
Magnetic Field Effect in Bimolecular Rate Constant of Radical Recombination



Abstract
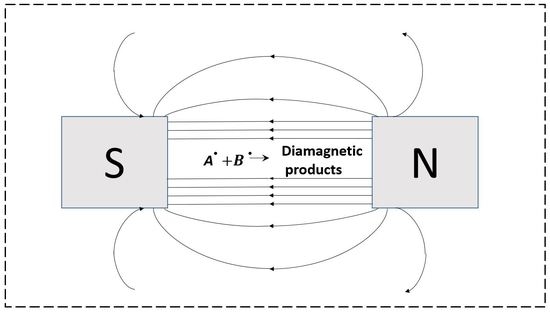
{kind=link}
{kind=link}
{kind=link}
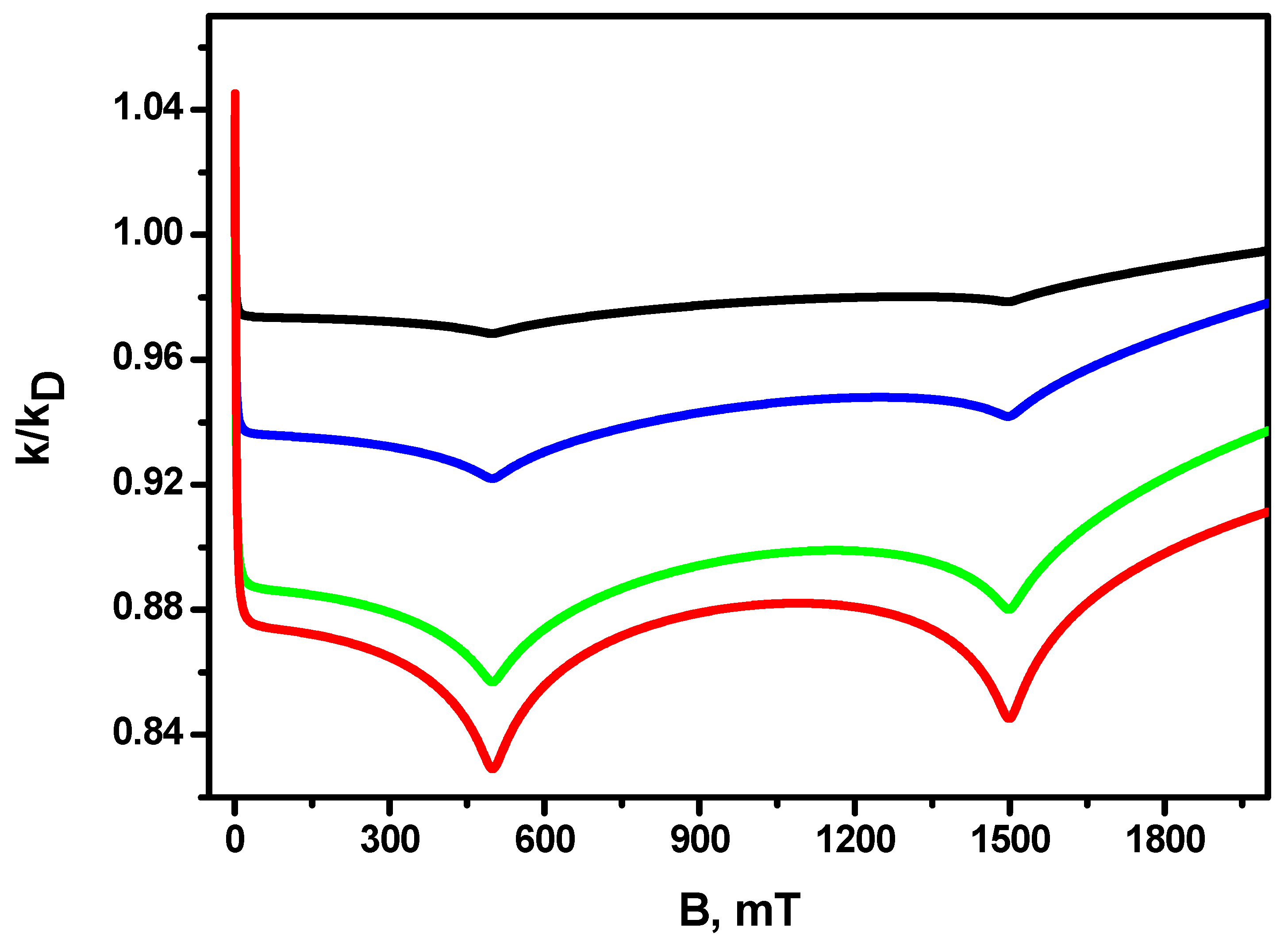{kind=link}
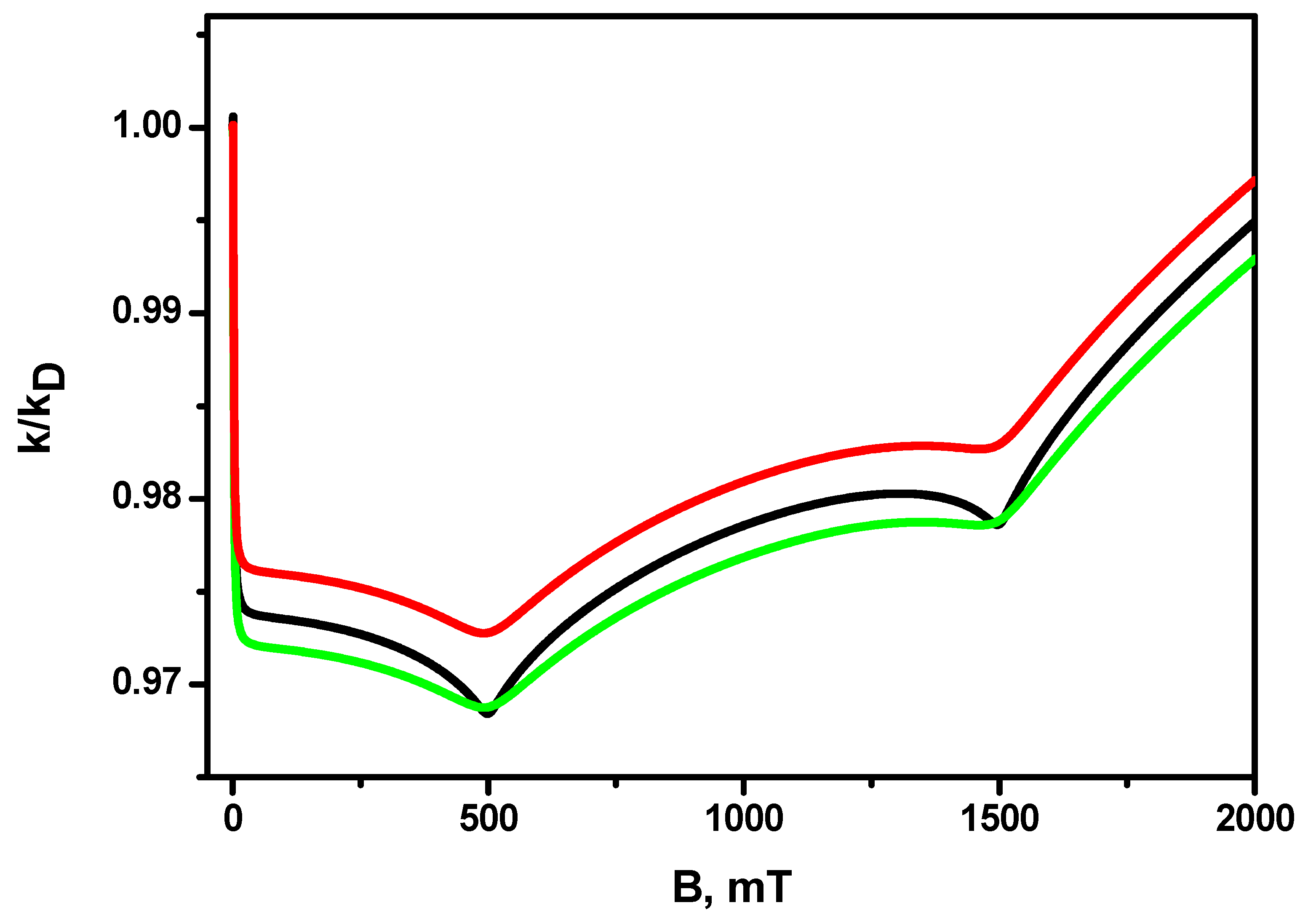{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Theory
2.2. Magnetic Field Effects Calculation Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salikhov, K.M.; Molin, Y.N.; Sagdeev, R.; Buchachenko, A. Spin Polarization and Magnetic Effects in Radical Reactions; Elsevier Science: Amsterdam, The Netherlands, 1984; p. 419. [Google Scholar]
- Steiner, U.E.; Ulrich, T. Magnetic field effects in chemical kinetics and related phenomena. Chem. Rev. 1989, 89, 51–147. [Google Scholar] [CrossRef]
- Nagakura, S.; Hayashi, H.; Azumi, T. Dynamic Spin Chemistry: Magnetic Controls and Spin Dynamics of Chemical Reactions; Kodansha-Wiley: Tokyo, NY, USA, 1998; p. 297. [Google Scholar]
- Buchachenko, A.L. Magnetic Effects in Chemical Reactions. Russ. Chem. Rev. 1976, 45, 375. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Thompson, J.M.T.; Timmel, C.R.; Henbest, K.B. A study of spin chemistry in weak magnetic fields. Philos. Trans. R. Soc. London. Ser. A Math. Phys. Eng. Sci. 2004, 362, 2573–2589. [Google Scholar]
- Ivanov, K.L.; Wagenpfahl, A.; Deibel, C.; Matysik, J. Spin-chemistry concepts for spintronics scientists. Beilstein J. Nanotechnol. 2017, 8, 1427–1445. [Google Scholar] [CrossRef] [PubMed]
- Hore, P.J.; Ivanov, K.L.; Wasielewski, M.R. Spin chemistry. J. Chem. Phys. 2020, 152, 120401. [Google Scholar] [CrossRef]
- Haberkorn, R. Density matrix description of spin-selective radical pair reactions. Mol. Phys. 1976, 32, 1491–1493. [Google Scholar] [CrossRef]
- Noyes, R.M. A Treatment of Chemical Kinetics with Special Applicability to Diffusion Controlled Reactions. J. Chem. Phys. 1954, 22, 1349–1359. [Google Scholar] [CrossRef]
- Closs, G.L. Mechanism explaining nuclear spin polarizations in radical combination reactions. J. Am. Chem. Soc. 1969, 91, 4552–4554. [Google Scholar] [CrossRef]
- Closs, G.L.; Trifunac, A.D. Theory of chemically induced nuclear spin polarization. V. Comparison of coupling reactions in singlet and triplet derived radical pairs and of radicals not generated in pairs. J. Am. Chem. Soc. 1970, 92, 2186–2187. [Google Scholar] [CrossRef]
