


Chronological Age and DNA Damage Accumulation in Blood Mononuclear Cells: A Linear Association in Healthy Humans after 50 Years of Age
 ,
,  , ,
, ,  , ,
, ,  ,
,
Abstract

1. Introduction
2. Results
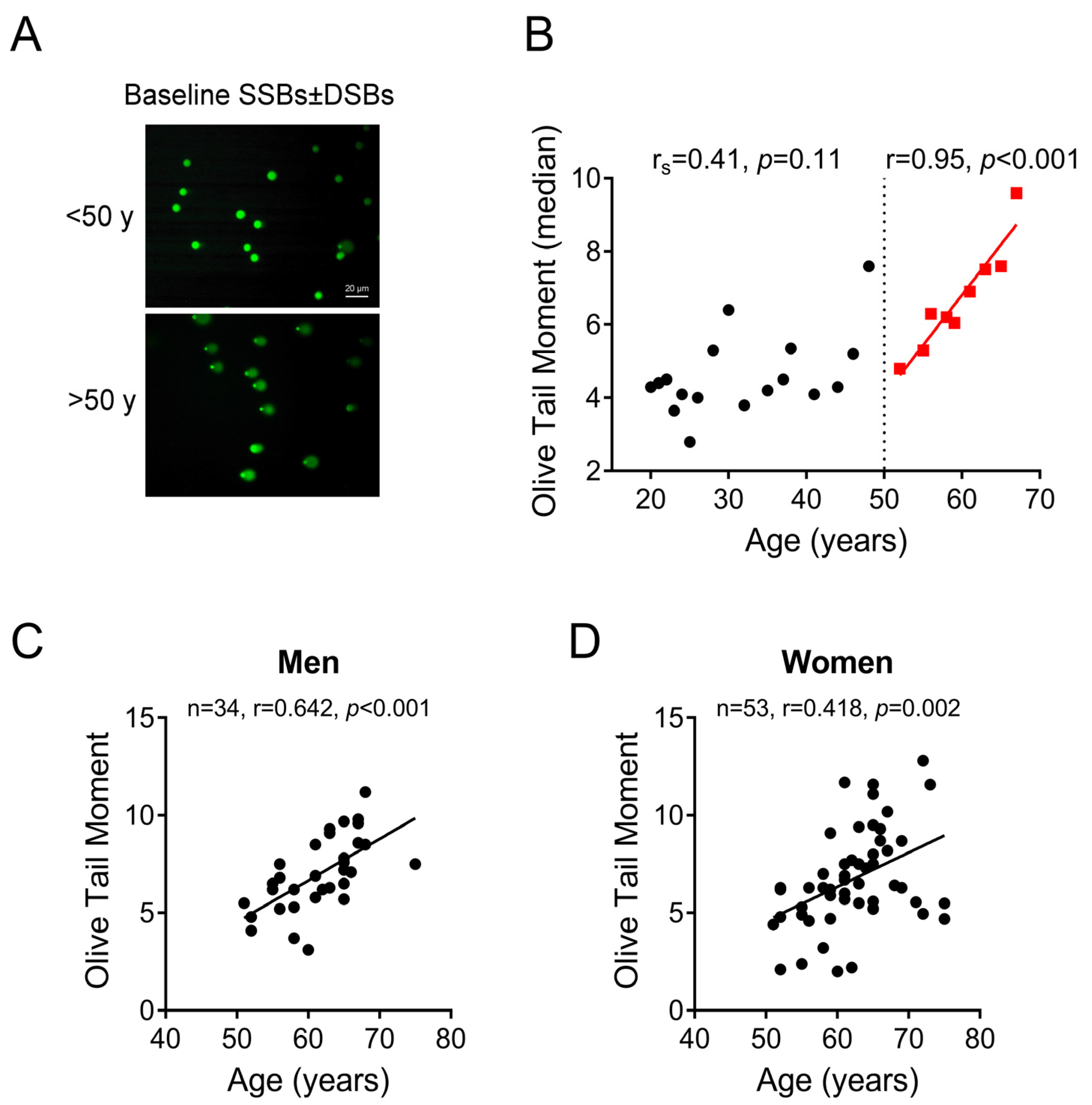
2.1. Association between Age and DNA SSBs/DSBs Accumulation in PBMCs: The Critical Threshold of 50 Years
2.2. DNA Damage Formation and Repair Capacity of DNA Double Strand Breaks Differs before and after 50 Years of Age
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Peripheral Blood Mononuclear Cell Isolation
4.3. Alkaline Comet Assay
4.4. Immunofluorescence Detection of γH2AX Foci
4.5. Assessment of DNA Damage Formation
4.6. Repair Capacity of DNA DSBs
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L. Understanding the Odd Science of Aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J.H.J. The Central Role of DNA Damage in the Ageing Process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banáth, J.P. The Comet Assay: A Method to Measure DNA Damage in Individual Cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.; Møller, P.; Gajski, G.; Vodenková, S.; Abdulwahed, A.; Anderson, D.; Bankoglu, E.E.; Bonassi, S.; Boutet-Robinet, E.; Brunborg, G.; et al. Measuring DNA Modifications with the Comet Assay: A Compendium of Protocols. Nat. Protoc. 2023, 18, 929–989. [Google Scholar] [CrossRef]
- Bankoglu, E.E.; Mukama, T.; Katzke, V.; Stipp, F.; Johnson, T.; Kühn, T.; Seyfried, F.; Godschalk, R.; Collins, A.; Kaaks, R.; et al. Short- and Long-Term Reproducibility of the COMET Assay for Measuring DNA Damage Biomarkers in Frozen Blood Samples of the EPIC-Heidelberg Cohort. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2022, 874–875, 503442. [Google Scholar] [CrossRef]
- Azqueta, A.; Ladeira, C.; Giovannelli, L.; Boutet-Robinet, E.; Bonassi, S.; Neri, M.; Gajski, G.; Duthie, S.; Del Bo’, C.; Riso, P.; et al. Application of the Comet Assay in Human Biomonitoring: An HCOMET Perspective. Mutat. Res. Rev. Mutat. Res. 2020, 783, 108288. [Google Scholar] [CrossRef]
- Collins, A.; Koppen, G.; Valdiglesias, V.; Dusinska, M.; Kruszewski, M.; Møller, P.; Rojas, E.; Dhawan, A.; Benzie, I.; Coskun, E.; et al. The Comet Assay as a Tool for Human Biomonitoring Studies: The ComNet Project. Mutat. Res. Rev. Mutat. Res. 2014, 759, 27–39. [Google Scholar] [CrossRef]
- Dusinska, M.; Collins, A.R. The Comet Assay in Human Biomonitoring: Gene-Environment Interactions. Mutagenesis 2008, 23, 191–205. [Google Scholar] [CrossRef]
- Soares, J.P.; Cortinhas, A.; Bento, T.; Leitão, J.C.; Collins, A.R.; Gaivão, I.; Mota, M.P. Aging and DNA Damage in Humans: A Meta-Analysis Study. Aging 2014, 6, 432–439. [Google Scholar] [CrossRef]
