
Azacitidine Is Synergistically Lethal with XPO1 Inhibitor Selinexor in Acute Myeloid Leukemia by Targeting XPO1/eIF4E/c-MYC Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
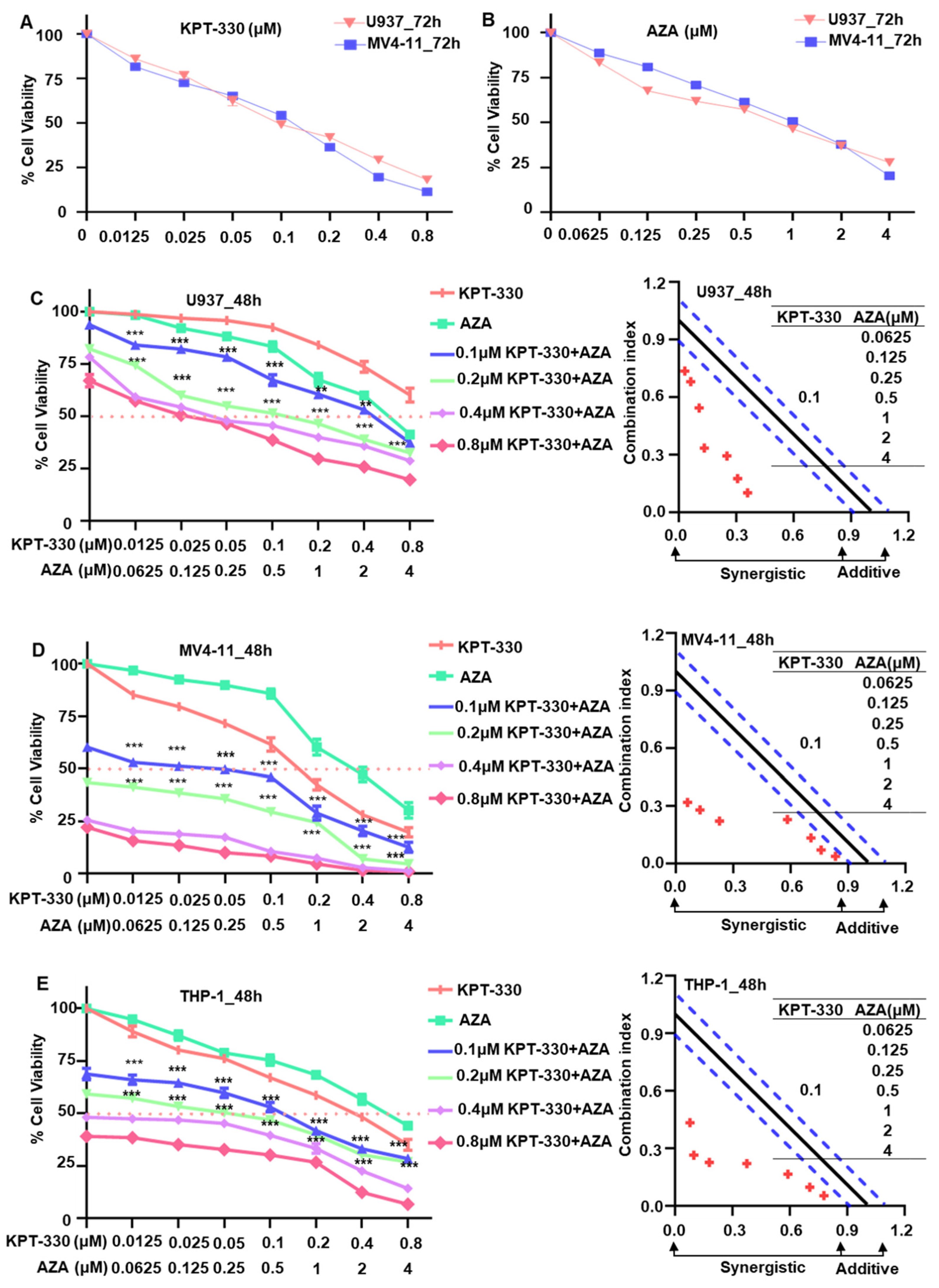
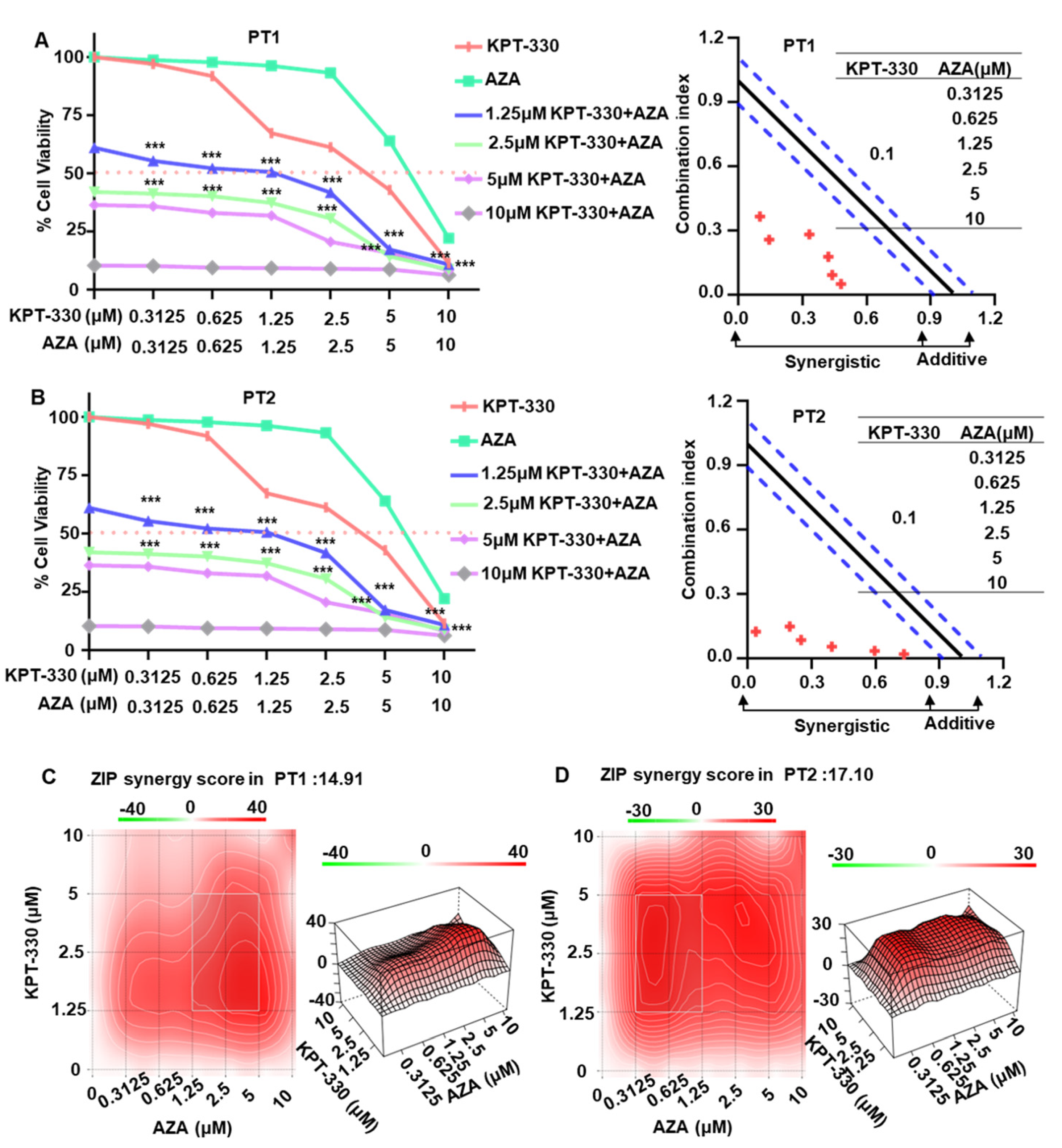