- Kaptein, R.; Oosterhoff, L. Chemically induced dynamic nuclear polarization III (anomalous multiplets of radical coupling and disproportionation products). Chem. Phys. Lett. 1969, 4, 214–216. [Google Scholar] [CrossRef]
- Suzuki, T.; Miura, T.; Maeda, K.; Arai, T. Spin Dynamics of the Radical Pair in a Low Magnetic Field Studied by the Transient Absorption Detected Magnetic Field Effect on the Reaction Yield and Switched External Magnetic Field. J. Phys. Chem. A 2005, 109, 9911–9918. [Google Scholar] [CrossRef] [PubMed]
- Buchachenko, A.L. Magnetic field-dependent molecular and chemical processes in biochemistry, genetics and medicine. Russ. Chem. Rev. 2014, 83, 1–12. [Google Scholar] [CrossRef]
- Hore, P.J.; Mouritsen, H. The Radical-Pair Mechanism of Magnetoreception. Annu. Rev. Biophys. 2016, 45, 299–344. [Google Scholar] [CrossRef] [PubMed]
- Tadjikov, B.M.; Stas, D.V.; Molin, Y.N. Study of radical cations ofcis- andtrans-decalin in nonpolar solutions by the MARY and optically detected ESR spectroscopy. Russ. Chem. Bull. 1997, 46, 928–933. [Google Scholar] [CrossRef]
- Shafirovich, V.Y.; Levin, P.P. Influence of the linking chain length on the recombination kinetics of covalently bonded triplet radical pairs and magnetic field effects. Russ. Chem. Bull. 2001, 50, 599–606. [Google Scholar] [CrossRef]
- Evans, E.W.; Kattnig, D.R.; Henbest, K.B.; Hore, P.J.; MacKenzie, S.R.; Timmel, C.R. Sub-millitesla magnetic field effects on the recombination reaction of flavin and ascorbic acid radicals. J. Chem. Phys. 2016, 145, 85101. [Google Scholar] [CrossRef]
- Pliss, E.M.; Grobov, A.M.; Kuzaev, A.K.; Buchachenko, A.L. Magnetic field effect on the oxidation of hydrocarbons by molecular oxygen. Mendeleev Commun. 2017, 27, 246–247. [Google Scholar] [CrossRef]
- Pliss, E.M.; Grobov, A.M.; Kuzaev, A.K.; Buchachenko, A.L. Magnetic field effect on the oxidation of organic substances by molecular oxygen. J. Phys. Org. Chem. 2019, 32, e3915. [Google Scholar] [CrossRef]
- Hoang, H.M.; Pham, V.T.B.; Grampp, G.; Kattnig, D.R. Magnetic Field-Sensitive Radical Pair Dynamics in Polymethylene Ether-Bridged Donor–Acceptor Systems. ACS Omega 2018, 3, 10296–10305. [Google Scholar] [CrossRef]
- Sacher, M.; Grampp, G. Magnetic field effects on the luminescence of p-phenylenediamine derivatives. Ber. Der Bunsenges. Für Phys. Chem. 1997, 101, 971–974. [Google Scholar] [CrossRef]
- Justinek, M.; Grampp, G.; Landgraf, S. Determination of electron self-exchange rate constants with MARY spectroscopy: Dependence on the fluorophore. Phys. Chem. Chem. Phys. 2002, 4, 5550–5553. [Google Scholar] [CrossRef]
