Abstract
Here, we report a switching method of singlet oxygen (1O2) generation based on the adsorption/desorption of porphyrins to gold nanoparticles driven by sulfide (thiol or disulfide) compounds. The generation of 1O2 by photosensitization is effectively suppressed by the gold nanoparticles and can be restored by a sulfide ligand exchange reaction. The on/off ratio of 1O2 quantum yield (ΦΔ) reached 7.4. By examining various incoming sulfide compounds, it was found that the ligand exchange reaction on the gold nanoparticle surface could be thermodynamically or kinetically controlled. The remaining gold nanoparticles in the system still suppress the generation of 1O2, which can be precipitated out simultaneously with porphyrin desorption by the proper polarity choice of the incoming sulfide to restore the 1O2 generation.
1. Introduction
Singlet oxygen (1O2), a kind of reactive oxygen species (ROS), has been employed for a myriad of applications due to its mild reactivity, long lifetime and unique electron spin properties. Among the methods for generating 1O2, a photosensitizing reaction is used in many research fields [1,2,3,4,5,6,7]. This reaction is based on energy transfer from an excited triplet state photosensitizer molecule to a ground state triplet molecular oxygen (3O2), in which porphyrins are representative and highly efficient photosensitizers. Switchable photosensitization triggered by specific external stimuli is a potential requirement [8,9,10,11,12], as other photo-processes such as fluorescence, phosphorescence and photochemical reactions [13,14,15]. To achieve the switching, a molecular design that enables activation/deactivation of the photosensitization under the desired conditions is required.
We have previously shown that a gold nanoparticle (AuNP) efficiently quenches the excited state of porphyrin when they are covalently linked to form a conjugate [16,17,18]. In the porphyrin–AuNP conjugates, [16] 1O2 generation was almost quenched (singlet oxygen generation quantum yield, ΦΔ = 0.01−0.08) from corresponding unbound porphyrin (ΦΔ = 0.70 for meso-tetraphenyl porphyrin (TPP) [19], at a partial pressure of oxygen pO2 = 0.21 atm). Recently, we have established a switchable photosensitization system for 1O2 generation driven by an acid–base reaction that incorporates a supramolecular architecture [18]. This switch is based on a reversible change of the distance between the porphyrin and the AuNP, a so-called shuttling motion [20]. The porphyrin is topologically interlocked with the AuNP shuttles between two ‘stations’ placed on an axle molecule, which is introduced perpendicular to the AuNP surface via a S–Au covalent linkage. This supramolecular switch has an advantage in that it is possible to design a switch that responds to a desired stimulus through the molecular design of the supramolecular part, and reversible switching is also possible. However, the on/off ratio of ΦΔ was only 1.9 (ΦΔon/ΦΔoff = 0.097/0.052), suggesting that the excitation energy transfer is still dominant in the on-state (where the porphyrin is located at the station away from the AuNP).
Here, we demonstrate a method to switch the generation of 1O2 by sulfide ligand exchange reactions on the porphyrin–AuNP conjugates. The ligand exchange reaction between the conjugates with sulfide compounds (thiol or disulfide) resulted in the porphyrin desorption from the AuNP quencher surface and restored ΦΔ. The on–off ratio depends on the type of incoming sulfide, suggesting that the exchange reaction is thermodynamically or kinetically controlled. Even after complete porphyrin desorption, light absorption by AuNPs in the system still suppressed the 1O2 generation. With proper molecular design of the incoming sulfide, the AuNPs can be precipitated simultaneously with the porphyrin desorption to restore ΦΔ.
2. Results and Discussion
2.1. Synthesis of Porphyrin–AuNP Conjugates Via Disulfide/Thiolate Ligand Exchange Reaction
Common methods for introducing desired functional groups onto the surface of thiolate-protected AuNPs can be classified into two approaches [21]: (i) a direct synthesis method in which thiols and gold ions (typically Au+ or Au3+) are allowed to coexist for reduction, and (ii) a post-synthetic method in which separately synthesized thiolate-protected AuNPs are mixed with a functional group-appended sulfide compound to allow for a sulfide ligand exchange reaction. Scheme 1 shows a schematic diagram of the sulfide ligand exchange reaction [22]. In the reaction between thiol (R1SH) and thiolate-protected AuNP (R2S–AuNP, only one ligand is shown for simplicity), the thiol is introduced onto the AuNP surface via a proton exchange reaction with the thiolate ligand (R2S) on the AuNP surface (thiol–thiolate (T–T) exchange reaction, Scheme 1A). A similar exchange reaction occurs when the disulfide (R1SSR1) is used instead of the thiol (disulfide–thiolate (D–T) exchange reaction, Scheme 1B). The D–T exchange reaction is much slower than the T–T exchange reaction, so it is a method that is not often used, but it has the advantage of being easy to operate due to the stability of disulfides against oxidation [23]. The disulfide R1SSR1 reacts nucleophilically with the thiolate ligand R2S on the AuNP surface to give the asymmetric disulfide R1SSR2. Thus, the atom economy of this reaction step is 0.5, but the remaining asymmetric disulfide can again react with the thiolate on the AuNP surface to achieve a thermodynamically determined equilibrium. Since the T–T exchange reaction is fast, it usually proceeds at room temperature, while the D–T reaction often requires heating. A 1:1 stoichiometry is assumed here for the incoming sulfide and the outgoing sulfide [24], which is known to not always be the case but will not be discussed here.