2.1. Synergistic Effect of KPT-330 Combined with AZA on Cell Proliferation of AML Cells
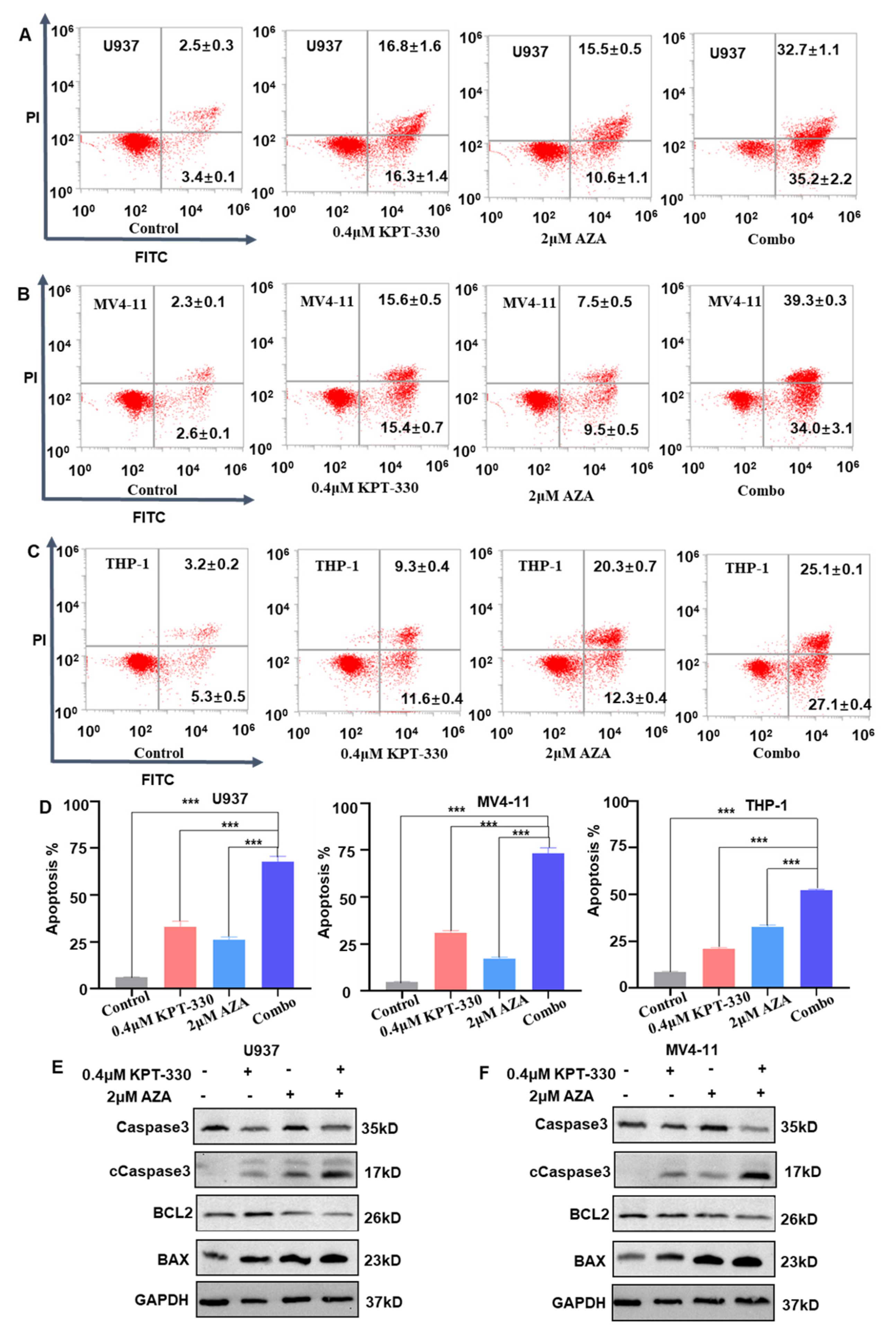