- Riese, S.; Brand, J.S.; Mims, D.; Holzapfel, M.; Lukzen, N.N.; Steiner, U.E.; Lambert, C. Giant magnetic field effects in donor–acceptor triads: On the charge separation and recombination dynamics in triarylamine–naphthalenediimide triads with bis-diyprrinato-palladium(II), porphodimethenato-palladium(II), and palladium(II)–porphyrin photosensitizers. J. Chem. Phys. 2020, 153, 054306. [Google Scholar]
- Mims, D.; Herpich, J.; Lukzen, N.N.; Steiner, U.E.; Lambert, C. Readout of spin quantum beats in a charge-separated radical pair by pump-push spectroscopy. Science 2021, 374, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.H.; Schmidt, D.; Steiner, U.E.; Lambert, C. Complete Monitoring of Coherent and Incoherent Spin Flip Domains in the Recombination of Charge-Separated States of Donor-Iridium Complex-Acceptor Triads. J. Am. Chem. Soc. 2015, 137, 11011–11021. [Google Scholar] [CrossRef] [PubMed]
- Victor, A.B.; Vsevolod, I.B.; Yuri, N.M. Quantum beats in radical pairs. Russ. Chem. Rev. 2007, 76, 493. [Google Scholar]
- Molin, Y.N. Spin oscillations as a new tool to study recombining radical ion pairs. Mendeleev Commun. 2004, 14, 85–88. [Google Scholar] [CrossRef]
- Maeda, K.; Henbest, K.B.; Cintolesi, F.; Kuprov, I.; Rodgers, C.T.; Liddell, P.A.; Gust, D.; Timmel, C.R.; Hore, P.J. Chemical compass model of avian magnetoreception. Nature 2008, 453, 387–390. [Google Scholar] [CrossRef]
- Stovbun, S.V.; Zlenko, D.V.; Bukhvostov, A.A.; Vedenkin, A.A.; Skoblin, A.A.; Kuznetsov, D.A.; Buchachenko, A.L. Magnetic field and nuclear spin influence on the DNA synthesis rate. Sci. Rep. 2023, 13, 465. [Google Scholar] [CrossRef]
- Maeda, K.; Neil, S.R.T.; Henbest, K.B.; Weber, S.; Schleicher, E.; Hore, P.J.; Mackenzie, S.R.; Timmel, C.R. Following Radical Pair Reactions in Solution: A Step Change in Sensitivity Using Cavity Ring-Down Detection. J. Am. Chem. Soc. 2011, 133, 17807–17815. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; Mosquera-Vázquez, S.; Lovy, D.; Sherin, P.; Markovic, V.; Vauthey, E. Broadband ultraviolet-visible transient absorption spectroscopy in the nanosecond to microsecond time domain with sub-nanosecond time resolution. Rev. Sci. Instrum. 2013, 84, 73107. [Google Scholar] [CrossRef] [PubMed]
