


A Protocol for Simultaneous In Vivo Imaging of Cardiac and Neuroinflammation in Dystrophin-Deficient MDX Mice Using [18F]FEPPA PET

 , ,
, ,
Abstract
1. Introduction
2. Results
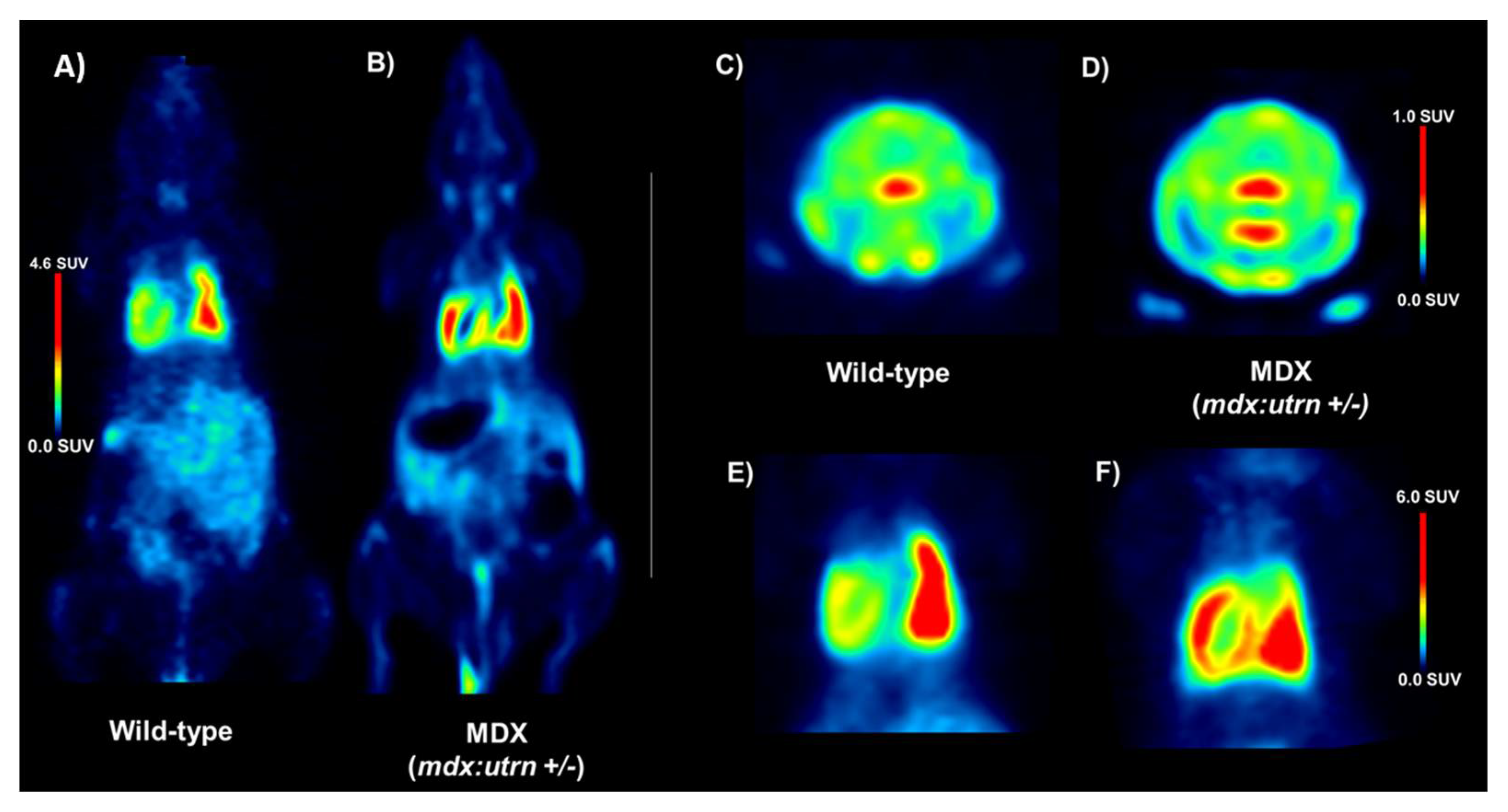
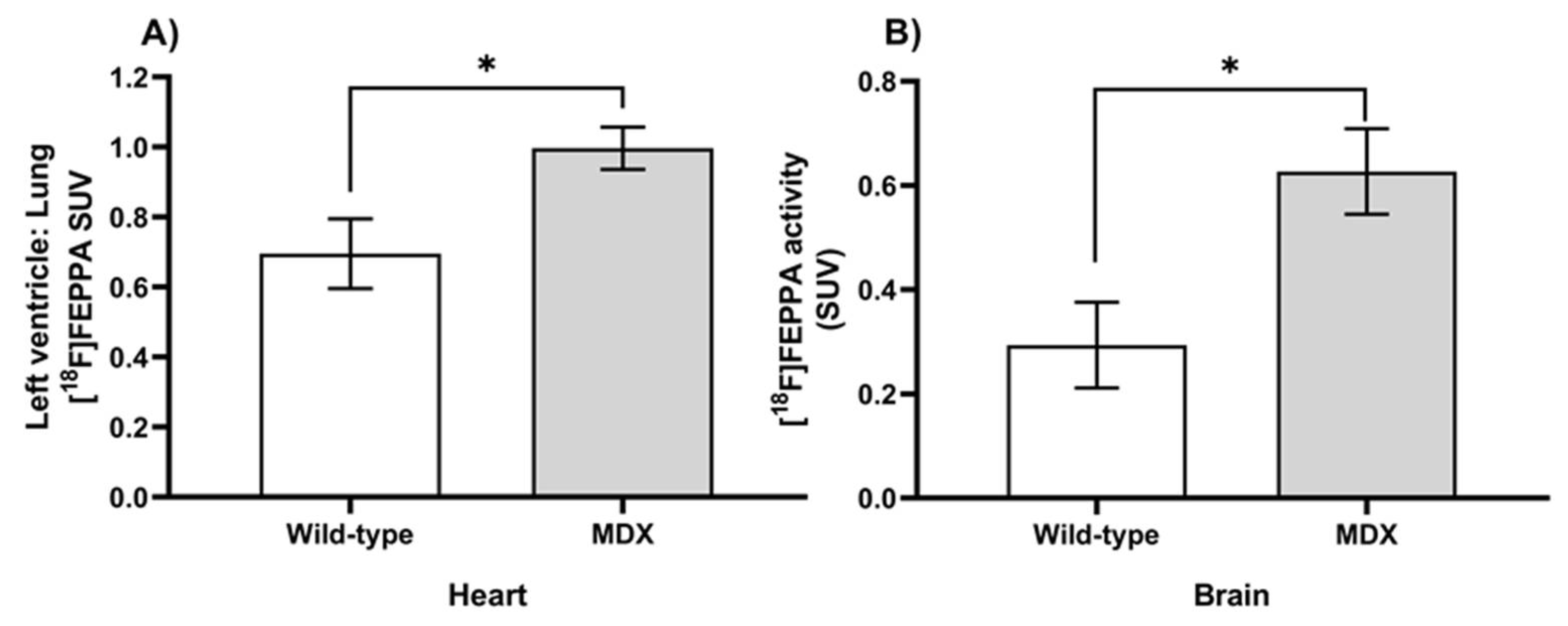
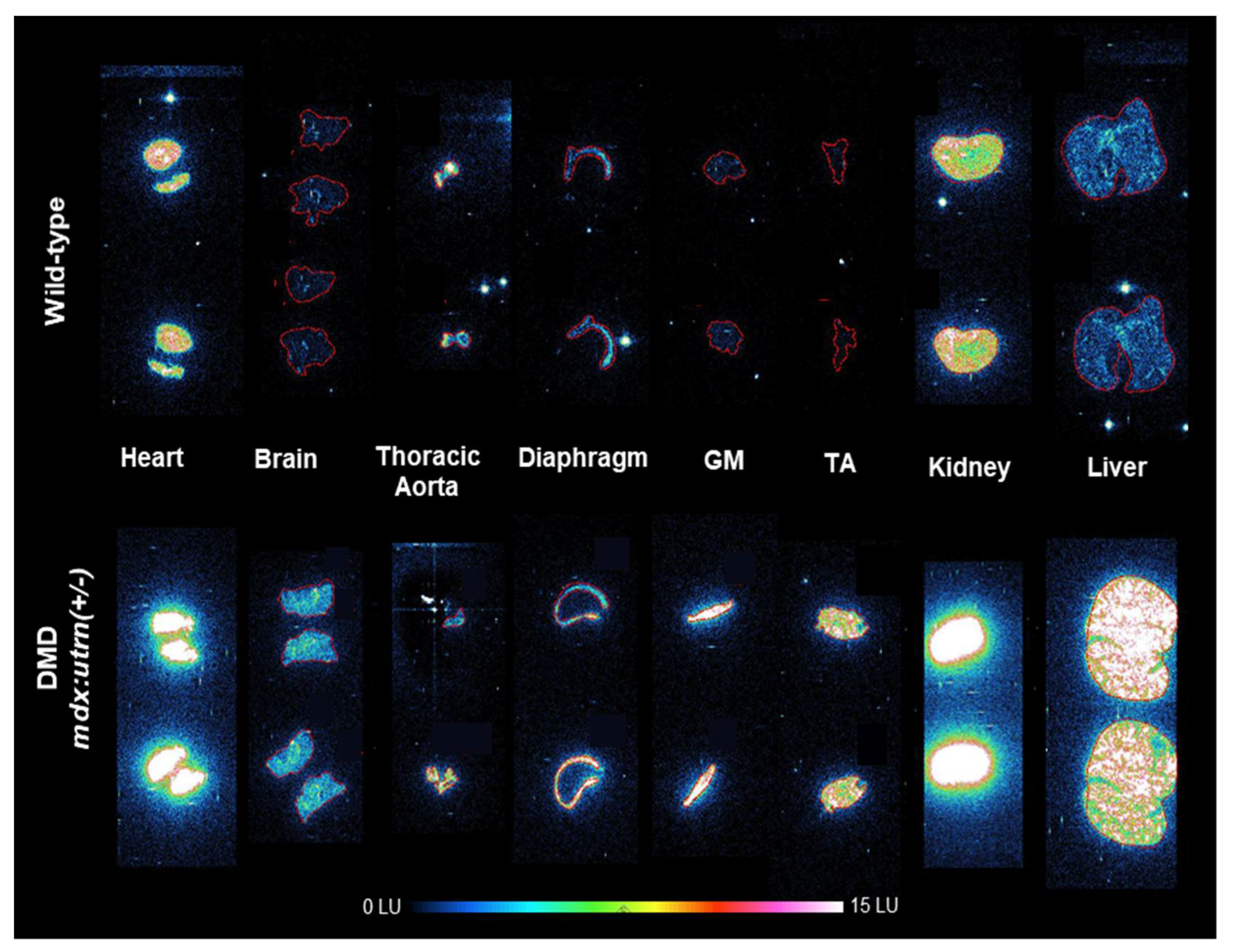
2.1. Elevation of In Vivo Inflammation-Targeted Radiotracer Binding in DMD Models
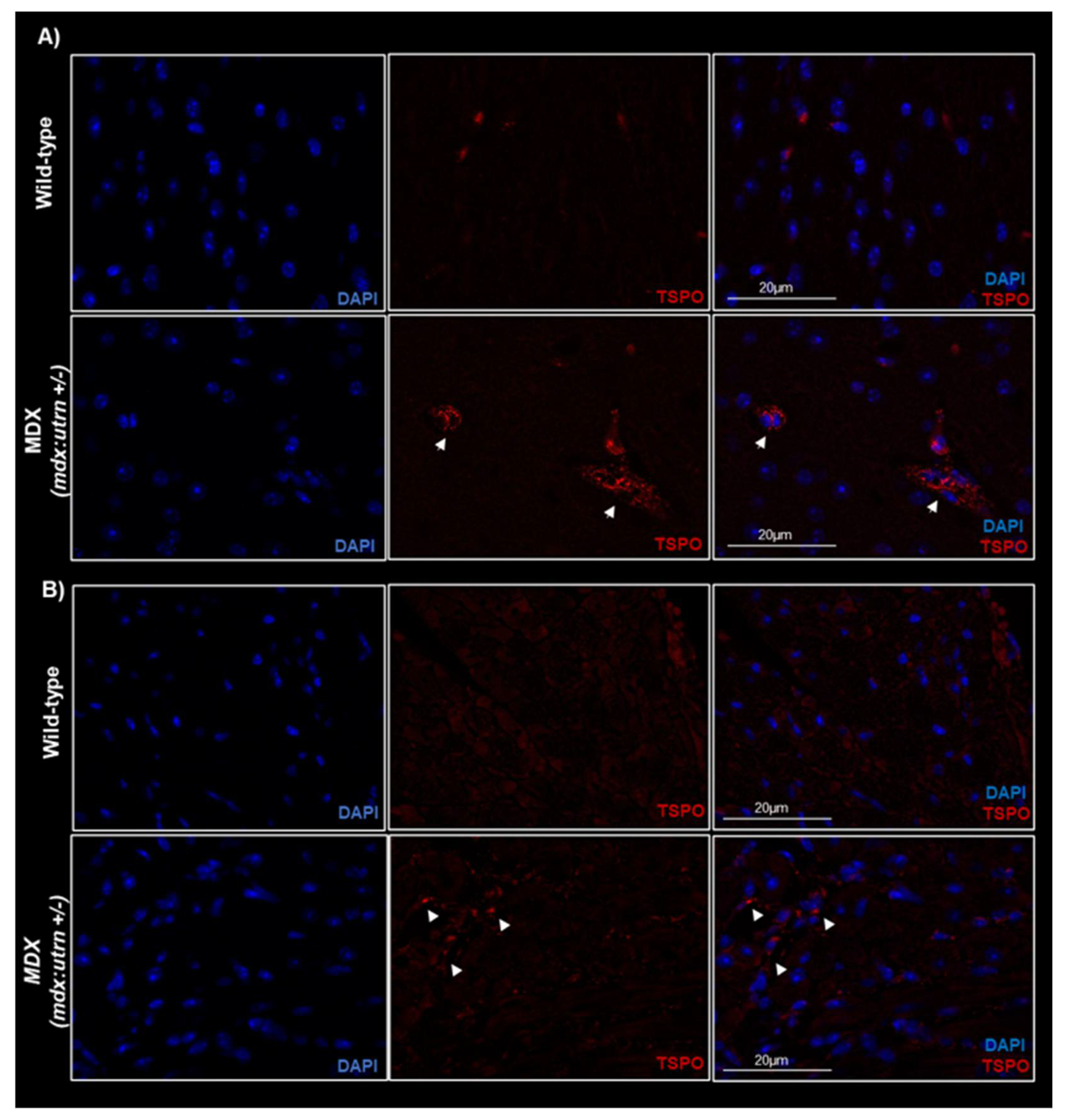
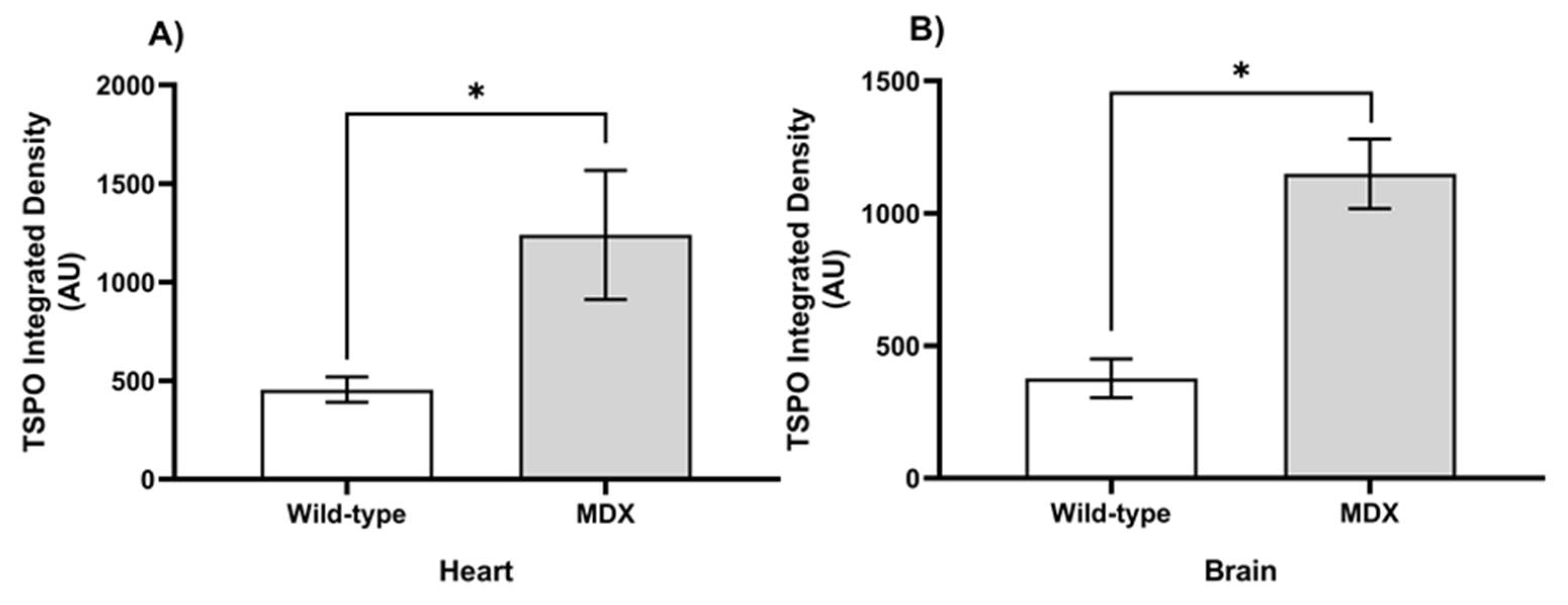
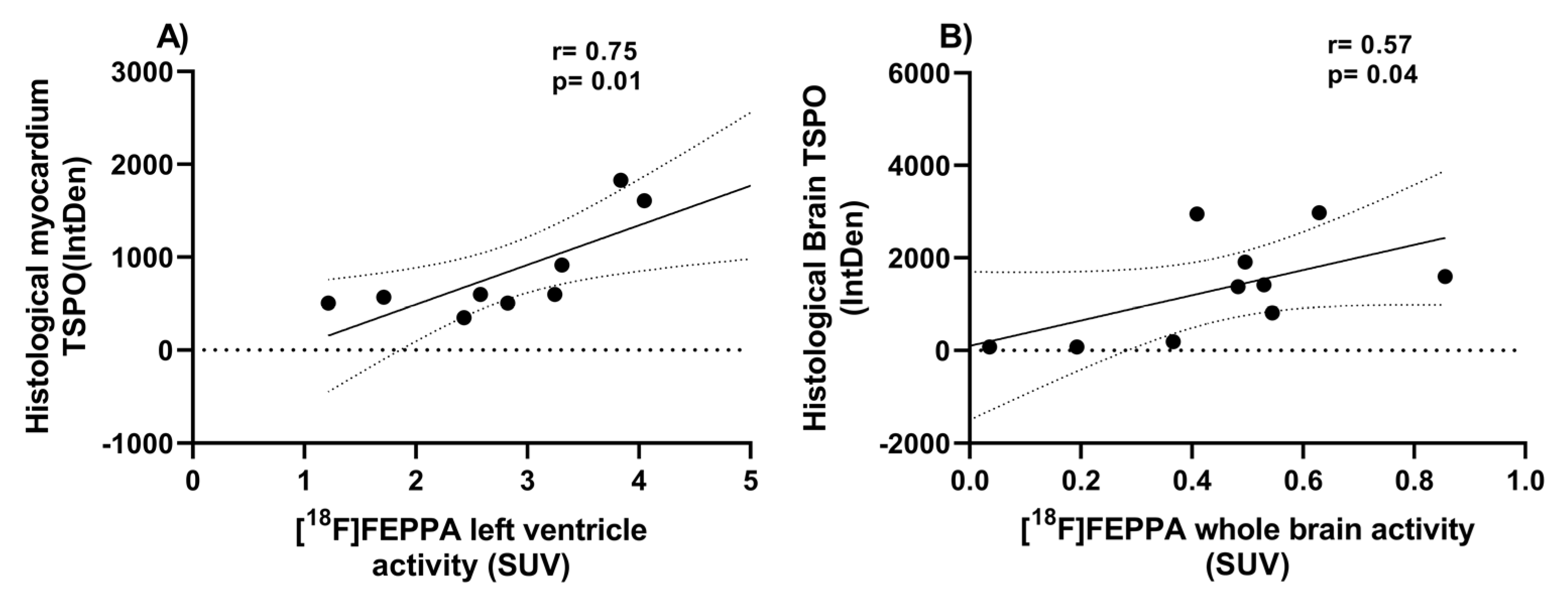
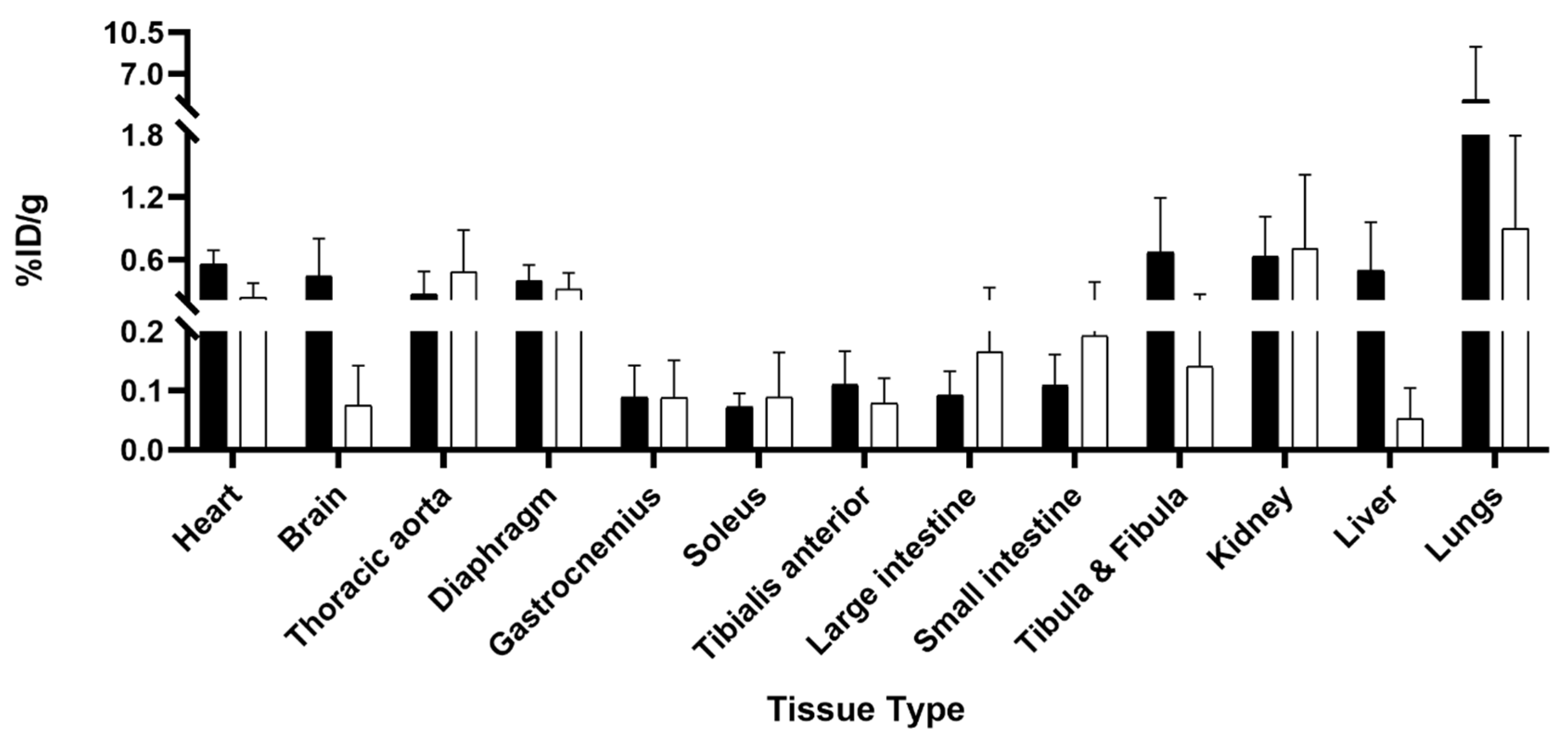
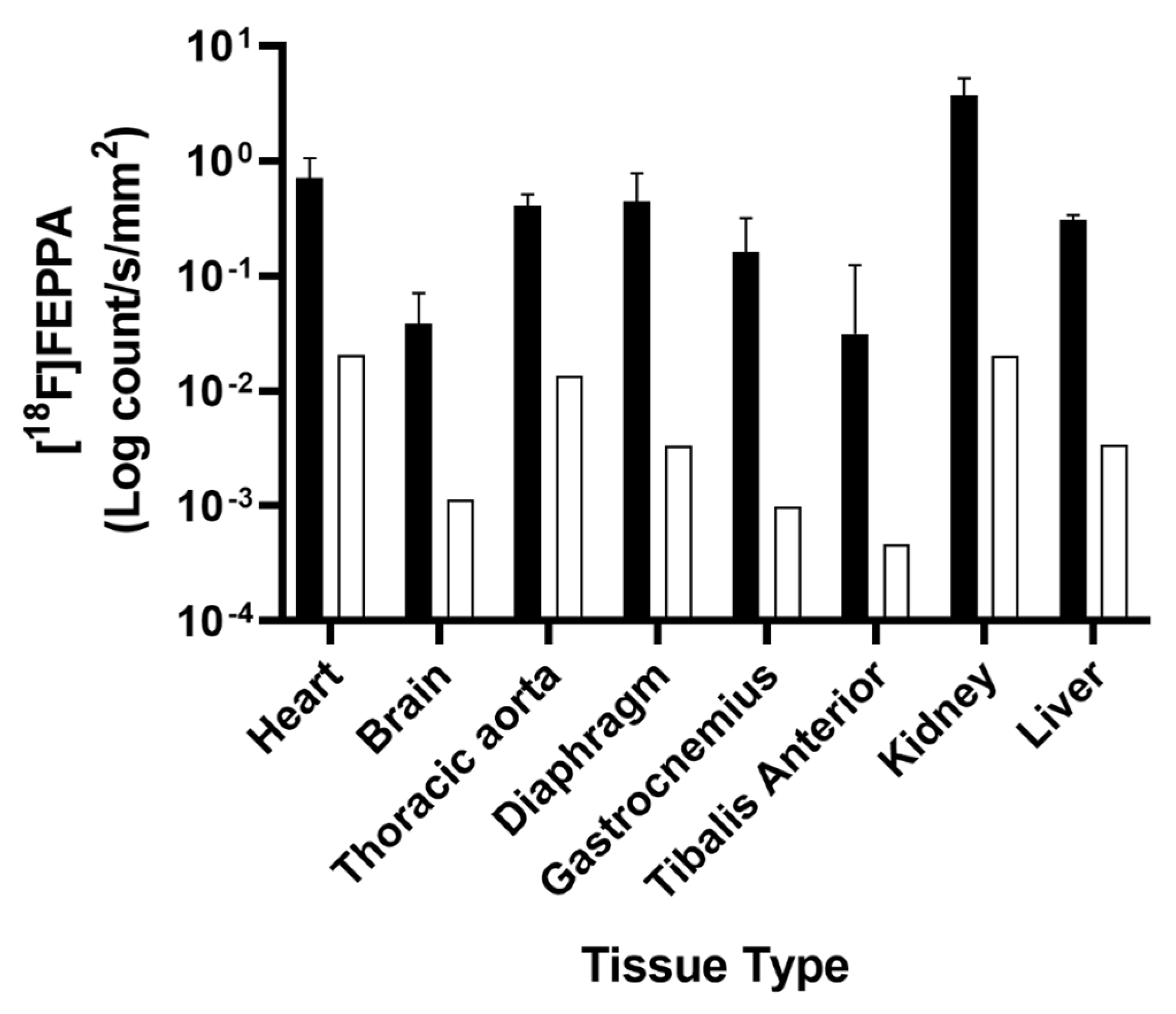
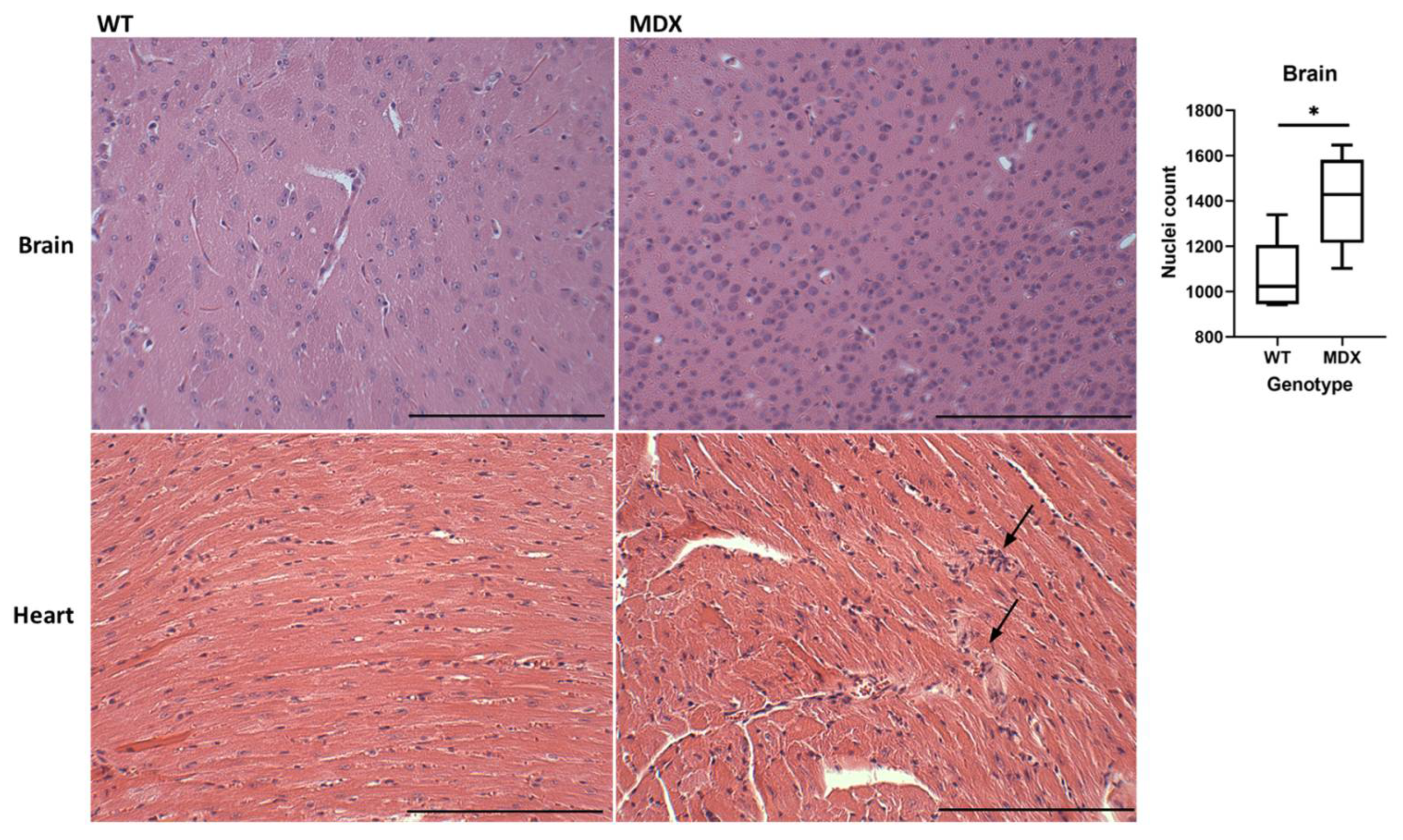
2.2. Ex Vivo TSPO Signal Indicates Heightened Cardiac and Neuroinflammation in DMD
2.3. Sample Size Estimation
3. Discussion
4. Materials and Methods
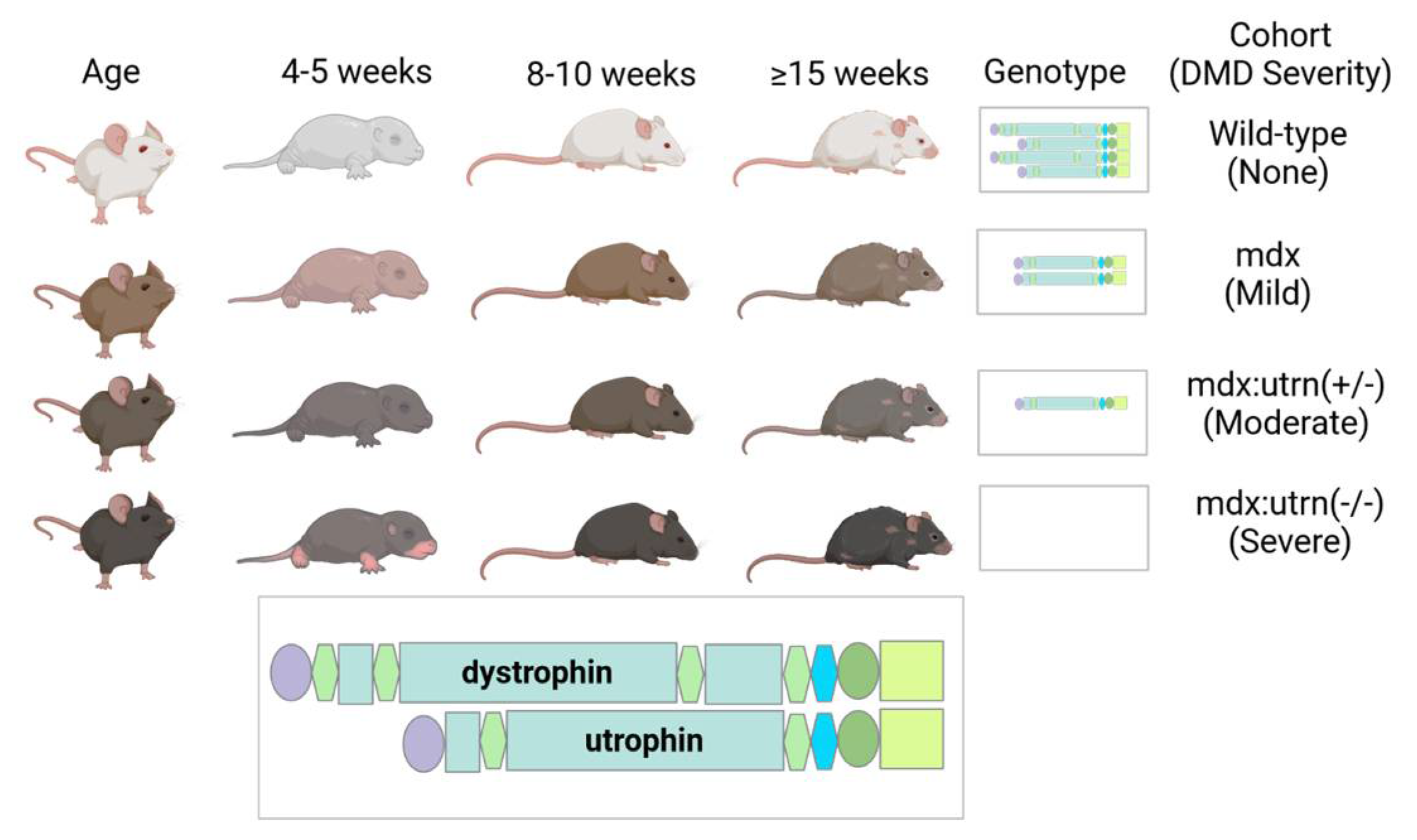
4.1. Study Population
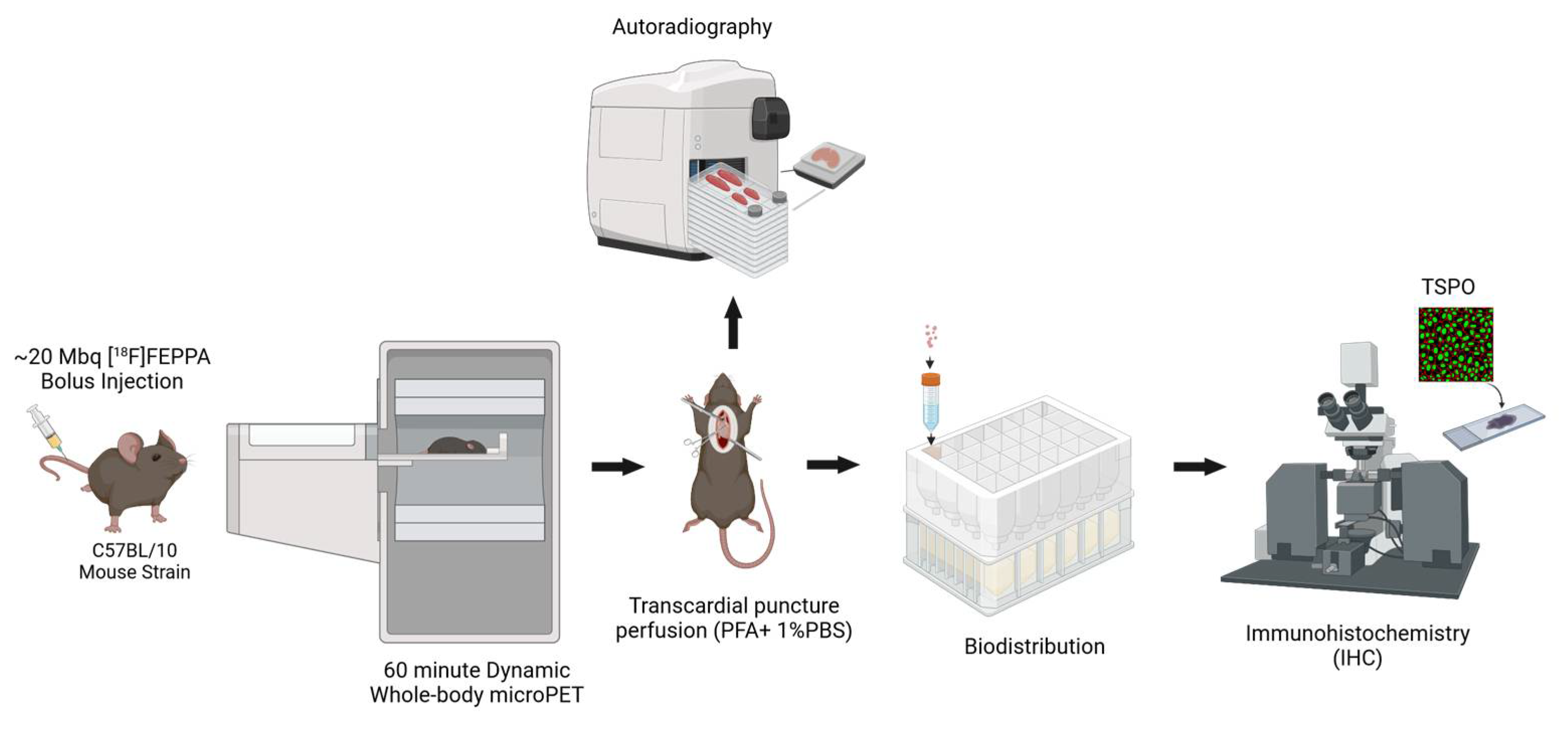
4.2. PET Imaging Protocol
4.3. PET Image Analysis
4.4. Biodistribution and Autoradiography
4.5. Histology Tissue Preparation
4.6. Immunohistochemistry Protocol
4.7. Microscopy and Image Analysis
4.8. Hematoxylin and Eosin (H&E) Staining
4.9. Statistical Analysis
4.10. Sample Size Estimation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A




Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PET | Biodistribution | Autoradiography | Immunohistochemistry | H&E | |
|---|---|---|---|---|---|
| Wild-type C57Bl10 | 6 | 2 | 2 | 3 | 1 |
| MDX mdx:utrn(+/−) | 4 | 4 | 1 | 3 | 1 |
References
- Dooley, J.; Gordon, K.E.; Dodds, L.; Macsween, J. Duchenne muscular dystrophy: A 30-year population-based incidence study. Clin. Pediatr. (Phila) 2010, 49, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y. Cognitive Function and Quality of Life of Muscular Dystrophy. In Muscular Dystrophies; Sakuma, K., Ed.; IntechOpen: Rijeka, Croatia, 2019; p. 3. [Google Scholar] [CrossRef]
- Doorenweerd, N. Combining genetics, neuropsychology and neuroimaging to improve understanding of brain involvement in Duchenne muscular dystrophy—A narrative review. Neuromuscul. Disord. 2020, 30, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef]
