
A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer



Abstract
1. Introduction
2. Background on Breast Cancer
2.1. Classification
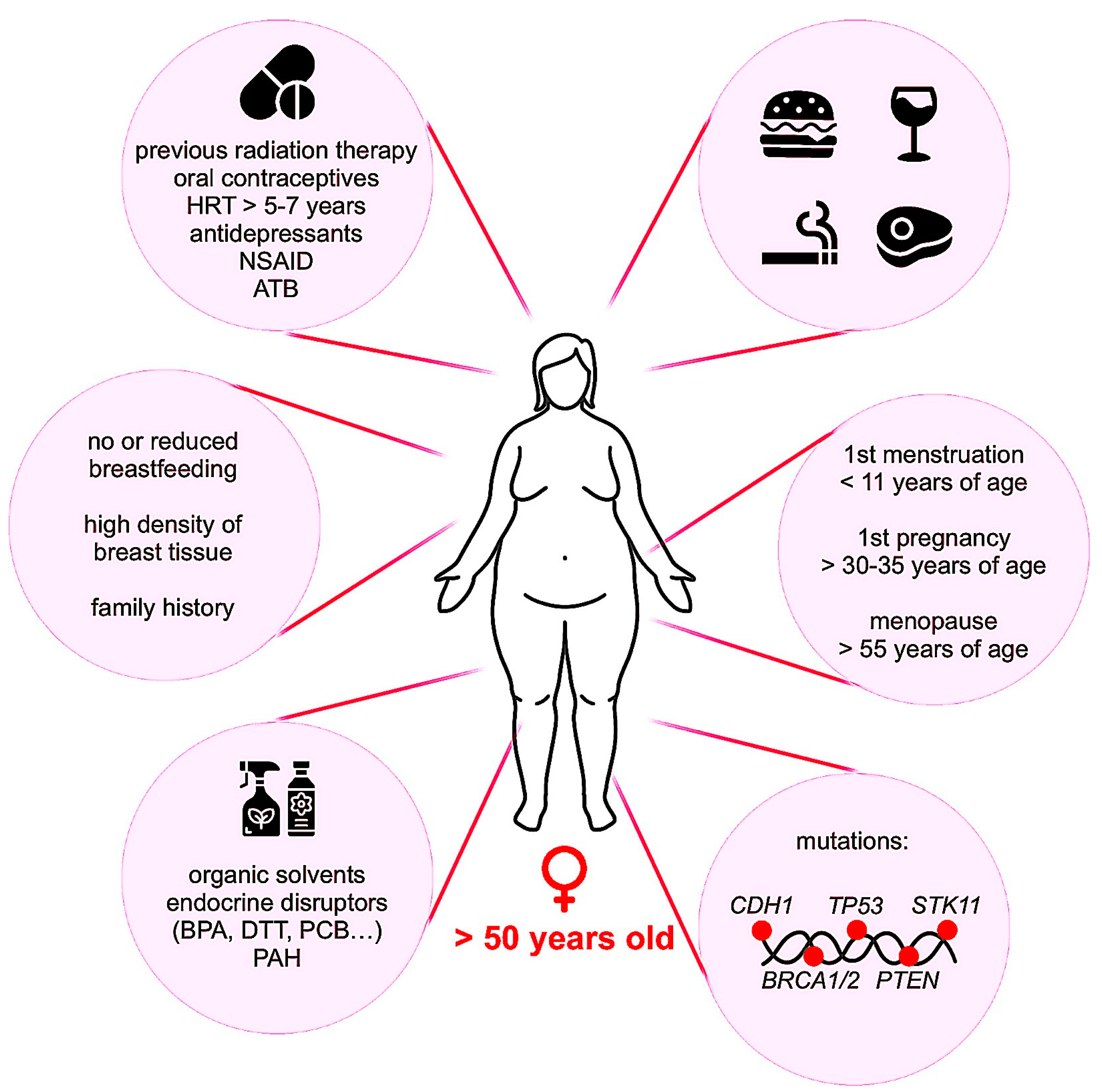
2.2. Occurrence and Risk Factors
2.3. Diagnosis and Treatments
3. ER-Positive Breast Cancer
3.1. Molecular Mechanism of ER
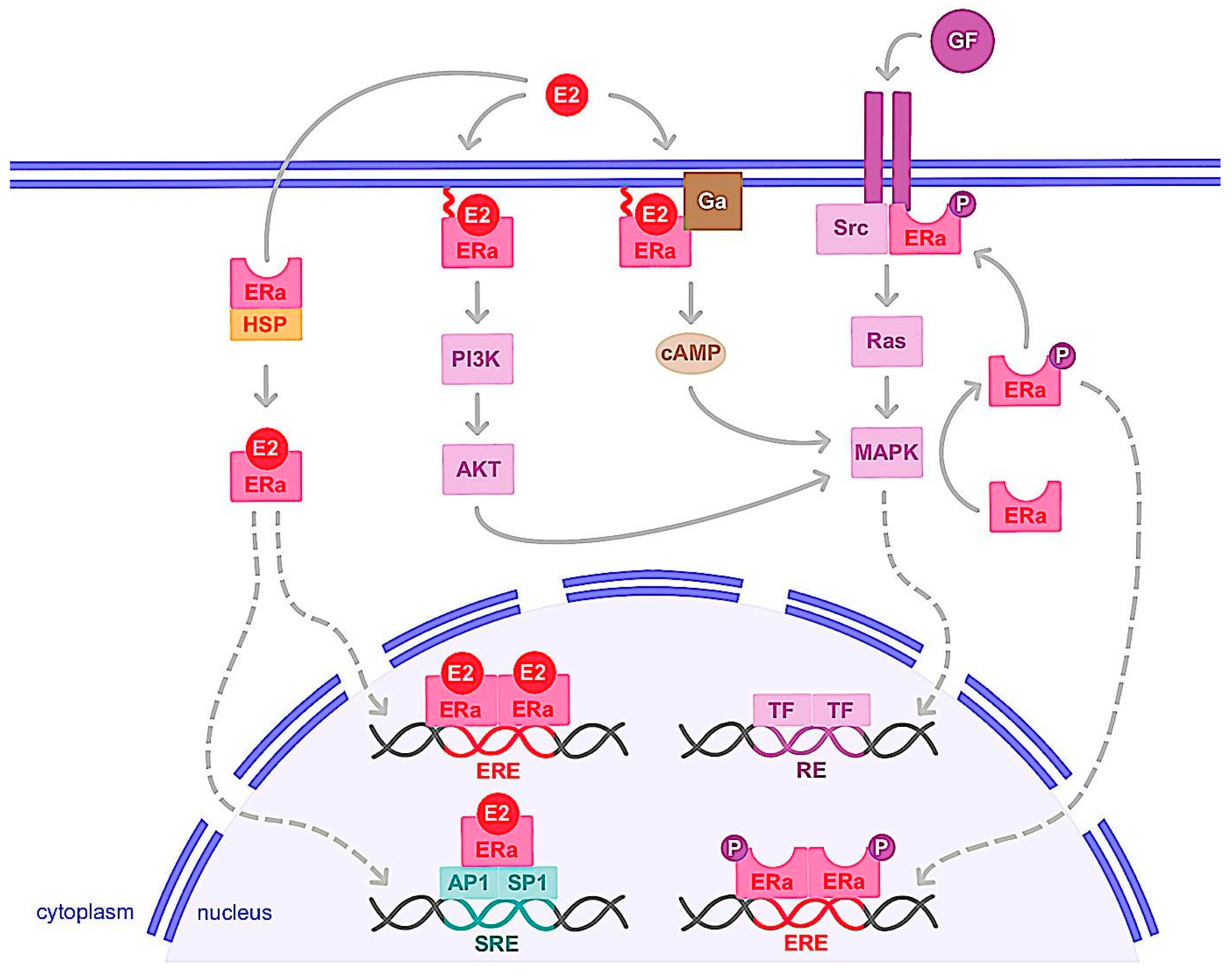
3.1.1. Genomic Action
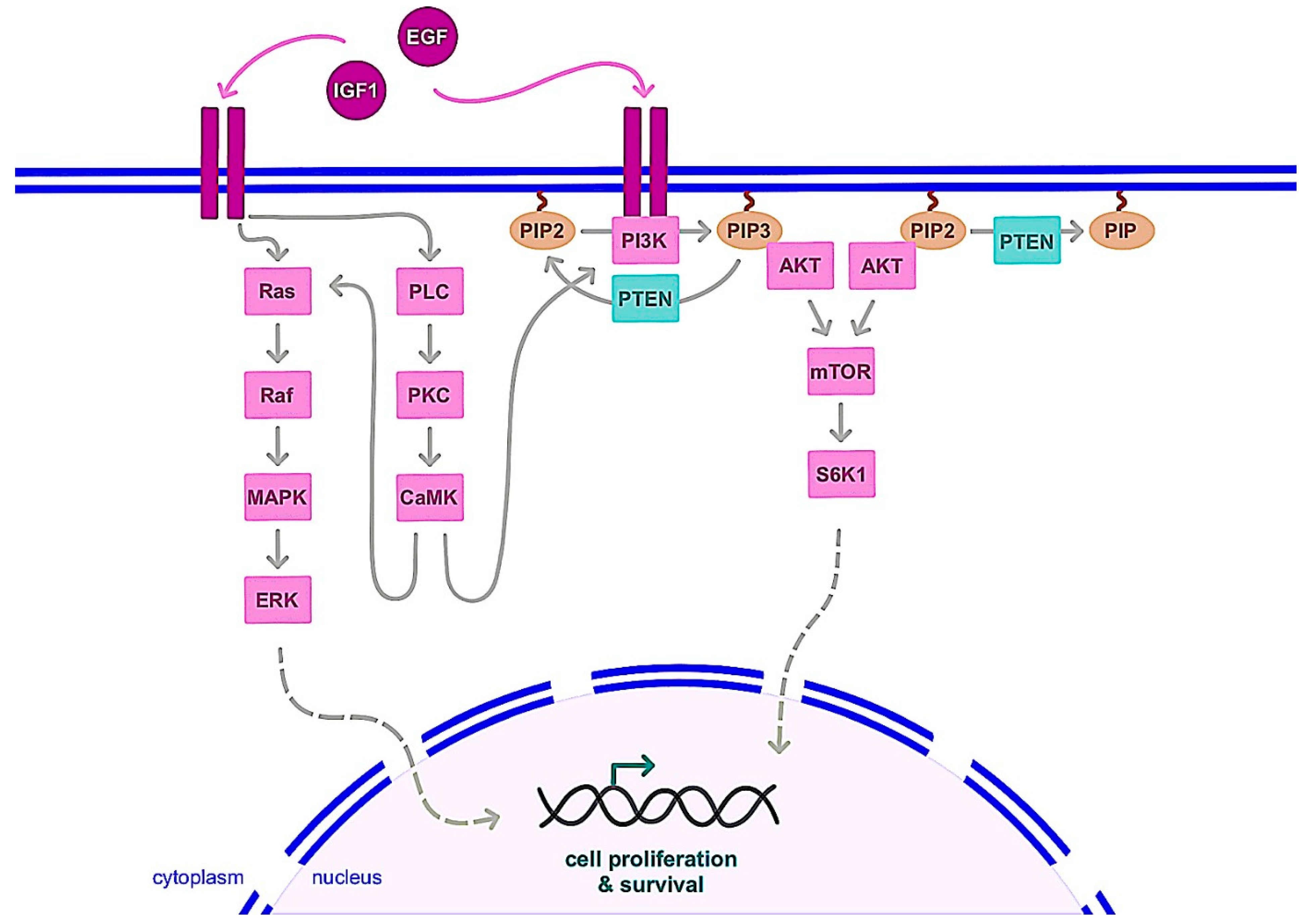
3.1.2. Nongenomic Action
3.2. ERα Variants and Mutations
3.3. Hormone Therapy and Resistance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Pandya, S.; Moore, R.G. Breast Development and Anatomy. Clin. Obstet. Gynecol. 2011, 54, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Pellacani, D.; Tan, S.; Lefort, S.; Eaves, C.J. Transcriptional Regulation of Normal Human Mammary Cell Heterogeneity and Its Perturbation in Breast Cancer. EMBO J. 2019, 38, e100330. [Google Scholar] [CrossRef] [PubMed]
- Fridriksdottir, A.J.; Kim, J.; Villadsen, R.; Klitgaard, M.C.; Hopkinson, B.M.; Petersen, O.W.; Rønnov-Jessen, L. Propagation of Oestrogen Receptor-Positive and Oestrogen-Responsive Normal Human Breast Cells in Culture. Nat. Commun. 2015, 6, 8786. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Boulan, E.; Macara, I.G. Organization and Execution of the Epithelial Polarity Programme. Nat. Rev. Mol. Cell Biol. 2014, 15, 225–242. [Google Scholar] [CrossRef]
- Ciarloni, L.; Mallepell, S.; Brisken, C. Amphiregulin Is an Essential Mediator of Estrogen Receptor α Function in Mammary Gland Development. Proc. Natl. Acad. Sci. USA 2007, 104, 5455–5460. [Google Scholar] [CrossRef]
- Troyer, K.L.; Lee, D.C. Regulation of Mouse Mammary Gland Development and Tumorigenesis by the ERBB Signaling Network. J. Mammary Gland Biol. Neoplasia 2001, 6, 7–21. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic Heterogeneity of Breast Cancer: Molecular Mechanism and Potential Therapeutic Targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Primer 2019, 5, 66. [Google Scholar] [CrossRef]
- Tan, P.H.; Ellis, I.; Allison, K.; Brogi, E.; Fox, S.B.; Lakhani, S.; Lazar, A.J.; Morris, E.A.; Sahin, A.; Salgado, R.; et al. The 2019 World Health Organization Classification of Tumours of the Breast. Histopathology 2020, 77, 181–185. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer—Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies—An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Prat, A.; Cheang, M.C.U.; Martín, M.; Parker, J.S.; Carrasco, E.; Caballero, R.; Tyldesley, S.; Gelmon, K.; Bernard, P.S.; Nielsen, T.O.; et al. Prognostic Significance of Progesterone Receptor-Positive Tumor Cells within Immunohistochemically Defined Luminal A Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 203–209. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical Implications of the Intrinsic Molecular Subtypes of Breast Cancer. Breast Edinb. Scotl. 2015, 24 (Suppl. S2), S26–S35. [Google Scholar] [CrossRef]
- Slepicka, P.F.; Cyrill, S.L.; dos Santos, C.O. Pregnancy and Breast Cancer: Pathways to Understand Risk and Prevention. Trends Mol. Med. 2019, 25, 866–881. [Google Scholar] [CrossRef]
- Winters, S.; Martin, C.; Murphy, D.; Shokar, N.K. Breast Cancer Epidemiology, Prevention, and Screening. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2017; Volume 151, pp. 1–32. ISBN 978-0-12-812772-8. [Google Scholar]
- Calaf, G.; Ponce-Cusi, R.; Aguayo, F.; Muñoz, J.; Bleak, T. Endocrine Disruptors from the Environment Affecting Breast Cancer. Oncol. Lett. 2020, 20, 19–32. [Google Scholar] [CrossRef]
- Koual, M.; Tomkiewicz, C.; Cano-Sancho, G.; Antignac, J.-P.; Bats, A.-S.; Coumoul, X. Environmental Chemicals, Breast Cancer Progression and Drug Resistance. Environ. Health 2020, 19, 117. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, M.; Xu, Y. Understanding the Mechanisms Underlying Obesity in Remodeling the Breast Tumor Immune Microenvironment: From the Perspective of Inflammation. Cancer Biol. Med. 2023, 20, 20220547. [Google Scholar] [CrossRef]
- Godet, I.; Gilkes, D.M. BRCA1 and BRCA2 Mutations and Treatment Strategies for Breast Cancer. Integr. Cancer Sci. Ther. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group on Hormonal Factors in Breast Cancer. Menarche, Menopause, and Breast Cancer Risk: Individual Participant Meta-Analysis, Including 118 964 Women with Breast Cancer from 117 Epidemiological Studies. Lancet Oncol. 2012, 13, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Pfeiffer, R.M.; Gierach, G.L.; Falk, R.T. Use of Postmenopausal Hormone Therapies and Risk of Histology- and Hormone Receptor-Defined Breast Cancer: Results from a 15-Year Prospective Analysis of NIH-AARP Cohort. Breast Cancer Res. BCR 2020, 22, 129. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Bilancio, A.; Migliaccio, A. The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2018, 9, 492. [Google Scholar] [CrossRef]