- Dodson, C.A.; Wedge, C.J.; Murakami, M.; Maeda, K.; Wallace, M.I.; Hore, P.J. Fluorescence-detected magnetic field effects on radical pair reactions from femtolitre volumes. Chem. Commun. 2015, 51, 8023–8026. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.B.; Shokhirev, N.V.; Salikhov, K.M. The influence of singlet-triplet transitions on non-stationary kinetics of radical recombination in homogeneous solutions. II. Numerical case studies. Chem. Phys. 1989, 137, 207–219. [Google Scholar] [CrossRef]
- Shokhirev, N.V.; Krissinel, E.B.; Salikhov, K.M. The influence of singlet-triplet transitions on non-stationary kinetics of radical recombination in homogeneous solutions. I. General theory and analytical model treatment. Chem. Phys. 1989, 137, 197–205. [Google Scholar] [CrossRef]
- Gorelik, E.V.; Lukzen, N.N.; Sagdeev, R.Z.; Murai, H. Application of integral encounter theory to account for spin effects in radical reactions: Photochemical generation and spin selective recombination of radical ions. Phys. Chem. Chem. Phys. 2003, 5, 5438–5443. [Google Scholar] [CrossRef]
- Nenashev, A.V.; Jansson, F.; Baranovskii, S.D.; Österbacka, R.; Dvurechenskii, A.; Gebhard, F. Role of diffusion in two-dimensional bimolecular recombination. Appl. Phys. Lett. 2010, 96, 213304. [Google Scholar] [CrossRef]
- Shushin, A.I. Magnetic field effects on electron-hole recombination in disordered organic semiconductors. Phys. Rev. B 2011, 84, 115212. [Google Scholar] [CrossRef]
- Shushin, A.I.; Sakun, V.P. Anomalous migration of polarons in disordered organic semiconductors and its manifestation in magnetic-field effects. Russ. J. Phys. Chem. B 2015, 9, 120–126. [Google Scholar] [CrossRef]
- Sampson, C.; Keens, R.H.; Kattnig, D.R. On the magnetosensitivity of lipid peroxidation: Two- versus three-radical dynamics. Phys. Chem. Chem. Phys. 2019, 21, 13526–13538. [Google Scholar] [CrossRef]
- Lukzen, N.N.; Ivanov, K.L.; Sadovsky, V.M.; Sagdeev, R.Z. Magnetic field effect on recombination of radicals diffusing on a two-dimensional plane. J. Chem. Phys. 2020, 152, 34103. [Google Scholar] [CrossRef]
- Gorelik, E.; Lukzen, N.; Sagdeev, R.; Steiner, U. Application of integral encounter theory to account for the spin effects in radical reactions: I. Δg and spin relaxation effects on recombination kinetics of free radicals. Chem. Phys. 2000, 262, 303–323. [Google Scholar] [CrossRef]
- Kipriyanov, A.; Purtov, P. Exactly solvable many-particle model of bulk recombination of coupled radical pairs with allowance for singlet–triplet transitions. Chem. Phys. 2009, 355, 1–13. [Google Scholar] [CrossRef]