- D’amario, D.; Amodeo, A.; Adorisio, R.; Tiziano, F.D.; Leone, A.M.; Perri, G.; Bruno, P.; Massetti, M.; Ferlini, A.; Pane, M.; et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Nigro, G.; Comi, L.; Politano, L.; Bain, R. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Ballard, E.; Grey, N.; Jungbluth, H.; Wraige, E.; Kapetanakis, S.; Davidson, C.; Hart, N. Observation cohort study of cause of death in patients with Duchenne muscular dystrophy (DMD). Eur. Respir. J. 2012, 40 (Suppl. 56), P1720. [Google Scholar]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Belosludtsev, K.N. Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy. Int. J. Mol. Sci. 2023, 24, 2229. [Google Scholar] [CrossRef]
- Johnstone, V.P.A.; Viola, H.M.; Hool, L.C. Dystrophic Cardiomyopathy—Potential Role of Calcium in Pathogenesis, Treatment and Novel Therapies. Genes 2017, 8, 108. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Takeda, S.; Okada, T. Inflammatory predisposition predicts disease phenotypes in muscular dystrophy. Inflamm. Regen. 2016, 36, 14. [Google Scholar] [CrossRef]
- Meyers, T.A.; Townsend, D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef]
- Snow, W.M.; Anderson, J.E.; Jakobson, L.S. Neuropsychological and neurobehavioral functioning in Duchenne muscular dystrophy: A review. Neurosci. Biobehav. Rev. 2013, 37, 743–752. [Google Scholar] [CrossRef]
- Bagdatlioglu, E.; Porcari, P.; Greally, E.; Blamire, A.M.; Straub, V.W. Cognitive impairment appears progressive in the mdx mouse. Neuromuscul. Disord. 2020, 30, 368–388. [Google Scholar] [CrossRef]
- Winterholler, M.; Holländer, C.; Kerling, F.; Weber, I.; Dittrich, S.; Türk, M.; Schröder, R. Stroke in Duchenne Muscular Dystrophy: A Retrospective Longitudinal Study in 54 Patients. Stroke 2016, 47, 2123–2126. [Google Scholar] [CrossRef]
- Mavrogeni, S.; Papavasiliou, A.; Spargias, K.; Constandoulakis, P.; Papadopoulos, G.; Karanasios, E.; Georgakopoulos, D.; Kolovou, G.; Demerouti, E.; Polymeros, S.; et al. Myocardial inflammation in Duchenne Muscular Dystrophy as a precipitating factor for heart failure: A prospective study. BMC Neurol. 2010, 10, 33. [Google Scholar] [CrossRef]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Aarli, J.A. Role of cytokines in neurological disorders. Curr. Med. Chem. 2003, 10, 1931–1937. [Google Scholar] [CrossRef]