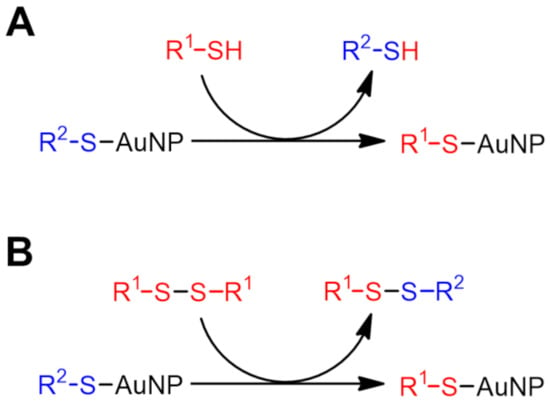
Scheme 1.
Schematic illustration of sulfide ligand exchange reaction on AuNPs. (A) thiol–thiolate (T–T) exchange reaction and (B) disulfide–thiolate (D–T) exchange reaction. Only one exchange site is shown here for simplicity.
Scheme 2 shows the chemical structure of the present porphyrin–AuNP conjugate. We designed a disulfide-type porphyrin ligand (1) to introduce porphyrin onto the surface of AuNPs via a D–T exchange reaction. This compound is stable to air and moisture, so it can be handled without special precautions, unlike thiols. The AuNPs, into which porphyrin is going to be introduced, were synthesized by the two-phase method reported by Brust et al. [25], where a gold ion (Au3+) is reduced in the presence of 1-dodecanethiol (2). The resulting 1-dodecanethiolate-protected AuNPs (2@AuNP) are highly soluble in common organic solvents which allow ligand exchange reactions to proceed in homogeneous solutions. The nanoparticle diameter could be predicted to be ca. 2.5 nm because the AuNPs with an average composition of Au400(C12H25S)126 were synthesized under identical conditions [26].
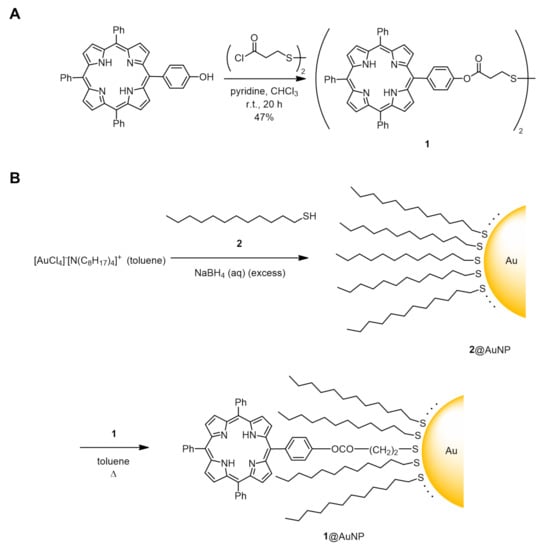
Scheme 2.
Synthetic route to porphyrin–AuNP conjugate 1@AuNP. (A) Synthesis of disulfide porphyrin ligand (1). (B) Disulfide–thiolate ligand exchange reaction between 1 and 1-dodecanethiolate-protected AuNPs (2@AuNP, average core diameter = 2.5 nm). Number of porphyrin ligands was estimated to be 12.3 per AuNP.
When 1 was mixed with 2@AuNP in toluene (ca. 6.15:1 in mole, see details in experimental section), the D–T exchange reaction was not observed at room temperature. On the other hand, a higher reaction temperature (at 60 °C and 80 °C) allowed for the introduction of porphyrins onto the AuNP surface by promoting the exchange reactions. Figure 1A,B show the UV–vis absorption spectra of the porphyrin–AuNP conjugates (1@AuNP) after purification by reprecipitation. Judging from the characteristic Soret band (around 420 nm), the exchange reaction was not complete even after 24 h at 60 °C, while the reaction reached equilibrium in about 10 h at 80 °C (Figure 1C). These D–T exchange reactions were well approximated as pseudo-first-order reactions, and the rate constants were k60°C = 1.1 × 10−5 and k80°C = 9.9 × 10−5 [s−1], respectively. These values are smaller than known D–T exchange reactions, possibly due to the bulkiness of the porphyrins and the short linkers [27,28].
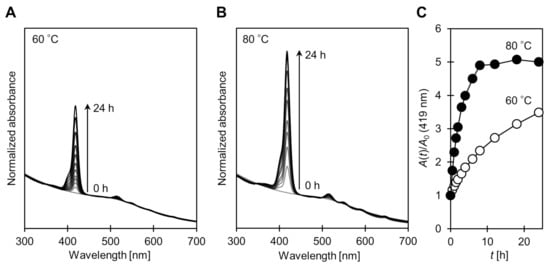
Figure 1.
Change in UV–vis absorption spectra on disulfide/thiolate ligand exchange reaction between 1 and 2@AuNP at (A) 60 °C and (B) 80 °C. Conditions: [1]0 = 2.95 mmol/L, [2@AuNP]0 = 0.48 mmol/L. Spectra were normalized at 450 nm where absorption of porphyrin is negligible. (C) Relative absorbance of Soret band (419 nm): ⭘ 60 °C; ⬤ 80 °C.
In the purification process by reprecipitation, the supernatant after centrifugation contained almost no porphyrin after 10 h when the D–T exchange reaction was performed at 80 °C (judging from the characteristic violet color of porphyrin), indicating that the introduction of 1 is thermodynamically favorable. This seems surprising given the highly sterically hindered structure of 1, but may be influenced by the thermodynamic stability of the remaining disulfide (i.e., didodecyl disulfide). Assuming that all porphyrins have been introduced, approximately 12.3 porphyrin ligands have been introduced per AuNP. In subsequent experiments, 1@AuNP which reacted at 80 °C for 24 h was used.