- Feskov, S.V.; Burshtein, A.I.; Ivanov, A.I. Magnetic Field Effects in Fluorescence of Exciplex and Fluorophore for the Weller Schemes I and II: Similarities and Differences. J. Phys. Chem. C 2014, 118, 21365–21376. [Google Scholar] [CrossRef]
- Mikhailov, S.A.; Purtov, P.A.; Doktorov, A.B. Theory of geminate recombination of radical pairs with instantaneously changing spin Hamiltonian. III. Radical recombination in switched high magnetic field. Chem. Phys. 1992, 166, 35–49. [Google Scholar] [CrossRef]
- Doktorov, A.B.; Mikhailov, S.A.; Purtov, P.A. Theory of geminate recombination of radical pairs with instantaneously changing spin-Hamiltonian. II. Method of summation of re-encounter contributions. Chem. Phys. 1992, 160, 239–254. [Google Scholar] [CrossRef]
- Mikhailov, S.A.; Purtov, P.A. Kinematic approximation in the theory of stimulated nuclear polarization in radical recombination. Theor. Exp. Chem. 1989, 24, 504–510. [Google Scholar] [CrossRef]
- Purtov, P.A.; Salikhov, K.M. Semiclassical theory of magnetic effects in the recombination of radicals. Theor. Exp. Chem. 1981, 16, 413–418. [Google Scholar] [CrossRef]
- Purtov, P.A.; Salikhov, K.M. Magnetic effects and nuclear polarization in the recombination of radical pairs with one magnetic nucleus with spin I = 1/2. Theor. Exp. Chem. 1981, 16, 530–538. [Google Scholar] [CrossRef]
- Doktorov, A.B. The influence of spin relaxation and locally strong spin exchange on magneto-spin effects in radical pairs in high magnetic fields. Eur. Phys. J. Plus 2021, 136, 992. [Google Scholar] [CrossRef]
- Kuz’Min, V.A.; Levin, P. Influence of paramagnetic additives on the magnetic effect in the recombination of phenoxyl and semiquinone radicals in micelles. Russ. Chem. Bull. 1986, 35, 1291–1293. [Google Scholar] [CrossRef]
- Mints, R.; Pukhov, A. The influence of paramagnetic impurities on magnetic effects in radical reactions. Chem. Phys. 1985, 87, 467–472. [Google Scholar] [CrossRef]
- Karogodina, T.Y.; Dranov, I.G.; Sergeeva, S.V.; Stass, D.V.; Steiner, U.E. Kinetic Magnetic-Field Effect Involving the Small Biologically Relevant Inorganic Radicals NO and O2.−. ChemPhysChem 2011, 12, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, K.R. Diffusion-controlled reactions in two-dimensional fluids: Discussion of measurements of lateral diffusion of lipids in biological membranes. Chem. Phys. Lett. 1974, 28, 280–284. [Google Scholar] [CrossRef]