- McNamara, K.M.; Moore, N.L.; Hickey, T.E.; Sasano, H.; Tilley, W.D. Complexities of Androgen Receptor Signalling in Breast Cancer. Endocr. Relat. Cancer 2014, 21, T161–T181. [Google Scholar] [CrossRef]
- Berrino, F.; Pasanisi, P.; Bellati, C.; Venturelli, E.; Krogh, V.; Mastroianni, A.; Berselli, E.; Muti, P.; Secreto, G. Serum Testosterone Levels and Breast Cancer Recurrence. Int. J. Cancer 2005, 113, 499–502. [Google Scholar] [CrossRef]
- Praud, D.; Deygas, F.; Amadou, A.; Bouilly, M.; Turati, F.; Bravi, F.; Xu, T.; Grassot, L.; Coudon, T.; Fervers, B. Traffic-Related Air Pollution and Breast Cancer Risk: A Systematic Review and Meta-Analysis of Observational Studies. Cancers 2023, 15, 927. [Google Scholar] [CrossRef]
- Lecomte, S.; Habauzit, D.; Charlier, T.D.; Pakdel, F. Emerging Estrogenic Pollutants in the Aquatic Environment and Breast Cancer. Genes 2017, 8, 229. [Google Scholar] [CrossRef]
- E Huff, J.; Haseman, J.K.; DeMarini, D.M.; Eustis, S.; Maronpot, R.R.; Peters, A.C.; Persing, R.L.; E Chrisp, C.; Jacobs, A.C. Multiple-Site Carcinogenicity of Benzene in Fischer 344 Rats and B6C3F1 Mice. Environ. Health Perspect. 1989, 82, 125–163. [Google Scholar] [CrossRef]
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef]
- McAnulty, J.; DiFeo, A. The Molecular ‘Myc-Anisms’ behind Myc-Driven Tumorigenesis and the Relevant Myc-Directed Therapeutics. Int. J. Mol. Sci. 2020, 21, 9486. [Google Scholar] [CrossRef]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]
- Steelman, L.S.; Chappell, W.H.; Akula, S.M.; Abrams, S.L.; Cocco, L.; Manzoli, L.; Ratti, S.; Martelli, A.M.; Montalto, G.; Cervello, M.; et al. Therapeutic Resistance in Breast Cancer Cells Can Result from Deregulated EGFR Signaling. Adv. Biol. Regul. 2020, 78, 100758. [Google Scholar] [CrossRef]
- Christopoulos, P.F.; Msaouel, P.; Koutsilieris, M. The Role of the Insulin-like Growth Factor-1 System in Breast Cancer. Mol. Cancer 2015, 14, 43. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. How Do Mutations Affecting the Breast Cancer Genes BRCA1 and BRCA2 Cause Cancer Susceptibility? DNA Repair 2019, 81, 102668. [Google Scholar] [CrossRef]
- Silwal-Pandit, L.; Langerød, A.; Børresen-Dale, A.-L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef]
- Csolle, M.P.; Ooms, L.M.; Papa, A.; Mitchell, C.A. PTEN and Other PtdIns(3,4,5)P3 Lipid Phosphatases in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9189. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, C.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 4490. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Cardoso, F.; Bedard, P.L.; Winer, E.P.; Pagani, O.; Senkus-Konefka, E.; Fallowfield, L.J.; Kyriakides, S.; Costa, A.; Cufer, T.; Albain, K.S.; et al. International Guidelines for Management of Metastatic Breast Cancer: Combination vs Sequential Single-Agent Chemotherapy. JNCI J. Natl. Cancer Inst. 2009, 101, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A.; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in Postmenopausal Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Shen, G.; Fang, Q.; Xin, Y.; Huo, X.; Li, J.; Zhao, F.; Ren, D.; Liu, Z.; Li, Z.; et al. Cyclin-Dependent Kinase 4 and 6 Inhibitors in Combination with Neoadjuvant Endocrine Therapy in Estrogen Receptor-Positive Early Breast Cancer: A Systematic Review and Meta-Analysis. Clin. Exp. Med. 2022; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Bisi, J.E.; Sorrentino, J.A.; Jordan, J.L.; Darr, D.D.; Roberts, P.J.; Tavares, F.X.; Strum, J.C. Preclinical Development of G1T38: A Novel, Potent and Selective Inhibitor of Cyclin Dependent Kinases 4/6 for Use as an Oral Antineoplastic in Patients with CDK4/6 Sensitive Tumors. Oncotarget 2017, 8, 42343–42358. [Google Scholar] [CrossRef]