- Milić, M.; Ceppi, M.; Bruzzone, M.; Azqueta, A.; Brunborg, G.; Godschalk, R.; Koppen, G.; Langie, S.; Møller, P.; Teixeira, J.P.; et al. The HCOMET Project: International Database Comparison of Results with the Comet Assay in Human Biomonitoring. Baseline Frequency of DNA Damage and Effect of Main Confounders. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108371. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Souliotis, V.L.; Vlachogiannis, N.I.; Pappa, M.; Argyriou, A.; Ntouros, P.A.; Sfikakis, P.P. DNA Damage Response and Oxidative Stress in Systemic Autoimmunity. Int. J. Mol. Sci. 2019, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Dodds, R.M.; Syddall, H.E.; Cooper, R.; Benzeval, M.; Deary, I.J.; Dennison, E.M.; Der, G.; Gale, C.R.; Inskip, H.M.; Jagger, C.; et al. Grip Strength across the Life Course: Normative Data from Twelve British Studies. PLoS ONE 2014, 9, e113637. [Google Scholar] [CrossRef]
- Franceschi, C.; Ostan, R.; Mariotti, S.; Monti, D.; Vitale, G. The Aging Thyroid: A Reappraisal within the Geroscience Integrated Perspective. Endocr. Rev. 2019, 40, 1250–1270. [Google Scholar] [CrossRef] [PubMed]
- Lambrinoudaki, I.; Armeni, E.; Goulis, D.; Bretz, S.; Ceausu, I.; Durmusoglu, F.; Erkkola, R.; Fistonic, I.; Gambacciani, M.; Geukes, M.; et al. Menopause, Wellbeing and Health: A Care Pathway from the European Menopause and Andropause Society. Maturitas 2022, 163, 1–14. [Google Scholar] [CrossRef]
- Kaufman, J.-M.; Lapauw, B.; Mahmoud, A.; T’Sjoen, G.; Huhtaniemi, I.T. Aging and the Male Reproductive System. Endocr. Rev. 2019, 40, 906–972. [Google Scholar] [CrossRef]
- Endo, Y.; Nourmahnad, A.; Sinha, I. Optimizing Skeletal Muscle Anabolic Response to Resistance Training in Aging. Front. Physiol. 2020, 11, 874. [Google Scholar] [CrossRef]
- Distefano, G.; Goodpaster, B.H. Effects of Exercise and Aging on Skeletal Muscle. Cold Spring Harb. Perspect. Med. 2018, 8, a029785. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A Novel Biomarker for DNA Double-Strand Breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Valdiglesias, V.; Giunta, S.; Fenech, M.; Neri, M.; Bonassi, S. ΓH2AX as a Marker of DNA Double Strand Breaks and Genomic Instability in Human Population Studies. Mutat. Res. 2013, 753, 24–40. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yao, L.; Brown, C.; Rizzo, C.J.; Turesky, R.J. Quantitation of Apurinic/Apyrimidinic Sites in Isolated DNA and in Mammalian Tissue with a Reduced Level of Artifacts. Anal. Chem. 2019, 91, 7403–7410. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Wilson, D.M. Overview of Base Excision Repair Biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef]
- De Bont, R.; van Larebeke, N. Endogenous DNA Damage in Humans: A Review of Quantitative Data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Walker, V.E.; Upton, P.B.; Chiang, S.Y.; Kow, Y.W.; Swenberg, J.A. Highly Sensitive Apurinic/Apyrimidinic Site Assay Can Detect Spontaneous and Chemically Induced Depurination under Physiological Conditions. Cancer Res. 1998, 58, 222–225. [Google Scholar]