- Comim, C.M.; Ventura, L.; Freiberger, V.; Dias, P.; Bragagnolo, D.; Dutra, M.L.; Amaral, R.A.; Camargo-Fagundes, A.L.S.; Reis, P.A.; Castro-Faria-Neto, H.C.; et al. Neurocognitive Impairment in mdx Mice. Mol. Neurobiol. 2019, 56, 7608–7616. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, J.T.; Hupe, H.C.; Wang, Y.; Bankstahl, J.P.; Berding, G.; Ross, T.L.; Bauersachs, J.; Wollert, K.C.; Bengel, F.M. Myocardial Inflammation Predicts Remodeling and Neuroinflammation After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.A.; Garcia, A.; Parkes, J.; McCormick, P.; Stephenson, K.A.; Houle, S.; Vasdev, N. Radiosynthesis and initial evaluation of [18F]-FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl. Med. Biol. 2008, 35, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, J.T.; Bengel, F. Molecular Imaging of Myocardial Inflammation with Positron Emission Tomography Post-Ischemia: A Determinant of Subsequent Remodeling or Recovery. JACC Cardiovasc. Imaging 2018, 11, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Cuhlmann, S.; Gsell, W.; Van der Heiden, K.; Habib, J.; Tremoleda, J.L.; Khalil, M.; Turkheimer, F.; Meens, M.J.; Kwak, B.R.; Bird, J.; et al. In vivo mapping of vascular inflammation using the translocator protein tracer 18F-FEDAA1106. Mol. Imaging 2014, 13, 1–10. [Google Scholar] [CrossRef]
- Largeau, B.; Dupont, A.-C.; Guilloteau, D.; Santiago-Ribeiro, M.-J.; Arlicot, N. TSPO PET Imaging: From Microglial Activation to Peripheral Sterile Inflammatory Diseases? Contrast Media Mol. Imaging 2017, 2017, 6592139. [Google Scholar] [CrossRef]
- Gerhard, A.; Neumaier, B.; Elitok, E.; Glatting, G.; Ries, V.; Tomczak, R.; Ludolph, A.C.; Reske, S.N. In vivo imaging of activated microglia using [11 C]PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport 2000, 11, 2957–2960. [Google Scholar] [CrossRef]
- Schneider, S.M.; Sridhar, V.; Bettis, A.K.; Heath-Barnett, H.; Balog-Alvarez, C.J.; Guo, L.-J.; Johnson, R.; Jaques, S.; Vitha, S.; Glowcwski, A.C.; et al. Glucose Metabolism as a Pre-clinical Biomarker for the Golden Retriever Model of Duchenne Muscular Dystrophy. Mol. Imaging Biol. 2018, 20, 780–788. [Google Scholar] [CrossRef]
- Lee, J.S.; Pfund, Z.; Juhász, C.; Behen, M.E.; Muzik, O.; Chugani, D.C.; Nigro, M.A.; Chugani, H.T. Altered regional brain glucose metabolism in Duchenne muscular dystrophy: A pet study. Muscle Nerve 2002, 26, 506–512. [Google Scholar] [CrossRef]
- Bresolin, N.; Castelli, E.; Comi, G.; Felisari, G.; Bardoni, A.; Perani, D.; Grassi, F.; Turconi, A.C.; Mazzucchelli, F.; Gallotti, D.; et al. Cognitive impairment in Duchenne muscular dystrophy. Neuromuscul. Disord. 1994, 4, 359–369. [Google Scholar] [CrossRef]