2.2. Ligand Exchange Reaction with 1-Dodecanethiol
2.2.1. Desorption Monitoring by UV–Vis Absorption Spectra
To investigate the applicability of the sulfide ligand exchange reaction to the switch in 1O2 generation, we preliminarily investigated whether porphyrin desorption is possible using the T–T exchange reaction between 1@AuNP and 2. In the UV–vis absorption spectrum, a slight broadening of the Soret band is observed when porphyrin is introduced onto the AuNP surface. Similar broadening is also widely observed in other dye–AuNP conjugates and is mainly attributed to the exciton coupling and restricted molecular motion of the chromophore [29,30,31]. Using this property, the desorption of the porphyrin by the ligand exchange reaction can be monitored by the absorption spectra. When a large excess 2 was added to 1@AuNP in toluene at room temperature, the Soret band (the absorption maxima = 419 nm) sharpened with time, suggesting the desorption of the porphyrin (Figure 2). The full width at half maximum was asymptotic from 13.1 nm to 12.0 nm, in which the latter is equivalent to that of TPP [18], in about 10 h. This observation suggests that almost all porphyrin ligands were desorbed from the AuNP surface.
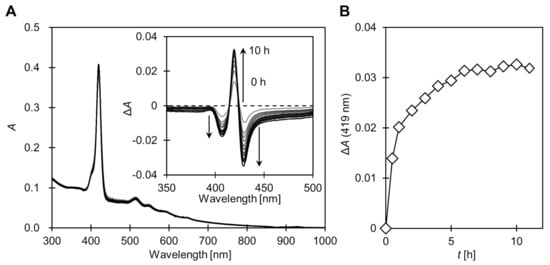
Figure 2.
(A) Change in UV–vis absorption spectra of 1@AuNP after addition of large excess 1-dodecanethiol (2) in toluene at room temperature. Inset shows difference spectra ΔA = A(t) − A0. (B) Change in absorption maxima at Soret band (419 nm). Conditions: [1@AuNP] = 0.07 μmol/L, [2] = 135 mmol/L.
2.2.2. Concentration Dependence on Singlet Oxygen Generation Quantum Yield (ΦΔ)
The quantum yield of the 1O2 generation (ΦΔ) after the T–T exchange reaction with 2 at different stoichiometries ([2] = 0–50 mmol/L, up to 2500 equivalent) was determined by the chemical quencher method (details are given in the experimental section). ΦΔ can be obtained by monitoring the decomposition of 1,3-diphenylisobenzofuran (DPBF) by light irradiation in the presence of photosensitizer [19]. A long-pass filter with a cut-on wavelength of 500 nm was inserted into the optical path to avoid direct photodegradation of DPBF. Therefore, the photoexcitation of the porphyrins occurred only in the Q-band (500–700 nm, corresponding to the S0→S1 transition), and the Soret band (around 420 nm, S0→S2 transition) did not contribute to the generation of 1O2.
The ΦΔ of the 1@AuNP before ligand exchange was determined to be 0.08, which is close to the results for our previously reported porphyrin–AuNP conjugates [16]. In contrast, a clear restoration of ΦΔ was observed after the ligand exchange reaction with 2 (Figure 3). This is because the porphyrin was desorbed from the AuNP surface by the ligand exchange reaction, and the excitation energy transfer from porphyrin to the AuNP was eliminated. Under the conditions investigated ([1@AuNP] = 0.20 μmol/L, room temperature, 10 h), ΦΔ was asymptotic to about 0.4 at the concentration of 2, which was about 10 mmol/L, suggesting that the porphyrin ligands from the AuNP surfaces were completely desorbed above this concentration. However, this ΦΔ value is still only about half that of the TPP (ΦΔ = 0.70). This may be due to the following factors. As apparent from the absorption spectrum, in the irradiation wavelength region (>500 nm), the irradiated light is absorbed by the AuNPs rather than the porphyrins. For example, the absorbance of AuNPs at 515 nm at this concentration is approximately 0.2, which corresponds to a transmittance of 63%. In contrast, porphyrins have an absorbance of only about 0.05. This is where the porphyrin contribution is greatest (highest Q-band molar absorption coefficient), so a significant fraction of the light is absorbed by the AuNPs over the entire wavelength range. Another possibility is that there is excitation energy transfer from the unbound porphyrins to the AuNPs, but this would make a small contribution under such dilute conditions.
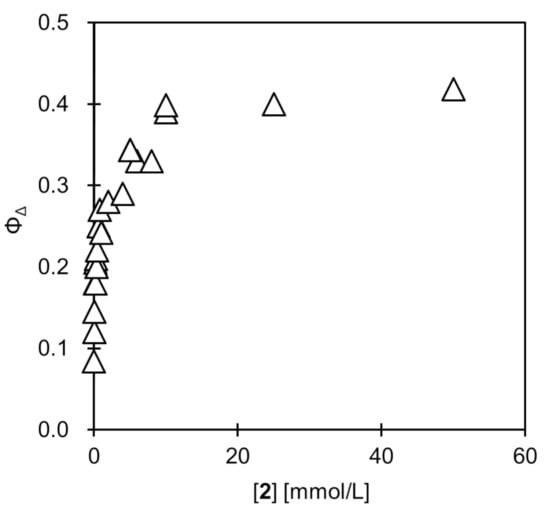
Figure 3.