- Kow, Y.W.; Bao, G.; Minesinger, B.; Jinks-Robertson, S.; Siede, W.; Jiang, Y.L.; Greenberg, M.M. Mutagenic Effects of Abasic and Oxidized Abasic Lesions in Saccharomyces Cerevisiae. Nucleic Acids Res. 2005, 33, 6196–6202. [Google Scholar] [CrossRef]
- Boiteux, S.; Guillet, M. Abasic Sites in DNA: Repair and Biological Consequences in Saccharomyces Cerevisiae. DNA Repair 2004, 3, 1–12. [Google Scholar] [CrossRef]
- Genadieva-Stavric, S.; Cavallo, F.; Palumbo, A. New Approaches to Management of Multiple Myeloma. Curr. Treat. Options Oncol. 2014, 15, 157–170. [Google Scholar] [CrossRef]
- Episkopou, H.; Kyrtopoulos, S.A.; Sfikakis, P.P.; Fousteri, M.; Dimopoulos, M.A.; Mullenders, L.H.F.; Souliotis, V.L. Association between Transcriptional Activity, Local Chromatin Structure, and the Efficiencies of Both Subpathways of Nucleotide Excision Repair of Melphalan Adducts. Cancer Res. 2009, 69, 4424–4433. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA Interstrand Crosslink Repair and Cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Homologous Recombination in Cancer Development, Treatment and Development of Drug Resistance. Carcinogenesis 2010, 31, 955–960. [Google Scholar] [CrossRef]
- Møller, P. Effect of Age and Sex on the Level of DNA Strand Breaks and Oxidatively Damaged DNA in Human Blood Cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 838, 16–21. [Google Scholar] [CrossRef]
- Garm, C.; Moreno-Villanueva, M.; Bürkle, A.; Petersen, I.; Bohr, V.A.; Christensen, K.; Stevnsner, T. Age and Gender Effects on DNA Strand Break Repair in Peripheral Blood Mononuclear Cells. Aging Cell 2013, 12, 58–66. [Google Scholar] [CrossRef]
- Møller, P.; Azqueta, A.; Boutet-Robinet, E.; Koppen, G.; Bonassi, S.; Milić, M.; Gajski, G.; Costa, S.; Teixeira, J.P.; Costa Pereira, C.; et al. Minimum Information for Reporting on the Comet Assay (MIRCA): Recommendations for Describing Comet Assay Procedures and Results. Nat. Protoc. 2020, 15, 3817–3826. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P.; Durand, R.E. Heterogeneity in Radiation-Induced DNA Damage and Repair in Tumor and Normal Cells Measured Using the “Comet” Assay. Radiat. Res. 1990, 122, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.E.; Eskenazi, B.; Baumgartner, A.; Marchetti, F.; Young, S.; Weldon, R.; Anderson, D.; Wyrobek, A.J. The Effects of Male Age on Sperm DNA Damage in Healthy Non-Smokers. Hum. Reprod. Oxf. Engl. 2007, 22, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Souliotis, V.L.; Sfikakis, P.P. Increased DNA Double-Strand Breaks and Enhanced Apoptosis in Patients with Lupus Nephritis. Lupus 2015, 24, 804–815. [Google Scholar] [CrossRef]
- Souliotis, V.L.; Vougas, K.; Gorgoulis, V.G.; Sfikakis, P.P. Defective DNA Repair and Chromatin Organization in Patients with Quiescent Systemic Lupus Erythematosus. Arthritis Res. Ther. 2016, 18, 182. [Google Scholar] [CrossRef]