- Basu, S.; Zhuang, H.; Torigian, D.A.; Rosenbaum, J.; Chen, W.; Alavi, A. Functional imaging of inflammatory diseases using nuclear medicine techniques. Semin. Nucl. Med. 2009, 39, 124–145. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Yoon, H.J.; Kang, D.-Y. Assessment of change in glucose metabolism in white matter of amyloid-positive patients with Alzheimer disease using F-18 FDG PET. Medicine 2017, 96, e9042. [Google Scholar] [CrossRef]
- MacRitchie, N.; Frleta-Gilchrist, M.; Sugiyama, A.; Lawton, T.; McInnes, I.B.; Maffia, P. Molecular imaging of inflammation—Current and emerging technologies for diagnosis and treatment. Pharmacol. Ther. 2020, 211, 107550. [Google Scholar] [CrossRef]
- Lange, P.S.; Avramovic, N.; Frommeyer, G.; Wasmer, K.; Pott, C.; Eckardt, L.; Wenning, C. Routine 18F-FDG PET/CT does not detect inflammation in the left atrium in patients with atrial fibrillation. Int. J. Cardiovasc. Imaging 2017, 33, 1271–1276. [Google Scholar] [CrossRef]
- Brendel, M.; Probst, F.; Jaworska, A.; Overhoff, F.; Korzhova, V.; Albert, N.L.; Beck, R.; Lindner, S.; Gildehaus, F.-J.; Baumann, K.; et al. Glial Activation and Glucose Metabolism in a Transgenic Amyloid Mouse Model: A Triple-Tracer PET Study. J. Nucl. Med. 2016, 57, 954–960. [Google Scholar] [CrossRef]
- Zimmer, E.R.; Parent, M.J.; Souza, D.G.; Leuzy, A.; Lecrux, C.; Kim, H.-I.; Gauthier, S.; Pellerin, L.; Hamel, E.; Rosa-Neto, P. [18F]FDG PET signal is driven by astroglial glutamate transport. Nat. Neurosci. 2017, 20, 393–395. [Google Scholar] [CrossRef]
- Giatzakis, C.; Papadopoulos, V. Differential utilization of the promoter of peripheral-type benzodiazepine receptor by steroidogenic versus nonsteroidogenic cell lines and the role of Sp1 and Sp3 in the regulation of basal activity. Endocrinology 2004, 145, 1113–1123. [Google Scholar] [CrossRef]
- Gavish, M.; Bachman, I.; Shoukrun, R.; Katz, Y.; Veenman, L.; Weisinger, G.; Weizman, A. Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 1999, 51, 629–650. [Google Scholar]
- Kim, G.R.; Paeng, J.C.; Jung, J.H.; Moon, B.S.; Lopalco, A.; Denora, N.; Lee, B.C.; Kim, S.E. Assessment of TSPO in a Rat Experimental Autoimmune Myocarditis Model: A Comparison Study between [18F]Fluoromethyl-PBR28 and [18F]CB251. Int. J. Mol. Sci. 2018, 19, 276. [Google Scholar] [CrossRef]