Change in singlet oxygen quantum yield (ΦΔ) of 1@AuNP after ligand exchange reaction with 1-dodecanethiol (2) in toluene. Conditions: [1@AuNP] = 0.20 μmol/L, [2] = 0–50 mmol/L, room temperature, 10 h. Saturated with oxygen at pO2 = ca. 0.21 atm.
2.3. Ligand Exchange Reaction with Various Sulfide Compounds
To confirm that the recovery of ΦΔ is due to the ligand exchange reaction, we performed similar experiments with various sulfide compounds 3–12 (Figure 4). The concentration of the sulfide compound was set to 1.0 mmol/L to clarify the differences between the compounds, where the ligand exchange reaction was not completed in the case of 2 (other conditions are fixed: [1@AuNP] = 0.20 μmol/L, room temperature, 10 h) (Table 1). The ΦΔ of the ligand exchange reaction product with 2 was 0.24 ± 0.01, while that of the corresponding disulfide 3 was 0.05 ± 0.03, almost unchanged from 1@AuNP before the exchange reaction, indicating that no D–T exchange reaction occurred (D–T exchange reactions are much slower than corresponding T–T exchange reactions) [23]. This trend was similar for thiophenol (4) and the corresponding disulfide (5) (ΦΔ after ligand exchange reaction: 0.24 ± 0.01 (4); 0.14 ± 0.02 (5)).
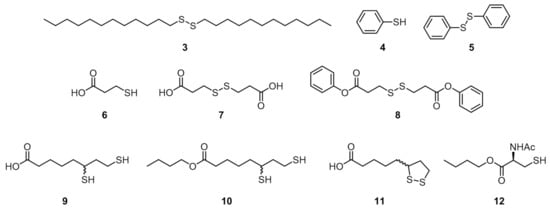
Figure 4.
Sulfide compounds 3–12 used for ligand exchange reactions.

Table 1.
Change in singlet oxygen quantum yield (ΦΔ) upon ligand exchange reactions with sulfide compounds 2–12.
In the ligand exchange reaction product with compounds 2–5, there was no significant change in the absorption spectra, except for the above-mentioned sharpening of the Soret band, suggesting that the AuNPs remain stably dispersed in the solution. On the other hand, in the absorption spectrum of the ligand exchange reaction product with 3-mercaptopropionic acid (6), the absorption band of the AuNPs was reduced indicating that the aggregation of the AuNPs took place (Figure 5). Black precipitates were formed and ΦΔ was restored to 0.42 ± 0.09, indicating that the light absorption by the remaining AuNPs was somewhat eliminated. The absorbance from the porphyrin did not decrease, indicating that the porphyrin ligands were not precipitated together with the AuNPs. Consequently, by introducing a polar functional group (i.e., carboxyl group, –COOH) to the terminal of the incoming thiol, the simultaneous porphyrin desorption and the precipitation removal of the AuNPs can be achieved (Scheme 3A). The disulfide (7) corresponding to 6 was not soluble in toluene, so its phenyl ester (8) was used. Similar to the other disulfides (3 and 5), no restoration of ΦΔ was observed (ΦΔ = 0.07 ± 0.01).
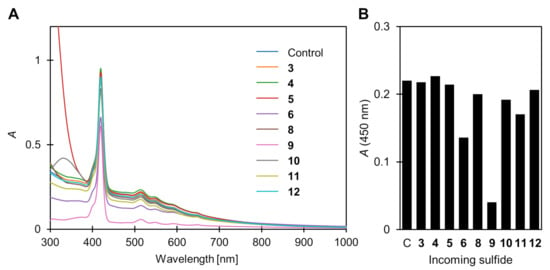
Figure 5.
(A) Change in UV–vis absorption spectra of 1@AuNP after ligand exchange reaction with sulfide compounds 3–12 in toluene (except 7, which has poor solubility). Conditions: [1@AuNP] = 0.20 μmol/L, [3–12] = 1 mmol/L, room temperature, 10 h. (B) Absorbance at 450 nm where absorption of porphyrin is negligible.
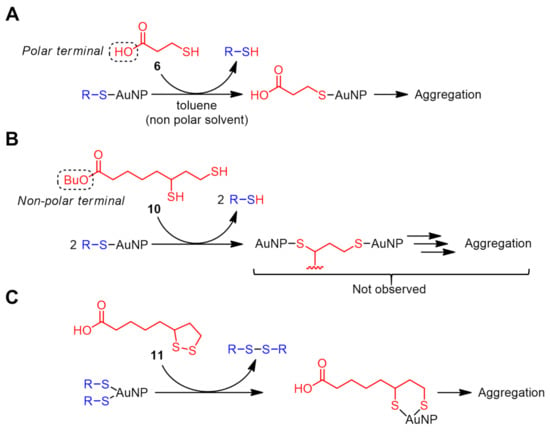
Scheme 3.
Possible ligand exchange mechanism with sulfide compounds: (A) 3-mercaptopropionic acid (6), (B) n-butyl dihydrolipoate (10), and (C) lipoic acid (11).
More prominent AuNP precipitation was observed in a ligand exchange reaction with a divalent thiol, dihydrolipoic acid (also called dihydrothioctic acid) (9) (Figure 5), where ΦΔ was restored to 0.59 ± 0.02 comparable to that of TPP. This is likely due to a more efficient ligand exchange reaction compared to the monovalent 6. In fact, when the carboxyl terminal was masked by an n-butyl group (i.e., compound 10), the ligand exchange product showed a ΦΔ of only 0.30 ± 0.01, which is slightly larger than the other non-polar thiols (i.e., 2 and 4). The crosslinking between the AuNPs by divalent thiols was also thought to be the cause of the precipitation (Scheme 3B) [32]. However, aggregation was not observed, probably because the distance (three carbon atoms) intramolecularly between the two thiols is insufficient to form the crosslinking.