- Souliotis, V.L.; Vlachogiannis, N.I.; Pappa, M.; Argyriou, A.; Sfikakis, P.P. DNA Damage Accumulation, Defective Chromatin Organization and Deficient DNA Repair Capacity in Patients with Rheumatoid Arthritis. Clin. Immunol. 2019, 203, 28–36. [Google Scholar] [CrossRef]
- Vlachogiannis, N.I.; Pappa, M.; Ntouros, P.A.; Nezos, A.; Mavragani, C.P.; Souliotis, V.L.; Sfikakis, P.P. Association between DNA Damage Response, Fibrosis and Type I Interferon Signature in Systemic Sclerosis. Front. Immunol. 2020, 11, 582401. [Google Scholar] [CrossRef]
- Ntouros, P.A.; Vlachogiannis, N.I.; Pappa, M.; Nezos, A.; Mavragani, C.P.; Tektonidou, M.G.; Souliotis, V.L.; Sfikakis, P.P. Effective DNA Damage Response after Acute but Not Chronic Immune Challenge: SARS-CoV-2 Vaccine versus Systemic Lupus Erythematosus. Clin. Immunol. 2021, 229, 108765. [Google Scholar] [CrossRef]
- Vlachogiannis, N.I.; Ntouros, P.A.; Pappa, M.; Verrou, K.-M.; Arida, A.; Souliotis, V.L.; Sfikakis, P.P. Deregulated DNA Damage Response Network in Behcet’s Disease. Clin. Immunol. 2023, 246, 109189. [Google Scholar] [CrossRef]
- Alsaleh, G.; Richter, F.C.; Simon, A.K. Age-Related Mechanisms in the Context of Rheumatic Disease. Nat. Rev. Rheumatol. 2022, 18, 694–710. [Google Scholar] [CrossRef]
- Pezone, A.; Olivieri, F.; Napoli, M.V.; Procopio, A.; Avvedimento, E.V.; Gabrielli, A. Inflammation and DNA Damage: Cause, Effect or Both. Nat. Rev. Rheumatol. 2023, 19, 200–211. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Løhr, M.; Jensen, A.; Eriksen, L.; Grønbæk, M.; Loft, S.; Møller, P. Age and Metabolic Risk Factors Associated with Oxidatively Damaged DNA in Human Peripheral Blood Mononuclear Cells. Oncotarget 2015, 6, 2641–2653. [Google Scholar] [CrossRef]
- Li, H.; Mitchell, J.R.; Hasty, P. DNA Double-Strand Breaks: A Potential Causative Factor for Mammalian Aging? Mech. Ageing Dev. 2008, 129, 416–424. [Google Scholar] [CrossRef]
- Ju, Y.-J.; Lee, K.-H.; Park, J.-E.; Yi, Y.-S.; Yun, M.-Y.; Ham, Y.-H.; Kim, T.-J.; Choi, H.M.; Han, G.J.; Lee, J.-H.; et al. Decreased Expression of DNA Repair Proteins Ku70 and Mre11 Is Associated with Aging and May Contribute to the Cellular Senescence. Exp. Mol. Med. 2006, 38, 686–693. [Google Scholar] [CrossRef]
- Frasca, D.; Barattini, P.; Tirindelli, D.; Guidi, L.; Bartoloni, C.; Errani, A.; Costanzo, M.; Tricerri, A.; Pierelli, L.; Doria, G. Effect of Age on DNA Binding of the Ku Protein in Irradiated Human Peripheral Blood Mononuclear Cells (PBMC). Exp. Gerontol. 1999, 34, 645–658. [Google Scholar] [CrossRef]