- Qi, X.; Xu, J.; Wang, F.; Xiao, J. Translocator protein (18 kDa): A promising therapeutic target and diagnostic tool for cardiovascular diseases. Oxid. Med. Cell. Longev. 2012, 2012, 162934. [Google Scholar] [CrossRef]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2008, 18, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Bridges, L. The association of cardiac muscle necrosis and inflammation with the degenerative and persistent myopathy of MDX mice. J. Neurol. Sci. 1986, 72, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Chiyo, T.; Nishiyama, A.; Okada, H.; Takeda, S.; Okada, T. Dystrophic mdx mice develop severe cardiac and respiratory dysfunction following genetic ablation of the anti-inflammatory cytokine IL-10. Hum. Mol. Genet. 2013, 23, 3990–4000. [Google Scholar] [CrossRef] [PubMed]
- Hainsey, T.; Senapati, S.; Kuhn, D.; Rafael, J. Cardiomyopathic features associated with muscular dystrophy are independent of dystrophin absence in cardiovasculature. Neuromuscul. Disord. 2003, 13, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, C.; Loch, D.; Laws, N.; Trebbin, A.; Hoey, A.J. Timeline of cardiac dystrophy in 3-18-month-old MDX mice. Muscle Nerve 2010, 42, 504–513. [Google Scholar] [CrossRef]
- Yucel, N.; Chang, A.C.; Day, J.W.; Rosenthal, N.; Blau, H.M. Humanizing the mdx mouse model of DMD: The long and the short of it. NPJ Regen. Med. 2018, 3, 4. [Google Scholar] [CrossRef]
- Gutpell, K.M.; Hrinivich, W.T.; Hoffman, L.M. Skeletal muscle fibrosis in the mdx/utrn+/− mouse validates its suitability as a murine model of Duchenne muscular dystrophy. PLoS ONE 2015, 10, e0117306. [Google Scholar] [CrossRef]
- Verhaart, I.E.; van Duijn, R.J.; Adel, B.D.; Roest, A.A.; Verschuuren, J.J.; Aartsma-Rus, A.; van der Weerd, L. Assessment of cardiac function in three mouse dystrophinopathies by magnetic resonance imaging. Neuromuscul. Disord. 2012, 22, 418–426. [Google Scholar] [CrossRef]
- Ballmann, C.; Denney, T.S.; Beyers, R.J.; Quindry, T.; Romero, M.; Amin, R.; Selsby, J.T.; Quindry, J.C. Lifelong quercetin enrichment and cardioprotection in Mdx/Utrn+/− mice. Am. J. Physiol. Circ. Physiol. 2017, 312, H128–H140. [Google Scholar] [CrossRef]
- Zammit, M.; Tao, Y.; Olsen, M.E.; Metzger, J.; Vermilyea, S.C.; Bjornson, K.; Slesarev, M.; Block, W.F.; Fuchs, K.; Phillips, S.; et al. [18F]FEPPA PET imaging for monitoring CD68-positive microglia/macrophage neuroinflammation in nonhuman primates. EJNMMI Res. 2020, 10, 93. [Google Scholar] [CrossRef]