The case of the intramolecular disulfide (dithiolane) corresponding to 9 (compound 11) is a little more complicated. Similar to other disulfide compounds (3, 5, and 8), no significant restoration of ΦΔ was observed (ΦΔ = 0.10 ± 0.03). However, slight precipitation of the AuNPs was found in the ligand exchange reaction product (Figure 5), suggesting that the D–T exchange reaction occurs at room temperature, and a small amount of introduced carboxylic groups promote the aggregation. Based on the mechanism of the D–T exchange reaction in Scheme 1, the porphyrin ligand can be retained as a disulfide on the AuNP surface via simultaneous or successive exchange reaction(s) (Scheme 3C).
From the above results, the only structural requirement of the incoming ligand for efficient ΦΔ restoration is the thiol group (–SH). The cysteine derivative 12 could also be involved in the T–T exchange reaction. However, the ΦΔ after the exchange reaction was 0.15 ± 0.01, which was the smallest restoration among thiols examined in this study, indicating that the ligand exchange reaction does not proceed efficiently due to bulkiness around the thiol group [28].
3. Materials and Methods
All chemicals were obtained from commercial sources and used without purification unless otherwise noted. Hydrogen chloride solution in n-butanol was prepared by passing dry hydrogen chloride through n-butanol. 1,3-Diphenylisobenzofuran (DPBF) was purchased from Sigma-Aldrich (St. Louis, MO, USA). For the spectra measurement and singlet oxygen generation experiment, freshly distilled and air-saturated toluene was used. The concentration of oxygen in the air-saturated toluene is 1.93 mM at atmospheric pressure (pO2 = 0.21 atm) [33].
3.1. Synthesis
1: To a mixture of 5-(4-hydroxyphenyl)-10,15,20-triphenylporphyrin (315 mg, 0.50 mmol), dry pyridine (1 mL) and dry chloroform (100 mL), 3,3′-dithiodipropionyl dichloride (173 mg, 0.70 mmol) in dry chloroform (1.4 mL) was gradually added. After 20 h of stirring, the mixture was washed with water, dried over sodium sulfate and concentrated in vacuo. The residue was purified by column chromatography (Wakogel C-200, methylene chloride) and subsequent reprecipitation was carried out from toluene/hexane to obtain 1 as a purple powder (0.170 g, 47%). CAS No. 2093202-53-8. 1H NMR (400 MHz, chloroform-d, SiMe4, RT): δ/ppm −2.80 (brs, 4H, NH), 3.29–3.33 (m, 8H, CH2), 7.56 (m, 4H, Ar), 7.65–7.80 (m, 18H, Ar), 8.13–8.26 (m, 16H, Ar), 8.78–8.26 (m, 16H, pyrrole).
1-Dodecanethiolate-protected AuNP (2@AuNP): synthesized from tetrachloroauric acid and 2 according to the literature [25].
1@AuNP: Two hundred microliters each toluene solution of 2@AuNP (50 mg/mL = 0.48 mmol/L, when assuming chemical composition of Au400(C12H25S)126 = 1.04 × 105 kg/mol) and 1 (4.23 mg/mL = 2.95 mmol/L) were placed in a glass vial together with a stir bar, and heated and stirred in an oil bath at 60 °C or 80 °C. At the certain time of reaction, an aliquot (10 μL) was transferred into a 1.5 mL centrifugal tube to which acetone (1 mL) was added, and then the contents were mixed rapidly by finger tapping. The conjugates were precipitated by centrifugation (10,000× g, 1 min) and the supernatant was discarded. After washing the precipitate by repeated redispersing and sonicating it in acetone, the precipitate was finally dried under reduced pressure.
Didodecyl disulfide (3): To a solution of 1-dodecanethiol (2) (1.0 g, 4.9 mmol) in ethanol (25 mL), elemental iodine crystals were slowly added until the color of the solution turned to pale yellow. The resulting white crystals were collected by suction filtration and the filtrate was concentrated to around half the volume. The second crystals were collected, and the combined crystals were recrystallized from chloroform/methanol to afford 3 as white crystals (0.872 g, 88%). CAS No. 2757-37-1. 1H NMR (400 MHz, chloroform-d, SiMe4, RT): δ/ppm 0.88 (t, 6H, CH3), 1.20–1.45 (m, 36H, CH2), 1.67 (quintet, 4H, 2-CH2), 2.68 (t, 4H, 1-CH2).
Diphenyl 3,3′-dithiodipropionate (8): To a mixture of phenol (0.94 g, 10 mmol) and dry pyridine (2.9 mL, 36 mmol) in dry chloroform (40 mL), a solution of 3,3′-dithiodipropionyl dichloride (2.0 g, 8 mmol) in dry chloroform (40 mL) was added over 2 h. The mixture was warmed up to room temperature and stirred for another 1 h. After the removal of the solvent, the residue was purified by column chromatography (Wakogel C-200, hexane:methylene chloride = 1:1) to afford 8 as a yellow solid (1.27 g, 70%). CAS No. 2376483-56-4. 1H NMR (400 MHz, chloroform-d, SiMe4, RT): δ/ppm 2.95–3.10 (m, 8H, CH2), 7.09 (m, 4H, ortho-H), 7.22 (m, 2H, para-H), 7.36 (m, 4H, meta-H).