- Fisher, B.; Anderson, S.; Bryant, J.; Margolese, R.G.; Deutsch, M.; Fisher, E.R.; Jeong, J.-H.; Wolmark, N. Twenty-Year Follow-up of a Randomized Trial Comparing Total Mastectomy, Lumpectomy, and Lumpectomy plus Irradiation for the Treatment of Invasive Breast Cancer. N. Engl. J. Med. 2002, 347, 1233–1241. [Google Scholar] [CrossRef]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating Tumour DNA Analysis to Direct Therapy in Advanced Breast Cancer (PlasmaMATCH): A Multicentre, Multicohort, Phase 2a, Platform Trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- Kodahl, A.R.; Ehmsen, S.; Pallisgaard, N.; Jylling, A.M.B.; Jensen, J.D.; Laenkholm, A.-V.; Knoop, A.S.; Ditzel, H.J. Correlation between Circulating Cell-Free PIK3CA Tumor DNA Levels and Treatment Response in Patients with PIK3CA-Mutated Metastatic Breast Cancer. Mol. Oncol. 2018, 12, 925–935. [Google Scholar] [CrossRef]
- Manoochehri, M.; Borhani, N.; Gerhäuser, C.; Assenov, Y.; Schönung, M.; Hielscher, T.; Christensen, B.C.; Lee, M.K.; Gröne, H.-J.; Lipka, D.B.; et al. DNA Methylation Biomarkers for Noninvasive Detection of Triple-Negative Breast Cancer Using Liquid Biopsy. Int. J. Cancer 2023, 152, 1025–1035. [Google Scholar] [CrossRef]
- Bortolini Silveira, A.; Bidard, F.-C.; Tanguy, M.-L.; Girard, E.; Trédan, O.; Dubot, C.; Jacot, W.; Goncalves, A.; Debled, M.; Levy, C.; et al. Multimodal Liquid Biopsy for Early Monitoring and Outcome Prediction of Chemotherapy in Metastatic Breast Cancer. Npj Breast Cancer 2021, 7, 115. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.N.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of Estrogen Up- and Down-Regulated Gene Expression in Human Breast Cancer Cells: Insights into Gene Networks and Pathways Underlying Estrogenic Control of Proliferation and Cell Phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar] [CrossRef]
- Frasor, J.; Stossi, F.; Danes, J.M.; Komm, B.; Lyttle, C.R.; Katzenellenbogen, B.S. Selective Estrogen Receptor Modulators: Discrimination of Agonistic versus Antagonistic Activities by Gene Expression Profiling in Breast Cancer Cells. Cancer Res. 2004, 64, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.N.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-Wide Analysis of Estrogen Receptor Alpha DNA Binding and Tethering Mechanisms Identifies Runx1 as a Novel Tethering Factor in Receptor-Mediated Transcriptional Activation. Mol. Cell. Biol. 2010, 30, 3943–3955. [Google Scholar] [CrossRef]
- Mal, R.; Magner, A.; David, J.; Datta, J.; Vallabhaneni, M.; Kassem, M.; Manouchehri, J.; Willingham, N.; Stover, D.; Vandeusen, J.; et al. Estrogen Receptor Beta (ERβ): A Ligand Activated Tumor Suppressor. Front. Oncol. 2020, 10, 587386. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kim, H.; Pollack, S. ERβ Isoforms Have Differential Clinical Significance in Breast Cancer Subtypes and Subgroups. Curr. Issues Mol. Biol. 2022, 44, 1564–1586. [Google Scholar] [CrossRef]
- Choi, Y. Estrogen Receptor β Expression and Its Clinical Implication in Breast Cancers: Favorable or Unfavorable? J. Breast Cancer 2022, 25, 75. [Google Scholar] [CrossRef]
- Girault, I.; Lerebours, F.; Amarir, S.; Tozlu, S.; Tubiana-Hulin, M.; Lidereau, R.; Bièche, I. Expression Analysis of Estrogen Receptor Alpha Coregulators in Breast Carcinoma: Evidence That NCOR1 Expression Is Predictive of the Response to Tamoxifen. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1259–1266. [Google Scholar]
- Ranganathan, P.; Nadig, N.; Nambiar, S. Non-Canonical Estrogen Signaling in Endocrine Resistance. Front. Endocrinol. 2019, 10, 708. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X. The Role of Estrogen Receptor Beta in Breast Cancer. Biomark. Res. 2020, 8, 39. [Google Scholar] [CrossRef]
- Curtis, S.W.; Washburn, T.; Sewall, C.; DiAugustine, R.; Lindzey, J.; Couse, J.F.; Korach, K.S. Physiological Coupling of Growth Factor and Steroid Receptor Signaling Pathways: Estrogen Receptor Knockout Mice Lack Estrogen-like Response to Epidermal Growth Factor. Proc. Natl. Acad. Sci. USA 1996, 93, 12626–12630. [Google Scholar] [CrossRef]
- El-Tanani, M.K.; Green, C.D. Two Separate Mechanisms for Ligand-Independent Activation of the Estrogen Receptor. Mol. Endocrinol. Baltim. Md 1997, 11, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Pace, P.E.; Coombes, R.C.; Ali, S. Phosphorylation of Human Estrogen Receptor Alpha by Protein Kinase A Regulates Dimerization. Mol. Cell. Biol. 1999, 19, 1002–1015. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomas, R.S.; Sarwar, N.; Phoenix, F.; Coombes, R.C.; Ali, S. Phosphorylation at Serines 104 and 106 by Erk1/2 MAPK Is Important for Estrogen Receptor-Alpha Activity. J. Mol. Endocrinol. 2008, 40, 173–184. [Google Scholar] [CrossRef]
- Kato, S. Estrogen Receptor-Mediated Cross-Talk with Growth Factor Signaling Pathways. Breast Cancer Tokyo Jpn. 2001, 8, 3–9. [Google Scholar] [CrossRef]