- Smoluchowski, M.V. Versuch einer mathematischen Theorie der Koagulationskinetik kolloider Lösungen. Z. Für Phys. Chem. 1918, 92, 129–168. [Google Scholar] [CrossRef]
- Collins, F.C.; E Kimball, G. Diffusion-controlled reaction rates. J. Colloid Sci. 1949, 4, 425–437. [Google Scholar] [CrossRef]
- Doktorov, A. The impact approximation in the theory of bimolecular quasi-resonant processes. Phys. A Stat. Mech. Its Appl. 1978, 90, 109–136. [Google Scholar] [CrossRef]
- Kipriyanov, A.A.; Doktorov, A.B.; Burshtein, A.I. Binary theory of dephasing in liquid solutions. I. The non-markovian theory of encounters. Chem. Phys. 1983, 76, 149–162. [Google Scholar] [CrossRef]
- Kipriyanov, A.; Igoshin, O.; Doktorov, A. A new approach to the derivation of binary non-Markovian kinetic equations. Phys. A Stat. Mech. Its Appl. 1999, 268, 567–606. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Lukzen, N.N.; Doktorov, A.B.; Burshtein, A.I. Integral encounter theories of multistage reactions. I. Kinetic equations. J. Chem. Phys. 2001, 114, 1754–1762. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Lukzen, N.N.; Doktorov, A.B.; Burshtein, A.I. Integral encounter theories of multistage reactions. II. Reversible inter-molecular energy transfer. J. Chem. Phys. 2001, 114, 1763–1774. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Lukzen, N.N.; Doktorov, A.B.; Burshtein, A.I. Integral encounter theories of the multistage reactions. III. Reversible intramolecular energy transfer. J. Chem. Phys. 2001, 114, 5682–5690. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Lukzen, N.N.; Morozov, V.A.; Doktorov, A.B. Integral encounter theories of multistage reactions. IV. Account of internal quantum states of reactants. J. Chem. Phys. 2002, 117, 9413–9422. [Google Scholar] [CrossRef]
- Kipriyanov, A.; Doktorov, A. A many-particle derivation of non-Markovian kinetic equations of reversible reaction A+B⇌C in solutions based on the effective pairs approach. Phys. A Stat. Mech. Its Appl. 2003, 326, 105–128. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Lukzen, N.N.; Doktorov, A.B. The integral encounter theory of multistage reactions containing association-dissociation reaction stages. III. Taking account of quantum states of reactants. J. Chem. Phys. 2004, 121, 5115–5124. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Lukzen, N.N.; Kipriyanov, A.A.; Doktorov, A.B. The integral encounter theory of multistage reactions containing association–dissociation reaction stages Part I. Kinetic equations. Phys. Chem. Chem. Phys. 2004, 6, 1706–1718. [Google Scholar] [CrossRef]
