication: Correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. USA 1992, 89, 10537–10541. [Google Scholar] [CrossRef] [PubMed]
- Gomara, M.J.; Perez, Y.; Gomez-Gutierrez, P.; Herrera, C.; Ziprin, P.; Martinez, J.P.; Meyerhans, A.; Perez, J.J.; Haro, I. Importance of structure-based studies for the design of a novel HIV-1 inhibitor peptide. Sci. Rep. 2020, 10, 14430. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef]
- Robey, F.A.; Kelson-Harris, T.; Roller, P.P.; Robert-Guroff, M. A helical epitope in the C4 domain of HIV glycoprotein 120. J. Biol. Chem. 1995, 270, 23918–23921. [Google Scholar] [CrossRef] [PubMed]
- Sala, M.; Spensiero, A.; Esposito, F.; Scala, M.C.; Vernieri, E.; Bertamino, A.; Manfra, M.; Carotenuto, A.; Grieco, P.; Novellino, E.; et al. Development and Identification of a Novel Anti-HIV-1 Peptide Derived by Modification of the N-Terminal Domain of HIV-1 Integrase. Front. Microbiol. 2016, 7, 845. [Google Scholar] [CrossRef]
- Hu, W.; Zeng, S.; Li, C.; Jie, Y.; Li, Z.; Chen, L. Identification of hits as matrix-2 protein inhibitors through the focused screening of a small primary amine library. J. Med. Chem. 2010, 53, 3831–3834. [Google Scholar] [CrossRef] [PubMed]
- Watkins, L.C.; DeGrado, W.F.; Voth, G.A. Influenza A M2 Inhibitor Binding Understood through Mechanisms of Excess Proton Stabilization and Channel Dynamics. J. Am. Chem. Soc. 2020, 142, 17425–17433. [Google Scholar] [CrossRef] [PubMed]
- Kamali, A.; Holodniy, M. Influenza treatment and prophylaxis with neuraminidase inhibitors: A review. Infect. Drug Resist. 2013, 6, 187–198. [Google Scholar]
- Barniol-Xicota, M.; Gazzarrini, S.; Torres, E.; Hu, Y.; Wang, J.; Naesens, L.; Moroni, A.; Vazquez, S. Slow but Steady Wins the Race: Dissimilarities among New Dual Inhibitors of the Wild-Type and the V27A Mutant M2 Channels of Influenza A Virus. J. Med. Chem. 2017, 60, 3727–3738. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Fry, A.M.; Gubareva, L.V. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir. Ther. 2012, 17, 159–173. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, Y.; Xu, S.; Zhang, Y.; Musharrafieh, R.; Hau, R.K.; Ma, C.; Wang, J. In Vitro Pharmacokinetic Optimizations of AM2-S31N Channel Blockers Led to the Discovery of Slow-Binding Inhibitors with Potent Antiviral Activity against Drug-Resistant Influenza A Viruses. J. Med. Chem. 2018, 61, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Feng, S.; Xu, Y.; Huang, X.; Zhang, J.; Chen, J.; An, X.; Zhang, Y.; Ning, X. Discovery and characterization of a novel peptide inhibitor against influenza neuraminidase. RSC Med. Chem. 2020, 11, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Ammendolia, M.G.; Agamennone, M.; Pietrantoni, A.; Lannutti, F.; Siciliano, R.A.; De Giulio, B.; Amici, C.; Superti, F. Bovine lactoferrin-derived peptides as novel broad-spectrum inhibitors of influenza virus. Pathog. Glob. Health 2012, 106, 12–19. [Google Scholar] [CrossRef]
- Scala, M.C.; Sala, M.; Pietrantoni, A.; Spensiero, A.; Di Micco, S.; Agamennone, M.; Bertamino, A.; Novellino, E.; Bifulco, G.; Gomez-Monterrey, I.M.; et al. Lactoferrin-derived Peptides Active towards Influenza: Identification of Three Potent Tetrapeptide Inhibitors. Sci. Rep. 2017, 7, 10593. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Turpin, E.A.; Bultmann, H.; Brandt, C.R.; Schultz-Cherry, S. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J. Virol. 2006, 80, 11960–11967. [Google Scholar] [CrossRef]