- Tremblay, A.; Tremblay, G.B.; Labrie, F.; Giguère, V. Ligand-Independent Recruitment of SRC-1 to Estrogen Receptor Beta through Phosphorylation of Activation Function AF-1. Mol. Cell 1999, 3, 513–519. [Google Scholar] [CrossRef]
- Duplessis, T.T.; Williams, C.C.; Hill, S.M.; Rowan, B.G. Phosphorylation of Estrogen Receptor α at Serine 118 Directs Recruitment of Promoter Complexes and Gene-Specific Transcription. Endocrinology 2011, 152, 2517–2526. [Google Scholar] [CrossRef]
- Font de Mora, J.; Brown, M. AIB1 Is a Conduit for Kinase-Mediated Growth Factor Signaling to the Estrogen Receptor. Mol. Cell. Biol. 2000, 20, 5041–5047. [Google Scholar] [CrossRef]
- Tharakan, R.; Lepont, P.; Singleton, D.; Kumar, R.; Khan, S. Phosphorylation of Estrogen Receptor Alpha, Serine Residue 305 Enhances Activity. Mol. Cell. Endocrinol. 2008, 295, 70–78. [Google Scholar] [CrossRef]
- Arnold, S.F.; Vorojeikina, D.P.; Notides, A.C. Phosphorylation of Tyrosine 537 on the Human Estrogen Receptor Is Required for Binding to an Estrogen Response Element. J. Biol. Chem. 1995, 270, 30205–30212. [Google Scholar] [CrossRef]
- Kim, M.Y.; Woo, E.M.; Chong, Y.T.E.; Homenko, D.R.; Kraus, W.L. Acetylation of Estrogen Receptor Alpha by P300 at Lysines 266 and 268 Enhances the Deoxyribonucleic Acid Binding and Transactivation Activities of the Receptor. Mol. Endocrinol. Baltim. Md 2006, 20, 1479–1493. [Google Scholar] [CrossRef]
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; Nanayakkara, M.; Lombardi, M.; de Falco, A.; Bilancio, A.; Varricchio, L.; Ciociola, A.; Auricchio, F. Steroid Receptor Regulation of Epidermal Growth Factor Signaling through Src in Breast and Prostate Cancer Cells: Steroid Antagonist Action. Cancer Res 2005, 65, 10585–10593. [Google Scholar] [CrossRef]
- Genua, M.; Pandini, G.; Sisci, D.; Castoria, G.; Maggiolini, M.; Vigneri, R.; Belfiore, A. Role of Cyclic AMP Response Element–Binding Protein in Insulin-like Growth Factor-I Receptor Up-regulation by Sex Steroids in Prostate Cancer Cells. Cancer Res. 2009, 69, 7270–7277. [Google Scholar] [CrossRef]
- Song, R.X.-D.; Zhang, Z.; Santen, R.J. Estrogen Rapid Action via Protein Complex Formation Involving ERalpha and Src. Trends Endocrinol. Metab. TEM 2005, 16, 347–353. [Google Scholar] [CrossRef]
- Kelly, M.J.; Levin, E.R. Rapid Actions of Plasma Membrane Estrogen Receptors. Trends Endocrinol. Metab. TEM 2001, 12, 152–156. [Google Scholar] [CrossRef]
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; de Falco, A.; Bontempo, P.; Nola, E.; Auricchio, F. Tyrosine Kinase/P21ras/MAP-Kinase Pathway Activation by Estradiol- Receptor Complex in MCF-7 Cells. EMBO J. 1996, 15, 1292–1300. [Google Scholar] [CrossRef]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; de Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-Kinase in Concert with Src Promotes the S-Phase Entry of Oestradiol-Stimulated MCF-7 Cells. EMBO J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef]
- Cabodi, S.; Moro, L.; Baj, G.; Smeriglio, M.; Di Stefano, P.; Gippone, S.; Surico, N.; Silengo, L.; Turco, E.; Tarone, G.; et al. P130Cas Interacts with Estrogen Receptor Alpha and Modulates Non-Genomic Estrogen Signaling in Breast Cancer Cells. J. Cell Sci. 2004, 117, 1603–1611. [Google Scholar] [CrossRef]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-Dependent Estrogen Receptor Alpha Membrane Localization: Regulation by 17beta-Estradiol. Mol. Biol. Cell. 2005, 16, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Ascenzi, P.; Fabozzi, G.; Visca, P.; Marino, M. S-Palmitoylation Modulates Human Estrogen Receptor-Alpha Functions. Biochem. Biophys. Res. Commun. 2004, 316, 878–883. [Google Scholar] [CrossRef]
- Acconcia, F.; Barnes, C.J.; Kumar, R. Estrogen and Tamoxifen Induce Cytoskeletal Remodeling and Migration in Endometrial Cancer Cells. Endocrinology 2006, 147, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Le Romancer, M.; Treilleux, I.; Leconte, N.; Robin-Lespinasse, Y.; Sentis, S.; Bouchekioua-Bouzaghou, K.; Goddard, S.; Gobert-Gosse, S.; Corbo, L. Regulation of Estrogen Rapid Signaling through Arginine Methylation by PRMT1. Mol. Cell 2008, 31, 212–221. [Google Scholar] [CrossRef]
- Poulard, C.; Treilleux, I.; Lavergne, E.; Bouchekioua-Bouzaghou, K.; Goddard-Léon, S.; Chabaud, S.; Trédan, O.; Corbo, L.; Le Romancer, M. Activation of Rapid Oestrogen Signalling in Aggressive Human Breast Cancers. EMBO Mol. Med. 2012, 4, 1200–1213. [Google Scholar] [CrossRef] [PubMed]