Dihydrolipoic acid (9): A mixture of DL-lipoic acid (11) (0.525 g, 2.5 mmol) and sodium bicarbonate (0.210 g, 2.5 mmol) in water (12.5 mL) was sonicated until all the substances were completely dissolved. The mixture was cooled in an ice bath; sodium borohydride (0.475 g, 12.5 mmol) was added dropwise in 20 min intervals and stirred for 30 min. After additional stirring for 30 min, the mixture was acidified to pH 1 by adding 2 M HCl under nitrogen bubbling. The mixture was extracted by chloroform (3 × 10 mL), dried over magnesium sulfate, and then concentrated in vacuo to afford 9 as a colorless oil (0.427 g, 82%). CAS No. 462-20-4. 1H NMR (400 MHz, chloroform-d, SiMe4, RT): δ/ppm 1.31 (d, 1H, 6-SH), 1.36 (t, 1H, 8-SH), 1.40-1.96 (m, 8H, 3,4,5,7-CH2), 2.38 (t, 2H, 2-CH2), 2.62–2.80 (2H, 8-CH2), 2.93 (m, 1H, 6-CH), 10.56 (brs, 1H, COOH).
n-Butyl dihydrolipoate (10): Compound 9 (1.91 g, 9.2 mmol) was dissolved in hydrogen chloride (ca. 10% in n-butanol, 15 mL). After 3 h of stirring at room temperature under nitrogen, the mixture was concentrated in vacuo. The residue was dissolved in toluene, and concentrated in vacuo to remove n-butanol as an azeotrope, which was repeated three times. The residue was redissolved in methylene chloride, washed with water (3×), dried over sodium sulfate, and concentrated in vacuo to afford 10 as a colorless liquid (2.39 g, 99%). CAS No. 245112-97-4. 1H NMR (400 MHz, chloroform-d, SiMe4, RT): δ/ppm 0.94 (t, 3H, butyl CH3), 1.30 (d, 1H, 6-SH), 1.35 (t, 1H, 8-SH), 1.38 (m, 2H, butyl-CH2), 1.40–1.70 (m, 9H), 1.70–1.96 (m, 2H, 7-CH2), 2.32 (t, 2H, 2-CH2), 2.60-2.80 (m, 2H, 8-CH2), 2.92 (m, 1H, 6-CH2), 4.07 (t, 2H, butyl-CH2).
N-Acetyl-L-cysteine n-butyl ester (12): This was synthesized in a similar manner as 10 from N-acetyl-L-cysteine (1.63 g, 10 mmol). The compound was finally reprecipitated from diethyl ether/hexane, washed with cold hexane, and dried to obtain 12 as a white solid (1.24 g, 62%). CAS No. 34233-59-5. 1H NMR (400 MHz, chloroform-d, SiMe4, RT): δ/ppm 0.95 (t, 3H, CH3), 1.33 (t, 1H, SH), 1.40 (sextet, 2H, butyl-CH2), 1.66 (quintet, 2H, butyl-CH2), 2.08 (s, 3H, COCH3), 3.03 (dd, 2H, cysteine-CH2), 4.04–4.27 (m, 2H, butyl-CH2), 4.88 (m, 1H, systeine-CH), 6.41 (brs, 1H, NH).
3.2. Sulfide Ligand Exchange Reaction
The toluene solution of 1@AuNP (25 μg/mL) and sulfide compound 2–12 were mixed at a 4:1 volume ratio and stirred at room temperature to allow for the ligand exchange reaction. The mixture was used for subsequent characterization without further treatment.
3.3. Determination of Singlet Oxygen Quantum Yield (ΦΔ)
All ΦΔ measurements were obtained in a dark room at a maintained temperature (25 °C). To the sample solution, 1,3-diphenylisobenzofuran (DPBF) (1.2 mmol/L in toluene) was added to make its concentration 20 μmol/L. The sample was filled in a quartz cuvette (1 cm × 1 cm) and irradiated by visible light (500 W Xe short arc lamp, USHIO optical modulex. UV-blue light (<500 nm) from the light source was cut off by passing it through an optical filter Y-50 to avoid the undesired decomposition of DPBF). The light irradiation period (typically 3 s) was controlled by a shutter and the decay of the DPBF absorption at 416 nm was recorded by a UV–vis spectrometer. The irradiation cycles were repeated until ~10% of DPBF was decomposed. This procedure was repeated at least four times for each sample. meso-Tetraphenylporphyrin (TPP) was used as the reference compound (1.00 × 10−7 M in toluene, ΦΔ = 0.70 at pO2 = 0.21 atm) and the decay rate was measured by the same method used for the conjugates.
The singlet oxygen quantum yields (ΦΔ) were determined by the procedure previously reported [16,18,19]. First, to eliminate the effect of light absorption by AuNPs, the molar extinction coefficient arising from porphyrin ligands was deconvoluted from whole conjugate spectra using the following equation:
Here, A(λ) is the actual extinction spectrum of 1@AuNP. εPor(λ) and εAuNP(λ) are the separately measured molar absorption coefficients of TPP and 2@AuNP, respectively. The proportional coefficients a and b were determined by the non-linear least squares method (300 ≤ λ ≤ 800 nm), and the residue R(λ) was obtained. Combining the above results, ΦΔ were calculated by the following equation
where A is the area of porphyrin absorption spectra, r is the decomposition rate of DPBF measured by a UV–vis spectrometer, and subscript R refers to a standard with known ΦΔ (i.e., TPP, ΦΔ = 0.70), respectively.