- Rae, M.G.; O’Malley, D. Cognitive dysfunction in Duchenne muscular dystrophy: A possible role for neuromodulatory immune molecules. J. Neurophysiol. 2016, 116, 1304–1315. [Google Scholar] [CrossRef]
- Stephenson, K.A.; Rae, M.G.; O’Malley, D. Interleukin-6: A neuro-active cytokine contributing to cognitive impairment in Duchenne muscular dystrophy? Cytokine 2020, 133, 155134. [Google Scholar] [CrossRef]
- Sawada, M.; Kondo, N.; Suzumura, A.; Marunouchi, T. Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res. 1989, 491, 394–397. [Google Scholar] [CrossRef]
- Hanisch, U. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Shriver, L.P.; Maresz, K.; Dittel, B.N. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J. Neurosci. Res. 2005, 81, 374–389. [Google Scholar] [CrossRef]
- Malpetti, M.; Kievit, R.A.; Passamonti, L.; Jones, P.S.; Tsvetanov, K.A.; Rittman, T.; Mak, E.; Nicastro, N.; Bevan-Jones, W.R.; Su, L.; et al. Microglial activation and tau burden predict cognitive decline in Alzheimer’s disease. Brain 2020, 143, 1588–1602. [Google Scholar] [CrossRef]
- Cotton, S.; Voudouris, N.J.; Greenwood, K.M. Intelligence and Duchenne muscular dystrophy: Full-scale, verbal, and performance intelligence quotients. Dev. Med. Child Neurol. 2001, 43, 497–501. [Google Scholar] [CrossRef]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Quondamatteo, F.; Herken, R.; Errede, M.; Ribatti, D.; Svelto, M.; Roncali, L. Role of aquaporin-4 water channel in the development and integrity of the blood-brain barrier. J. Cell Sci. 2001, 114, 1297–1307. [Google Scholar] [CrossRef]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2019, 57, 1748–1767. [Google Scholar] [CrossRef]
- Nico, B.; Tamma, R.; Annese, T.; Mangieri, D.; De Luca, A.; Corsi, P.; Benagiano, V.; Longo, V.; Crivellato, E.; Salmaggi, A.; et al. Glial dystrophin-associated proteins, laminin and agrin, are downregulated in the brain of mdx mouse. Lab. Investig. 2010, 90, 1645–1660. [Google Scholar] [CrossRef]
- Nico, B.; Nicchia, G.P.; Frigeri, A.; Corsi, P.; Mangieri, D.; Ribatti, D.; Svelto, M.; Roncali, L. Altered blood–brain barrier development in dystrophic MDX mice. Neuroscience 2004, 125, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sato, N.; Ikimura, K.; Nishino, H.; Rakugi, H.; Morishita, R. Increased blood–brain barrier vulnerability to systemic inflammation in an Alzheimer disease mouse model. Neurobiol. Aging 2013, 34, 2064–2070. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Corsi, P.; Ribatti, D.; Quondamatteo, F.; Herken, R.; Girolamo, F.; Marzullo, A.; Svelto, M.; et al. Severe alterations of endothelial and glial cells in the blood-brain barrier of dystrophic mdx mice. Glia 2003, 42, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.G.; Banks, W.A.; Kastin, A.J. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J. Neuroimmunol. 1993, 47, 169–176. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef]
- Gutpell, K.M.; Tasevski, N.; Wong, B.; Hrinivich, W.T.; Su, F.; Hadway, J.; Desjardins, L.; Lee, T.-Y.; Hoffman, L.M. ANG1 treatment reduces muscle pathology and prevents a decline in perfusion in DMD mice. PLoS ONE 2017, 12, e0174315. [Google Scholar] [CrossRef]
- Fairweather, D.; Coronado, M.J.; Garton, A.E.; Dziedzic, J.L.; Bucek, A.; Cooper, L.; Brandt, J.; Alikhan, F.S.; Wang, H.; Endres, C.J.; et al. Sex differences in translocator protein 18 kDa (TSPO) in the heart: Implications for imaging myocardial inflammation. J. Cardiovasc. Transl. Res. 2014, 7, 192–202. [Google Scholar] [CrossRef]
- Tuisku, J.; Plavén-Sigray, P.; Gaiser, E.C.; Airas, L.; Al-Abdulrasul, H.; Brück, A.; Carson, R.E.; Chen, M.-K.; Cosgrove, K.P.; Ekblad, L.; et al. Effects of age, BMI and sex on the glial cell marker TSPO—A multicentre [11C]PBR28 HRRT PET study. Eur. J. Nucl. Med. 2019, 46, 2329–2338. [Google Scholar] [CrossRef]
- Patel, A.M.; Wierda, K.; Thorrez, L.; van Putten, M.; De Smedt, J.; Ribeiro, L.; Tricot, T.; Gajjar, M.; Duelen, R.; Van Damme, P.; et al. Dystrophin deficiency leads to dysfunctional glutamate clearance in iPSC derived astrocytes. Transl. Psychiatry 2019, 9, 200. [Google Scholar] [CrossRef]
- Notter, T.; Schalbetter, S.M.; Clifton, N.E.; Mattei, D.; Richetto, J.; Thomas, K.; Meyer, U.; Hall, J. Neuronal activity increases translocator protein (TSPO) levels. Mol. Psychiatry 2020, 26, 2025–2037. [Google Scholar] [CrossRef]
- Liu, Q.; Johnson, E.M.; Lam, R.K.; Wang, Q.; Ye, H.B.; Wilson, E.N.; Minhas, P.S.; Liu, L.; Swarovski, M.S.; Tran, S.; et al. Peripheral TREM1 responses to brain and intestinal immunogens amplify stroke severity. Nat. Immunol. 2019, 20, 1023–1034. [Google Scholar] [CrossRef]