- Choucair, A.; Pham, T.H.; Omarjee, S.; Jacquemetton, J.; Kassem, L.; Trédan, O.; Rambaud, J.; Marangoni, E.; Corbo, L.; Treilleux, I.; et al. The Arginine Methyltransferase PRMT1 Regulates IGF-1 Signaling in Breast Cancer. Oncogene 2019, 38, 4015–4027. [Google Scholar] [CrossRef] [PubMed]
- Varricchio, L.; Migliaccio, A.; Castoria, G.; Yamaguchi, H.; de Falco, A.; Di Domenico, M.; Giovannelli, P.; Farrar, W.; Appella, E.; Auricchio, F. Inhibition of Estradiol Receptor/Src Association and Cell Growth by an Estradiol Receptor Alpha Tyrosine-Phosphorylated Peptide. Mol. Cancer Res. MCR 2007, 5, 1213–1221. [Google Scholar] [CrossRef]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty Years of the G Protein-Coupled Estrogen Receptor GPER: Historical and Personal Perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef]
- Owman, C.; Blay, P.; Nilsson, C.; Lolait, S.J. Cloning of Human CDNA Encoding a Novel Heptahelix Receptor Expressed in Burkitt’s Lymphoma and Widely Distributed in Brain and Peripheral Tissues. Biochem. Biophys. Res. Commun. 1996, 228, 285–292. [Google Scholar] [CrossRef]
- Xu, T.; Ma, D.; Chen, S.; Tang, R.; Yang, J.; Meng, C.; Feng, Y.; Liu, L.; Wang, J.; Luo, H.; et al. High GPER Expression in Triple-Negative Breast Cancer Is Linked to pro- Metastatic Pathways and Predicts Poor Patient Outcomes. NPJ Breast Cancer 2022, 8, 100. [Google Scholar] [CrossRef]
- Luo, J.; Liu, D. Does GPER Really Function as a G Protein-Coupled Estrogen Receptor in Vivo? Front. Endocrinol. 2020, 11, 148. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB- EGF. Mol. Endocrinol. Baltim. Md 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.-A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.-E.; et al. Cytoplasmic GPER Translocation in Cancer-Associated Fibroblasts Mediates CAMP/PKA/CREB/Glycolytic Axis to Confer Tumor Cells with Multidrug Resistance. Oncogene 2017, 36, 2131–2145. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-T.; Lai, A.C.-Y.; Lin, R.-J.; Wang, Y.-H.; Wang, Y.-T.; Chang, W.-W.; Wu, H.-Y.; Lin, Y.-J.; Chang, W.-Y.; Wu, J.-C.; et al. GPER-Induced Signaling Is Essential for the Survival of Breast Cancer Stem Cells. Int. J. Cancer 2020, 146, 1674–1685. [Google Scholar] [CrossRef] [PubMed]
- Maggiolini, M.; Vivacqua, A.; Fasanella, G.; Recchia, A.G.; Sisci, D.; Pezzi, V.; Montanaro, D.; Musti, A.M.; Picard, D.; Andò, S. The G Protein-Coupled Receptor GPR30 Mediates c-Fos up-Regulation by 17beta-Estradiol and Phytoestrogens in Breast Cancer Cells. J. Biol. Chem. 2004, 279, 27008–27016. [Google Scholar] [CrossRef] [PubMed]
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Andò, S.; et al. G Protein-Coupled Receptor 30 (GPR30) Mediates Gene Expression Changes and Growth Response to 17beta-Estradiol and Selective GPR30 Ligand G-1 in Ovarian Cancer Cells. Cancer Res. 2007, 67, 1859–1866. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. GPER Is Involved in the Functional Liaison between Breast Tumor Cells and Cancer-Associated Fibroblasts (CAFs). J. Steroid Biochem. Mol. Biol. 2018, 176, 49–56. [Google Scholar] [CrossRef]
- Vivacqua, A.; Romeo, E.; De Marco, P.; De Francesco, E.M.; Abonante, S.; Maggiolini, M. GPER Mediates the Egr-1 Expression Induced by 17β-Estradiol and 4-Hydroxitamoxifen in Breast and Endometrial Cancer Cells. Breast Cancer Res. Treat. 2012, 133, 1025–1035. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER Mediates Activation of HIF1α/VEGF Signaling by Estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef]
- Kampa, M.; Lappano, R.; Grande, F.; Rizzuti, B.; Maggiolini, M.; Castanas, E.; Jacquot, Y. Promising Perspectives of the Antiproliferative GPER Inverse Agonist ERα17p in Breast Cancer. Cells 2023, 12, 653. [Google Scholar] [CrossRef]
- Catalano, S.; Giordano, C.; Panza, S.; Chemi, F.; Bonofiglio, D.; Lanzino, M.; Rizza, P.; Romeo, F.; Fuqua, S.A.W.; Maggiolini, M.; et al. Tamoxifen through GPER Upregulates Aromatase Expression: A Novel Mechanism Sustaining Tamoxifen-Resistant Breast Cancer Cell Growth. Breast Cancer Res. Treat. 2014, 146, 273–285. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between Notch, HIF-1α and GPER in Breast Cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011. [Google Scholar] [CrossRef]
- Talia, M.; De Francesco, E.M.; Rigiracciolo, D.C.; Muoio, M.G.; Muglia, L.; Belfiore, A.; Maggiolini, M.; Sims, A.H.; Lappano, R. The G Protein-Coupled Estrogen Receptor (GPER) Expression Correlates with Pro-Metastatic Pathways in ER-Negative Breast Cancer: A Bioinformatics Analysis. Cells 2020, 9, 622. [Google Scholar] [CrossRef]