4. Conclusions
By incorporating porphyrin ligands onto the AuNP surface via a covalent linkage, 1O2 generation was almost quenched due to efficient excited energy transfer from the porphyrin to the AuNP. The porphyrin ligands were desorbed from the surface of the AuNPs by the reverse ligand exchange reaction and showed restored 1O2 generation ability with ΦΔon/ΦΔoff of up to 7.4 (0.59/0.08, incoming sulfide = dihydrolipoic acid 9), which was significantly different depending on the type of sulfur compound used. This difference was influenced by the efficiency of the ligand exchange reaction, as well as the aggregation of the AuNPs governed by the polarity of the protecting ligand layers. These results indicate that the adsorption/desorption between photosensitizers and AuNPs can be significantly applicable to the switching of 1O2 generation.
Author Contributions
A.S. conducted the majority of the experiments. A.S. and H.S. analyzed the data and wrote the manuscript. H.S. was the principal investigator; he conceived the work and designed the experiments. All authors discussed the results and commented on the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Number JP 21K04674 (HS).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Acknowledgments
We thank Edward A. Neal (NIMS) for reading the manuscript and providing pedantic feedback. The present work has been conducted in line with AS’s doctoral dissertation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nowakowska, M.; Kępczyński, M. Polymeric Photosensitizers 2. Photosensitized Oxidation of Phenol in Aqueous Solution. J. Photochem. Photobiol. A Chem. 1998, 116, 251–256. [Google Scholar] [CrossRef]
- Gerdes, R.; Bartels, O.; Schneider, G.; Wöhrle, D.; Schulz-Ekloff, G. Photooxidations of Phenol, Cyclopentadiene and Citronellol with Photosensitizers Ionically Bound at a Polymeric Ion Exchanger. Polym. Adv. Technol. 2001, 12, 152–160. [Google Scholar] [CrossRef]
- Iliev, V.; Prahov, L.; Bilyarska, L.; Fischer, H.; Schulz-Ekloff, G.; Wöhrle, D.; Petrov, L. Oxidation and Photooxidation of Sulfide and Thiosulfate Ions Catalyzed by Transition Metal Chalcogenides and Phthalocyanine Complexes. J. Mol. Catal. A Chem. 2000, 151, 161–169. [Google Scholar] [CrossRef]
- Turconi, J.; Griolet, F.; Guevel, R.; Oddon, G.; Villa, R.; Geatti, A.; Hvala, M.; Rossen, K.; Göller, R.; Burgard, A. Semisynthetic Artemisinin, the Chemical Path to Industrial Production. Org. Process Res. Dev. 2014, 18, 417–422. [Google Scholar] [CrossRef]
- Rebeiz, C.A.; Reddy, K.N.; Nandihalli, U.B.; Velu, J. Tetrapyrrole-Dependent Photodynamic Herbicides. Photochem. Photobiol. 1990, 52, 1099–1117. [Google Scholar] [CrossRef]
- ben Amor, T.; Jori, G. Sunlight-Activated Insecticides: Historical Background and Mechanisms of Phototoxic Activity. Insect Biochem. Mol. Biol. 2000, 30, 915–925. [Google Scholar] [CrossRef]
- Rebeiz, C.A.; Gut, L.J.; Lee, K.; Juvik, J.A.; Rebeiz, C.C.; Bouton, C.E.; Towers, G.H.N. Photodynamics of Porphyric Insecticides. Crit. Rev. Plant Sci. 1995, 14, 329–366. [Google Scholar] [CrossRef]
- Marras, S.A.E.; Kramer, F.R.; Tyagi, S. Efficiencies of Fluorescence Resonance Energy Transfer and Contact-Mediated Quenching in Oligonucleotide Probes. Nucleic Acids Res. 2002, 30, e122. [Google Scholar] [CrossRef]
- Wasielewski, M.R. Photoinduced Electron Transfer in Supramolecular Systems for Artificial Photosynthesis. Chem. Rev. 1992, 92, 435–461. [Google Scholar] [CrossRef]
- Piotrowiak, P. Photoinduced Electron Transfer in Molecular Systems: Recent Developments. Chem. Soc. Rev. 1999, 28, 143–150. [Google Scholar] [CrossRef]
- Foote, C.S.; Chang, Y.C.; Denny, R.W. Chemistry of Singlet Oxygen. X. Carotenoid Quenching Parallels Biological Protection. J. Am. Chem. Soc. 1970, 92, 5216–5218. [Google Scholar] [CrossRef]
- Ouchi, A.; Aizawa, K.; Iwasaki, Y.; Inakuma, T.; Terao, J.; Nagaoka, S.; Mukai, K. Kinetic Study of the Quenching Reaction of Singlet Oxygen by Carotenoids and Food Extracts in Solution. Development of a Singlet Oxygen Absorption Capacity (SOAC) Assay Method. J. Agric. Food Chem. 2010, 58, 9967–9978. [Google Scholar] [CrossRef]
- Hulleman, C.N.; Huisman, M.; Moerland, R.J.; Grünwald, D.; Stallinga, S.; Rieger, B. Fluorescence Polarization Control for On-Off Switching of Single Molecules at Cryogenic Temperatures. Small Methods 2018, 2, 1700323. [Google Scholar] [CrossRef]