- Nicol, M.Q.; Ligertwood, Y.; Bacon, M.N.; Dutia, B.M.; Nash, A.A. A novel family of peptides with potent activity against influenza A viruses. J. Gen. Virol. 2012, 93, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, C.M.; Dabelic, R.; Waiboci, L.W.; Jager, L.D.; Heron, L.L.; Johnson, H.M. SOCS-1 mimetics protect mice against lethal poxvirus infection: Identification of a novel endogenous antiviral system. J. Virol. 2009, 83, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Alghrair, Z.K.; Fernig, D.G.; Ebrahimi, B. Enhanced inhibition of influenza virus infection by peptide-noble-metal nanoparticle conjugates. Beilstein J. Nanotechnol. 2019, 10, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Kadam, R.U.; Juraszek, J.; Brandenburg, B.; Buyck, C.; Schepens WB, G.; Kesteleyn, B.; Stoops, B.; Vreeken, R.J.; Vermond, J.; Goutier, W.; et al. Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. [Google Scholar] [CrossRef]
- Hejdanek, J.; Radilova, K.; Pachl, P.; Hodek, J.; Machara, A.; Weber, J.; Rezacova, P.; Konvalinka, J.; Kozisek, M. Structural characterization of the interaction between the C-terminal domain of the influenza polymerase PA subunit and an optimized small peptide inhibitor. Antiviral. Res. 2021, 185, 104971. [Google Scholar] [CrossRef]
- Obayashi, E.; Yoshida, H.; Kawai, F.; Shibayama, N.; Kawaguchi, A.; Nagata, K.; Tame, J.R.; Park, S.Y. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature 2008, 454, 1127–1131. [Google Scholar] [CrossRef]
- Radilova, K.; Zima, V.; Kral, M.; Machara, A.; Majer, P.; Hodek, J.; Weber, J.; Brynda, J.; Strmen, T.; Konvalinka, J.; et al. Thermodynamic and structural characterization of an optimized peptide-based inhibitor of the influenza polymerase PA-PB1 subunit interaction. Antiviral. Res. 2022, 208, 105449. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Rhodes, C.A.; McCroskey, L.C.; Wen, J.; Appiah-Kubi, G.; Wang, D.J.; Guttridge, D.C.; Pei, D. Enhancing the Cell Permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization. Angew. Chem. Int. Ed. 2017, 56, 1525–1529. [Google Scholar] [CrossRef]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wu, X.D.; Shi, M.D.; Yang, R.F.; He, Y.Y.; Bian, C.; Shi, T.L.; Yang, S.; Zhu, X.L.; Jiang, W.H.; et al. Synthetic peptides derived from SARS coronavirus S protein with diagnostic and therapeutic potential. FEBS Lett. 2005, 579, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.J.; Guan, Y.; Hez, M.L.; Sun, H.; Du, L.; Zheng, Y.; Wong, K.L.; Chen, H.; Chen, Y.; Lu, L.; et al. Synthetic peptides outside the spike protein heptad repeat regions as potent inhibitors of SARS-associated coronavirus. Antivir. Ther. 2005, 10, 393–403. [Google Scholar] [CrossRef]
- Hu, H.; Li, L.; Kao, R.Y.; Kou, B.; Wang, Z.; Zhang, L.; Zhang, H.; Hao, Z.; Tsui, W.H.; Ni, A.; et al. Screening and identification of linear B-cell epitopes and entry-blocking peptide of severe acute respiratory syndrome (SARS)-associated coronavirus using synthetic overlapping peptide library. J. Comb. Chem. 2005, 7, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Pomplun, S.; Loftis, A.; Loas, A.; Pentelute, B. The first-in-class peptide binder to the SARS-CoV-2 spike protein. bioRxiv 2020, 10, 19–999318. [Google Scholar]
- Calleja, D.J.; Lessene, G.; Komander, D. Inhibitors of SARS-CoV-2 PLpro. Front. Chem. 2022, 10, 876212. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell. Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Uyar, A.; Dickson, A. Perturbation of ACE2 Structural Ensembles by SARS-CoV-2 Spike Protein Binding. J. Chem. Theory Comput. 2021, 17, 5896–5906. [Google Scholar] [CrossRef] [PubMed]
- Sadremomtaz, A.; Al-Dahmani, Z.M.; Ruiz-Moreno, A.J.; Monti, A.; Wang, C.; Azad, T.; Bell, J.C.; Doti, N.; Velasco-Velazquez, M.A.; de Jong, D.; et al. Synthetic Peptides That Antagonize the Angiotensin-Converting Enzyme-2 (ACE-2) Interaction with SARS-CoV-2 Receptor Binding Spike Protein. J. Med. Chem. 2022, 65, 2836–2847. [Google Scholar] [CrossRef] [PubMed]