- Lappano, R.; Pisano, A.; Maggiolini, M. GPER Function in Breast Cancer: An Overview. Front. Endocrinol. 2014, 5, 66. [Google Scholar] [CrossRef]
- Molina, L.; Figueroa, C.D.; Bhoola, K.D.; Ehrenfeld, P. GPER-1/GPR30 a Novel Estrogen Receptor Sited in the Cell Membrane: Therapeutic Coupling to Breast Cancer. Expert Opin. Ther. Targets 2017, 21, 755–766. [Google Scholar] [CrossRef]
- La Rosa, P.; Pesiri, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation Regulates 17β-Estradiol-Induced Estrogen Receptor-α Degradation and Transcriptional Activity. Mol. Endocrinol. Baltim. Md 2012, 26, 762–774. [Google Scholar] [CrossRef]
- Acconcia, F.; Marino, M. Synergism between Genomic and Non Genomic Estrogen Action Mechanisms. IUBMB Life 2003, 55, 145–150. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Aitkenhead, M.; Hughes, C.C.W.; Levin, E.R. Integration of the Non-Genomic and Genomic Actions of Estrogen. Membrane-Initiated Signaling by Steroid to Transcription and Cell Biology. J. Biol. Chem. 2002, 277, 50768–50775. [Google Scholar] [CrossRef]
- Björnström, L.; Sjöberg, M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol. Endocrinol. Baltim. Md 2005, 19, 833–842. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Ventrella, R.; Petry, L.; Katzenellenbogen, B.S. Novel Roles for ERK5 and Cofilin as Critical Mediators Linking ERα-Driven Transcription, Actin Reorganization, and Invasiveness in Breast Cancer. Mol. Cancer Res. MCR 2014, 12, 714–727. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Kim, S.H.; Gong, P.; Zhao, Y.C.; Zhang, H.; Chambliss, K.L.; Carlson, K.E.; Mayne, C.G.; Shaul, P.W.; Korach, K.S.; et al. Design of Pathway Preferential Estrogens That Provide Beneficial Metabolic and Vascular Effects without Stimulating Reproductive Tissues. Sci. Signal. 2016, 9, ra53. [Google Scholar] [CrossRef]
- Jehanno, C.; Percevault, F.; Boujrad, N.; Le Goff, P.; Fontaine, C.; Arnal, J.-F.; Primig, M.; Pakdel, F.; Michel, D.; Métivier, R.; et al. Nuclear Translocation of MRTFA in MCF7 Breast Cancer Cells Shifts ERα Nuclear/Genomic to Extra-Nuclear/Non Genomic Actions. Mol. Cell. Endocrinol. 2021, 530, 111282. [Google Scholar] [CrossRef]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A Role for Estrogen Receptor Alpha36 in Cancer Progression. Front. Endocrinol. 2020, 11, 506. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, Cloning, and Expression of Human Estrogen Receptor-Alpha36, a Novel Variant of Human Estrogen Receptor-Alpha66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Haynes, M.P.; Bender, J.R. Plasma Membrane Localization and Function of the Estrogen Receptor Alpha Variant (ER46) in Human Endothelial Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4807–4812. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Dong, B.; Li, Z.; Lu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Expression of ER-α36, a Novel Variant of Estrogen Receptor α, and Resistance to Tamoxifen Treatment in Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 3423–3429. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Yang, S.; Wei, M.; Vlantis, A.C.; Chan, J.Y.K.; van Hasselt, C.A.; Li, D.; Zeng, X.; Xue, L.; Tong, M.C.F.; et al. The Isoforms of Estrogen Receptor Alpha and Beta in Thyroid Cancer. Front. Oncol. 2022, 12, 916804. [Google Scholar] [CrossRef] [PubMed]
- Flouriot, G.; Brand, H.; Denger, S.; Metivier, R.; Kos, M.; Reid, G.; Sonntag-Buck, V.; Gannon, F. Identification of a New Isoform of the Human Estrogen Receptor-Alpha (HER-Alpha) That Is Encoded by Distinct Transcripts and That Is Able to Repress HER-Alpha Activation Function 1. EMBO J. 2000, 19, 4688–4700. [Google Scholar] [CrossRef] [PubMed]
- Denger, S.; Reid, G.; Kos, M.; Flouriot, G.; Parsch, D.; Brand, H.; Korach, K.S.; Sonntag-Buck, V.; Gannon, F. ERalpha Gene Expression in Human Primary Osteoblasts: Evidence for the Expression of Two Receptor Proteins. Mol. Endocrinol. Baltim. Md 2001, 15, 2064–2077. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, J.; Wei, J.; Chong, C.C.N.; Yang, D.; He, Y.; Chen, G.G.; Lai, P.B.S. Alternative Splicing of Estrogen Receptor Alpha in Hepatocellular Carcinoma. BMC Cancer 2016, 16, 926. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Yin, L. Estrogen Receptor Alpha-36 (ER-A36): A New Player in Human Breast Cancer. Mol. Cell. Endocrinol. 2015, 418, 193–206. [Google Scholar] [CrossRef]