- Milic, M.; Frustaci, A.; Del Bufalo, A.; Sánchez-Alarcón, J.; Valencia-Quintana, R.; Russo, P.; Bonassi, S. DNA Damage in Non-Communicable Diseases: A Clinical and Epidemiological Perspective. Mutat. Res. 2015, 776, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.; Stopper, H.; Collins, A.R. Measurement of DNA Damage with the Comet Assay in High-Prevalence Diseases: Current Status and Future Directions. Mutagenesis 2020, 35, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Borisovs, V.; Ļeonova, E.; Baumane, L.; Kalniņa, J.; Mjagkova, N.; Sjakste, N. Blood Levels of Nitric Oxide and DNA Breaks Assayed in Whole Blood and Isolated Peripheral Blood Mononucleated Cells in Patients with Multiple Sclerosis. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 843, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Lamonaca, P.; Milic, M.; Rojas, E.; Prinzi, G.; Cardaci, V.; Vitiello, L.; Proietti, S.; Santoro, A.; Tomino, C.; et al. Biomarkers of DNA Damage in COPD Patients Undergoing Pulmonary Rehabilitation: Integrating Clinical Parameters with Genomic Profiling. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 843, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Vodenkova, S.; Opattova, A.; Vodickova, L. DNA Damage and Repair Measured by Comet Assay in Cancer Patients. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 843, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Bonassi, S.; Ceppi, M.; Møller, P.; Azqueta, A.; Milić, M.; Neri, M.; Brunborg, G.; Godschalk, R.; Koppen, G.; Langie, S.A.S.; et al. DNA Damage in Circulating Leukocytes Measured with the Comet Assay May Predict the Risk of Death. Sci. Rep. 2021, 11, 16793. [Google Scholar] [CrossRef]
- Geric, M.; Gajski, G.; Orešcanin, V.; Garaj-Vrhovac, V. Seasonal Variations as Predictive Factors of the Comet Assay Parameters: A Retrospective Study. Mutagenesis 2018, 33, 53–60. [Google Scholar] [CrossRef]
- Kravvariti, E.; Ntouros, P.A.; Vlachogiannis, N.I.; Pappa, M.; Souliotis, V.L.; Sfikakis, P.P. Geriatric Frailty Is Associated with Oxidative Stress, Accumulation and Defective Repair of DNA Double-Strand Breaks Independent of Age and Comorbidities. J. Gerontol. A. Biol. Sci. Med. Sci. 2023, 78, 603–610. [Google Scholar] [CrossRef]
- Ntouros, P.A.; Kravvariti, E.; Vlachogiannis, N.I.; Pappa, M.; Trougakos, I.P.; Terpos, E.; Tektonidou, M.G.; Souliotis, V.L.; Sfikakis, P.P. Oxidative stress and endogenous DNA damage in blood mononuclear cells may predict anti-SARS-CoV-2 antibody titers after vaccination in older adults. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166393. [Google Scholar] [CrossRef]
- Schurman, S.H.; Dunn, C.A.; Greaves, R.; Yu, B.; Ferrucci, L.; Croteau, D.L.; Seidman, M.M.; Bohr, V.A. Age-related disease association of endogenous γ-H2AX foci in mononuclear cells derived from leukapheresis. PLoS ONE 2012, 7, e45728. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Horikawa, I.; Redon, C.; Nakamura, A.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell 2008, 7, 89–100. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Pilch, D.R.; Redon, C.; Bonner, W.M. Histone H2AX in DNA damage and repair. Cancer Biol. Ther. 2003, 2, 233–235. [Google Scholar] [CrossRef]