- Li, C.G.; Tang, W.; Chi, X.J.; Dong, Z.M.; Wang, X.X.; Wang, X.J. A cholesterol tag at the N terminus of the relatively broad-spectrum fusion inhibitory peptide targets an earlier stage of fusion glycoprotein activation and increases the peptide’s antiviral potency in vivo. J. Virol. 2013, 87, 9223–9232. [Google Scholar] [CrossRef]
- Pessi, A.; Bixler, S.L.; Soloveva, V.; Radoshitzky, S.; Retterer, C.; Kenny, T.; Zamani, R.; Gomba, G.; Gharabeih, D.; Wells, J.; et al. Cholesterol-conjugated stapled peptides inhibit Ebola and Marburg viruses in vitro and in vivo. Antiviral. Res. 2019, 171, 104592. [Google Scholar] [CrossRef]
- Sidorova, M.V.; Bibilashvili, R.S.; Avdeev, D.V.; Kozhokar, U.S.; Palkeeva, M.E.; Ovchinnikov, M.V.; Molokoedov, A.S.; Shirokov, D.A.; Semyonova, A.V.; Uvarova, V.I.; et al. Properties and Activity of Peptide Derivatives of ACE2 Cellular Receptor and Their Interaction with SARS-CoV-2 S Protein Receptor-Binding Domain. Dokl. Biochem. Biophys. 2022, 507, 237–241. [Google Scholar] [CrossRef]
- Weissenborn, L.; Richel, E.; Huseman, H.; Welzer, J.; Beck, S.; Schafer, S.; Sticht, H.; Uberla, K.; Eichler, J. Smaller, Stronger, More Stable: Peptide Variants of a SARS-CoV-2 Neutralizing Miniprotein. Int. J. Mol. Sci. 2022, 23, 6309. [Google Scholar] [CrossRef]
- Zhou, G.; Shi, Z.; Luo, J.; Cao, L.; Yang, B.; Wan, Y.; Wang, L.; Song, R.; Ma, Y.; Tian, H.; et al. Preparation and epitope mapping of monoclonal antibodies against African swine fever virus P30 protein. Appl. Microbiol. Biotechnol. 2022, 106, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Lennard, K.R.; Gardner, R.M.; Doigneaux, C.; Castillo, F.; Tavassoli, A. Development of a Cyclic Peptide Inhibitor of the p6/UEV Protein-Protein Interaction. ACS Chem. Biol. 2019, 14, 1874–1878. [Google Scholar] [CrossRef]
- Barrett, C.E.; Koyama, A.K.; Alvarez, P.; Chow, W.; Lundeen, E.A.; Perrine, C.G.; Pavkov, M.E.; Rolka, D.B.; Wiltz, J.L.; Bull-Otterson, L.; et al. Risk for Newly Diagnosed Diabetes > 30 Days After SARS-CoV-2 Infection Among Persons Aged < 18 Years—United States, March 1, 2020–June 28, 2021. Morb. Mortal. Wkly. Rep. 2022, 71, 59–65. [Google Scholar]
- Rathmann, W.; Kuss, O.; Kostev, K. Incidence of newly diagnosed diabetes after COVID-19. Diabetologia 2022, 65, 949–954. [Google Scholar] [CrossRef]
- Nystrom, S.; Hammarstrom, P. Amyloidogenesis of SARS-CoV-2 Spike Protein. J. Am. Chem. Soc. 2022, 144, 8945–8950. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Laubscher, G.J.; Pretorius, E. A central role for amyloid fibrin microclots in long COVID/PASC: Origins and therapeutic implications. Biochem. J. 2022, 479, 537–559. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monti, A.; Vitagliano, L.; Caporale, A.; Ruvo, M.; Doti, N. Targeting Protein–Protein Interfaces with Peptides: The Contribution of Chemical Combinatorial Peptide Library Approaches. Int. J. Mol. Sci. 2023, 24, 7842. https://doi.org/10.3390/ijms24097842
Monti A, Vitagliano L, Caporale A, Ruvo M, Doti N. Targeting Protein–Protein Interfaces with Peptides: The Contribution of Chemical Combinatorial Peptide Library Approaches. International Journal of Molecular Sciences. 2023; 24(9):7842. https://doi.org/10.3390/ijms24097842
Chicago/Turabian StyleMonti, Alessandra, Luigi Vitagliano, Andrea Caporale, Menotti Ruvo, and Nunzianna Doti. 2023. "Targeting Protein–Protein Interfaces with Peptides: The Contribution of Chemical Combinatorial Peptide Library Approaches" International Journal of Molecular Sciences 24, no. 9: 7842. https://doi.org/10.3390/ijms24097842
APA StyleMonti, A., Vitagliano, L., Caporale, A., Ruvo, M., & Doti, N. (2023). Targeting Protein–Protein Interfaces with Peptides: The Contribution of Chemical Combinatorial Peptide Library Approaches. International Journal of Molecular Sciences, 24(9), 7842. https://doi.org/10.3390/ijms24097842