- Konan, H.-P.; Kassem, L.; Omarjee, S.; Surmieliova-Garnès, A.; Jacquemetton, J.; Cascales, E.; Rezza, A.; Trédan, O.; Treilleux, I.; Poulard, C.; et al. ERα-36 Regulates Progesterone Receptor Activity in Breast Cancer. Breast Cancer Res. BCR 2020, 22, 50. [Google Scholar] [CrossRef]
- Thiebaut, C.; Konan, H.-P.; Guerquin, M.-J.; Chesnel, A.; Livera, G.; Le Romancer, M.; Dumond, H. The Role of ERα36 in Development and Tumor Malignancy. Int. J. Mol. Sci. 2020, 21, 4116. [Google Scholar] [CrossRef]
- Rao, J.; Jiang, X.; Wang, Y.; Chen, B. Advances in the Understanding of the Structure and Function of ER-A36, a Novel Variant of Human Estrogen Receptor-Alpha. J. Steroid Biochem. Mol. Biol. 2011, 127, 231–237. [Google Scholar] [CrossRef]
- Zhang, X.T.; Kang, L.G.; Ding, L.; Vranic, S.; Gatalica, Z.; Wang, Z.-Y. A Positive Feedback Loop of ER-A36/EGFR Promotes Malignant Growth of ER-Negative Breast Cancer Cells. Oncogene 2011, 30, 770–780. [Google Scholar] [CrossRef]
- Zhang, J.; Li, G.; Li, Z.; Yu, X.; Zheng, Y.; Jin, K.; Wang, H.; Gong, Y.; Sun, X.; Teng, X.; et al. Estrogen-Independent Effects of ER-A36 in ER-Negative Breast Cancer. Steroids 2012, 77, 666–673. [Google Scholar] [CrossRef]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 Mutations in Breast Cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef]
- Clusan, L.; Le Goff, P.; Flouriot, G.; Pakdel, F. A Closer Look at Estrogen Receptor Mutations in Breast Cancer and Their Implications for Estrogen and Antiestrogen Responses. Int. J. Mol. Sci. 2021, 22, 756. [Google Scholar] [CrossRef]
- Gu, G.; Tian, L.; Herzog, S.K.; Rechoum, Y.; Gelsomino, L.; Gao, M.; Du, L.; Kim, J.-A.; Dustin, D.; Lo, H.C.; et al. Hormonal Modulation of ESR1 Mutant Metastasis. Oncogene 2021, 40, 997–1011. [Google Scholar] [CrossRef]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Arao, Y.; Korach, K.S. The Physiological Role of Estrogen Receptor Functional Domains. Essays Biochem. 2021, 65, 867–875. [Google Scholar] [CrossRef]
- Kharb, R.; Haider, K.; Neha, K.; Yar, M.S. Aromatase Inhibitors: Role in Postmenopausal Breast Cancer. Arch. Pharm. 2020, 353, 2000081. [Google Scholar] [CrossRef]
- Wittmann, B.M.; Sherk, A.; McDonnell, D.P. Definition of Functionally Important Mechanistic Differences among Selective Estrogen Receptor Down-Regulators. Cancer Res. 2007, 67, 9549–9560. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.K.; Bihani, T. Selective Estrogen Receptor Modulators (SERMs) and Selective Estrogen Receptor Degraders (SERDs) in Cancer Treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Wang, L.; Sharma, A. The Quest for Orally Available Selective Estrogen Receptor Degraders (SERDs). ChemMedChem 2020, 15, 2072–2097. [Google Scholar] [CrossRef] [PubMed]
- Kerdivel, G.; Flouriot, G.; Pakdel, F. Modulation of Estrogen Receptor Alpha Activity and Expression during Breast Cancer Progression. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2013; Volume 93, pp. 135–160. ISBN 978-0-12-416673-8. [Google Scholar]
- Cottu, P.; Ring, A.; Abdel-Razeq, H.; Marchetti, P.; Cardoso, F.; Salvador Bofill, J.; Martín, M.; Menon-Singh, L.; Wu, J.; De Laurentiis, M. Ribociclib plus Letrozole in Subgroups of Special Clinical Interest with Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: Subgroup Analysis of the Phase IIIb CompLEEment-1 Trial. Breast 2022, 62, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Im, S.-A.; Iwata, H.; Cortés, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus Fulvestrant versus Placebo plus Fulvestrant in Postmenopausal, Hormone Receptor-Positive, HER2-Negative, Advanced Breast Cancer (BELLE-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.-J.; Piekarz, R.L.; Smith, K.L.; Brown- Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor-Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 3171–3181. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus Exemestane for Postmenopausal Patients with Advanced, Hormone Receptor-Positive Breast Cancer (ACE): A Randomised, Double-Blind, Placebo- Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Tchakarska, G.; Sola, B. The Double Dealing of Cyclin D1. Cell Cycle 2019, 19, 163–178. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating Cancer with Selective CDK4/6 Inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Oronsky, B.; Oronsky, N.; Knox, S.; Fanger, G.; Scicinski, J. Episensitization: Therapeutic Tumor Resensitization by Epigenetic Agents: A Review and Reassessment. Anticancer Agents Med. Chem. 2014, 14, 1121–1127. [Google Scholar] [CrossRef]