- Ito, S. Recent Advances in Mechanochromic Luminescence of Organic Crystalline Compounds. Chem. Lett. 2021, 50, 649–660. [Google Scholar] [CrossRef]
- Casellas, J.; Alcover-Fortuny, G.; de Graaf, C.; Reguero, M. Phenylazopyridine as Switch in Photochemical Reactions. A Detailed Computational Description of the Mechanism of Its Photoisomerization. Materials 2017, 10, 1342. [Google Scholar] [CrossRef]
- Shinohara, A.; Shinmori, H. Controlled Generation of Singlet Oxygen by Porphyrin-Appended Gold Nanoparticles. Bull. Chem. Soc. Jpn. 2016, 89, 1341–1343. [Google Scholar] [CrossRef]
- Shinohara, A.; Shao, G.; Nakanishi, T.; Shinmori, H. Porphyrin Photoabsorption and Fluorescence Variation with Adsorptive Loading on Gold Nanoparticles. Front. Chem. 2021, 9, 777041. [Google Scholar] [CrossRef]
- Shinohara, A.; Pan, C.; Wang, L.; Shinmori, H. Acid–Base Controllable Singlet Oxygen Generation in Supramolecular Porphyrin–Gold Nanoparticle Composites Tethered by Rotaxane Linkers. J. Porphyr. Phthalocyanines 2020, 24, 171–180. [Google Scholar] [CrossRef]
- Wilkinson, F.; Helman, W.P.; Ross, A.B. Quantum Yields for the Photosensitized Formation of the Lowest Electronically Excited Singlet State of Molecular Oxygen in Solution. J. Phys. Chem. Ref. Data 1993, 22, 113–262. [Google Scholar] [CrossRef]
- Stoddart, J.F. Mechanically Interlocked Molecules (MIMs)-Molecular Shuttles, Switches, and Machines (Nobel Lecture). Angew. Chem. Int. Ed. 2017, 56, 11094–11125. [Google Scholar] [CrossRef]
- Dumur, F.; Dumas, E.; Mayer, C.R. Functionalization of Gold Nanoparticles by Inorganic Entities. Nanomaterials 2020, 10, 548. [Google Scholar] [CrossRef] [PubMed]
- Caragheorgheopol, A.; Chechik, V. Mechanistic Aspects of Ligand Exchange in Au Nanoparticles. Phys. Chem. Chem. Phys. 2008, 10, 5029. [Google Scholar] [CrossRef]
- Ionita, P.; Caragheorgheopol, A.; Gilbert, B.C.; Chechik, V. Mechanistic Study of a Place-Exchange Reaction of Au Nanoparticles with Spin-Labeled Disulfides. Langmuir 2004, 20, 11536–11544. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Hou, J.; Menin, L.; Ong, Q.K.; Stellacci, F. Evolution of the Ligand Shell Morphology during Ligand Exchange Reactions on Gold Nanoparticles. Angew. Chem. Int. Ed. 2017, 56, 13521–13525. [Google Scholar] [CrossRef]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of Thiol-Derivatised Gold Nanoparticles in a Two-Phase Liquid–Liquid System. J. Chem. Soc. Chem. Commun. 1994, 1994, 801–802. [Google Scholar] [CrossRef]
- Terrill, R.H.; Postlethwaite, T.A.; Chen, C.; Poon, C.-D.; Terzis, A.; Chen, A.; Hutchison, J.E.; Clark, M.R.; Wignall, G. Monolayers in Three Dimensions: NMR, SAXS, Thermal, and Electron Hopping Studies of Alkanethiol Stabilized Gold Clusters. J. Am. Chem. Soc. 1995, 117, 12537–12548. [Google Scholar] [CrossRef]
- Song, Y.; Murray, R.W. Dynamics and Extent of Ligand Exchange Depend on Electronic Charge of Metal Nanoparticles. J. Am. Chem. Soc. 2002, 124, 7096–7102. [Google Scholar] [CrossRef]
- Krommenhoek, P.J.; Wang, J.; Hentz, N.; Johnston-Peck, A.C.; Kozek, K.A.; Kalyuzhny, G.; Tracy, J.B. Bulky Adamantanethiolate and Cyclohexanethiolate Ligands Favor Smaller Gold Nanoparticles with Altered Discrete Sizes. ACS Nano 2012, 6, 4903–4911. [Google Scholar] [CrossRef]
- Ashjari, M.; Dehfuly, S.; Fatehi, D.; Shabani, R.; Koruji, M. Efficient Functionalization of Gold Nanoparticles Using Cysteine Conjugated Protoporphyrin IX for Singlet Oxygen Production in Vitro. RSC Adv. 2015, 5, 104621–104628. [Google Scholar] [CrossRef]
- Prasanna, S.W.; Poorani, G.; Kumar, M.S.; Aruna, P.; Ganesan, S. Photodynamic Efficacy of Rosebengal-Gold Nanoparticle Complex on Vero and HeLa Cell Lines. Mater. Express 2014, 4, 359–366. [Google Scholar] [CrossRef]
- Eisfeld, A.; Briggs, J.S. The J- and H-Bands of Organic Dye Aggregates. Chem. Phys. 2006, 324, 376–384. [Google Scholar] [CrossRef]
- Nayak, S.; Horst, N.; Zhang, H.; Wang, W.; Mallapragada, S.; Travesset, A.; Vaknin, D. Ordered Networks of Gold Nanoparticles Crosslinked by Dithiol-Oligomers. Part. Part. Syst. Charact. 2018, 35, 1800097. [Google Scholar] [CrossRef]
- Fischer, K.; Wilken, M. Experimental Determination of Oxygen and Nitrogen Solubility in Organic Solvents up to 10 MPa at Temperatures between 298 K and 398 K. J. Chem. Thermodyn. 2001, 33, 1285–1308. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).