- Grady, R.; Teng, H.; Nichol, M.C.; Cunningham, J.C.; Wilkinson, R.S.; Sanes, J.R. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell 1997, 90, 729–738. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; De Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, B.; Feng, Z.; Yu, S.; Bao, Y. A study on immunomodulatory mechanism of Polysaccharopeptide mediated by TLR4 signaling pathway. BMC Immunol. 2015, 16, 34. [Google Scholar] [CrossRef]
- Homma, S.; Kaul, S.; Boucher, C.A. Correlates of lung/heart ratio of thallium-201 in coronary artery disease. J. Nucl. Med. 1987, 28, 1531–1535. [Google Scholar]
- Gage, G.J.; Kipke, D.R.; Shain, W. Whole animal perfusion fixation for rodents. J. Vis. Exp. 2012, 65, e3564. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ; U.S. National Institutes of Health: Bethesda, MD, USA, 1997. Available online: https://imagej.nih.gov/ij/ (accessed on 25 February 2023).
- Borm, G.F.; Bloem, B.R.; Munneke, M.; Teerenstra, S. A simple method for calculating power based on a prior trial. J. Clin. Epidemiol. 2010, 63, 992–997. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.M.; McClennan, A.; Liu, L.; Hadway, J.; Ronald, J.A.; Hicks, J.W.; Hoffman, L.; Anazodo, U.C. A Protocol for Simultaneous In Vivo Imaging of Cardiac and Neuroinflammation in Dystrophin-Deficient MDX Mice Using [18F]FEPPA PET. Int. J. Mol. Sci. 2023, 24, 7522. https://doi.org/10.3390/ijms24087522
Tang JM, McClennan A, Liu L, Hadway J, Ronald JA, Hicks JW, Hoffman L, Anazodo UC. A Protocol for Simultaneous In Vivo Imaging of Cardiac and Neuroinflammation in Dystrophin-Deficient MDX Mice Using [18F]FEPPA PET. International Journal of Molecular Sciences. 2023; 24(8):7522. https://doi.org/10.3390/ijms24087522
Chicago/Turabian StyleTang, Joanne M., Andrew McClennan, Linshan Liu, Jennifer Hadway, John A. Ronald, Justin W. Hicks, Lisa Hoffman, and Udunna C. Anazodo. 2023. "A Protocol for Simultaneous In Vivo Imaging of Cardiac and Neuroinflammation in Dystrophin-Deficient MDX Mice Using [18F]FEPPA PET" International Journal of Molecular Sciences 24, no. 8: 7522. https://doi.org/10.3390/ijms24087522
APA StyleTang, J. M., McClennan, A., Liu, L., Hadway, J., Ronald, J. A., Hicks, J. W., Hoffman, L., & Anazodo, U. C. (2023). A Protocol for Simultaneous In Vivo Imaging of Cardiac and Neuroinflammation in Dystrophin-Deficient MDX Mice Using [18F]FEPPA PET. International Journal of Molecular Sciences, 24(8), 7522. https://doi.org/10.3390/ijms24087522