- Zucchetti, B.; Shimada, A.K.; Katz, A.; Curigliano, G. The Role of Histone Deacetylase Inhibitors in Metastatic Breast Cancer. Breast Edinb. Scotl. 2019, 43, 130–134. [Google Scholar] [CrossRef]
- Tomita, Y.; Lee, M.-J.; Lee, S.; Tomita, S.; Chumsri, S.; Cruickshank, S.; Ordentlich, P.; Trepel, J.B. The Interplay of Epigenetic Therapy and Immunity in Locally Recurrent or Metastatic Estrogen Receptor-Positive Breast Cancer: Correlative Analysis of ENCORE 301, a Randomized, Placebo-Controlled Phase II Trial of Exemestane with or without Entinostat. Oncoimmunology 2016, 5, e1219008. [Google Scholar] [CrossRef]
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist 2018, 23, 528–539. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Medici, N.; Bilancio, A.; Migliaccio, A.; Castoria, G. Breast Cancer Stem Cells: The Role of Sex Steroid Receptors. World J. Stem Cells 2019, 11, 594–603. [Google Scholar] [CrossRef]
- Patra, S.; Elahi, N.; Armorer, A.; Arunachalam, S.; Omala, J.; Hamid, I.; Ashton, A.W.; Joyce, D.; Jiao, X.; Pestell, R.G. Mechanisms Governing Metabolic Heterogeneity in Breast Cancer and Other Tumors. Front. Oncol. 2021, 11, 700629. [Google Scholar] [CrossRef]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic Characterization of Metastatic Patterns from Prospective Clinical Sequencing of 25,000 Patients. Cell 2022, 185, 563–575.e11. [Google Scholar] [CrossRef]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer- Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic Mechanisms in Breast Cancer Therapy and Resistance. Nat. Commun. 2021, 12, 1786. [Google Scholar] [CrossRef]
- Thiebaut, C.; Vlaeminck-Guillem, V.; Trédan, O.; Poulard, C.; Le Romancer, M. Non- Genomic Signaling of Steroid Receptors in Cancer. Mol. Cell. Endocrinol. 2021, 538, 111453. [Google Scholar] [CrossRef]
- Jiménez-Salazar, J.E.; Damian-Ferrara, R.; Arteaga, M.; Batina, N.; Damián- Matsumura, P. Non-Genomic Actions of Estrogens on the DNA Repair Pathways Are Associated with Chemotherapy Resistance in Breast Cancer. Front. Oncol. 2021, 11, 631007. [Google Scholar] [CrossRef]
- Silva, E.; Kabil, A.; Kortenkamp, A. Cross-Talk between Non-Genomic and Genomic Signalling Pathways—Distinct Effect Profiles of Environmental Estrogens. Toxicol. Appl. Pharmacol. 2010, 245, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Korach, K.S. Estrogen Receptors: New Directions in the New Millennium. Endocr. Rev. 2018, 39, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Rusidzé, M.; Adlanmérini, M.; Chantalat, E.; Raymond-Letron, I.; Cayre, S.; Arnal, J.-F.; Deugnier, M.-A.; Lenfant, F. Estrogen Receptor-α Signaling in Post-Natal Mammary Development and Breast Cancers. Cell. Mol. Life Sci. 2021, 78, 5681–5705. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Huangyang, P.; Wang, Y.; Xue, L.; Devericks, E.; Nguyen, H.G.; Yu, X.; Oses- Prieto, J.A.; Burlingame, A.L.; Miglani, S.; et al. ERα Is an RNA-Binding Protein Sustaining Tumor Cell Survival and Drug Resistance. Cell 2021, 184, 5215–5229.e17. [Google Scholar] [CrossRef]
- Grinshpun, A.; Chen, V.; Sandusky, Z.M.; Fanning, S.W.; Jeselsohn, R. ESR1 Activating Mutations: From Structure to Clinical Application. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188830. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Luminal A | Luminal B | HER2 Positive | Triple Negative | |
|---|---|---|---|---|
| Proportion of cases | 60% | 10% | 20% | 10% |
| ERα expression | ++ | + | - | - |
| PR expression | ++ | + | - | - |
| HER2 expression | - | +/- | + | - |
| Proliferation (Ki67) | Low | High | High | High |
| Prognosis | Good | Intermediate | Intermediate | Poor |
| Therapy | Endocrine therapy | Endocrine therapy | Anti-HER2 therapy | Chemotherapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clusan, L.; Ferrière, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. https://doi.org/10.3390/ijms24076834
Clusan L, Ferrière F, Flouriot G, Pakdel F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. International Journal of Molecular Sciences. 2023; 24(7):6834. https://doi.org/10.3390/ijms24076834
Chicago/Turabian StyleClusan, Léa, François Ferrière, Gilles Flouriot, and Farzad Pakdel. 2023. "A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer" International Journal of Molecular Sciences 24, no. 7: 6834. https://doi.org/10.3390/ijms24076834
APA StyleClusan, L., Ferrière, F., Flouriot, G., & Pakdel, F. (2023). A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. International Journal of Molecular Sciences, 24(7), 6834. https://doi.org/10.3390/ijms24076834