- Dodin, D.V.; Ivanov, A.I.; Burshtein, A.I. Magnetic field effect in fluorescence of excited fluorophore equilibrated with exciplex that reversibly dissociates into radical-ion pair undergoing the spin conversion. J. Chem. Phys. 2012, 137, 24511. [Google Scholar] [CrossRef]
- Burshtein, A.I.; Ivanov, A.I. Diffusion affected magnetic field effect in exciplex fluorescence. J. Chem. Phys. 2014, 141, 24508. [Google Scholar] [CrossRef]
- Ivanov, A.I.; Burshtein, A.I. Luminescence Quenching by Reversible Ionization or Exciplex Formation/Dissociation. J. Phys. Chem. A 2008, 112, 11547–11558. [Google Scholar] [CrossRef]
- Feskov, S.V.; Ivanov, A.I.; Burshtein, A.I. Integral encounter theory of strong electron transfer. J. Chem. Phys. 2005, 122, 124509. [Google Scholar] [CrossRef]
- Purtov, P.A.; Doktorov, A.B. The Green function method in the theory of nuclear and electron spin polarization. I. General theory, zero approximation and applications. Chem. Phys. 1993, 178, 47–65. [Google Scholar] [CrossRef]
- Hahn, S.; Kim, K.; Kim, K.; Hu, X.; Painter, T.; Dixon, I.; Kim, S.; Bhattarai, K.R.; Noguchi, S.; Jaroszynski, J.; et al. 45.5-tesla direct-current magnetic field generated with a high-temperature superconducting magnet. Nature 2019, 570, 496–499. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doktorov, A.B.; Lukzen, N.N. Magnetic Field Effect in Bimolecular Rate Constant of Radical Recombination. Int. J. Mol. Sci. 2023, 24, 7555. https://doi.org/10.3390/ijms24087555
Doktorov AB, Lukzen NN. Magnetic Field Effect in Bimolecular Rate Constant of Radical Recombination. International Journal of Molecular Sciences. 2023; 24(8):7555. https://doi.org/10.3390/ijms24087555
Chicago/Turabian StyleDoktorov, Alexander B., and Nikita N. Lukzen. 2023. "Magnetic Field Effect in Bimolecular Rate Constant of Radical Recombination" International Journal of Molecular Sciences 24, no. 8: 7555. https://doi.org/10.3390/ijms24087555
APA StyleDoktorov, A. B., & Lukzen, N. N. (2023). Magnetic Field Effect in Bimolecular Rate Constant of Radical Recombination. International Journal of Molecular Sciences, 24(8), 7555. https://doi.org/10.3390/ijms24087555