- Sánchez-Flores, M.; Pásaro, E.; Bonassi, S.; Laffon, B.; Valdiglesias, V. γH2AX assay as DNA damage biomarker for human population studies: Defining experimental conditions. Toxicol. Sci. 2015, 144, 406–413. [Google Scholar] [CrossRef]
- Møller, P.; Loft, S.; Ersson, C.; Koppen, G.; Dusinska, M.; Collins, A. On the Search for an Intelligible Comet Assay Descriptor. Front. Genet. 2014, 5, 217. [Google Scholar] [CrossRef]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. GammaH2AX: A Sensitive Molecular Marker of DNA Damage and Repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Bhogal, N.; Jalali, F.; Bristow, R.G. Microscopic imaging of DNA repair foci in irradiated normal tissues. Int. J. Radiat. Biol. 2009, 85, 732–746. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age Group | Male No. | Female No. |
|---|---|---|
| 18–29 | 36 (43.9) | 46 (56.1) |
| 30–49 | 29 (39.2) | 45 (60.8) |
| 50–69 | 33 (41.3) | 47 (58.8) |
| 70–75 | 1 (14.3) | 6 (85.7) |
| Sum | 99 (40.7) | 144 (59.3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlachogiannis, N.I.; Ntouros, P.A.; Pappa, M.; Kravvariti, E.; Kostaki, E.G.; Fragoulis, G.E.; Papanikolaou, C.; Mavroeidi, D.; Bournia, V.-K.; Panopoulos, S.; et al. Chronological Age and DNA Damage Accumulation in Blood Mononuclear Cells: A Linear Association in Healthy Humans after 50 Years of Age. Int. J. Mol. Sci. 2023, 24, 7148. https://doi.org/10.3390/ijms24087148
Vlachogiannis NI, Ntouros PA, Pappa M, Kravvariti E, Kostaki EG, Fragoulis GE, Papanikolaou C, Mavroeidi D, Bournia V-K, Panopoulos S, et al. Chronological Age and DNA Damage Accumulation in Blood Mononuclear Cells: A Linear Association in Healthy Humans after 50 Years of Age. International Journal of Molecular Sciences. 2023; 24(8):7148. https://doi.org/10.3390/ijms24087148
Chicago/Turabian StyleVlachogiannis, Nikolaos I., Panagiotis A. Ntouros, Maria Pappa, Evrydiki Kravvariti, Evangelia Georgia Kostaki, Georgios E. Fragoulis, Christina Papanikolaou, Dimitra Mavroeidi, Vasiliki-Kalliopi Bournia, Stylianos Panopoulos, and et al. 2023. "Chronological Age and DNA Damage Accumulation in Blood Mononuclear Cells: A Linear Association in Healthy Humans after 50 Years of Age" International Journal of Molecular Sciences 24, no. 8: 7148. https://doi.org/10.3390/ijms24087148
APA StyleVlachogiannis, N. I., Ntouros, P. A., Pappa, M., Kravvariti, E., Kostaki, E. G., Fragoulis, G. E., Papanikolaou, C., Mavroeidi, D., Bournia, V.-K., Panopoulos, S., Laskari, K., Arida, A., Gorgoulis, V. G., Tektonidou, M. G., Paraskevis, D., Sfikakis, P. P., & Souliotis, V. L. (2023). Chronological Age and DNA Damage Accumulation in Blood Mononuclear Cells: A Linear Association in Healthy Humans after 50 Years of Age. International Journal of Molecular Sciences, 24(8), 7148. https://doi.org/10.3390/ijms24087148