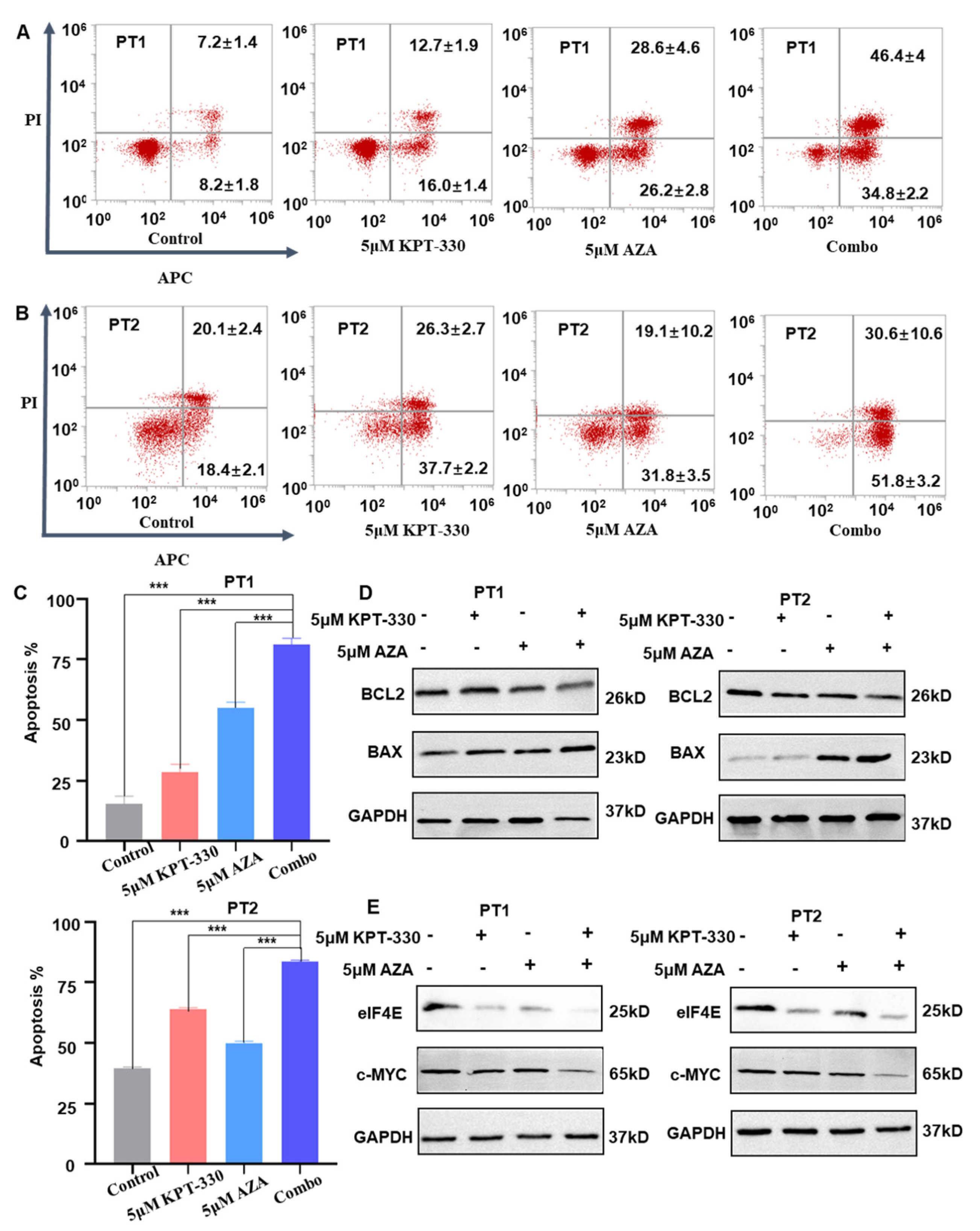
2.2. Synergistic Effect of KPT-330 with AZA on Apoptosis of AML Cells
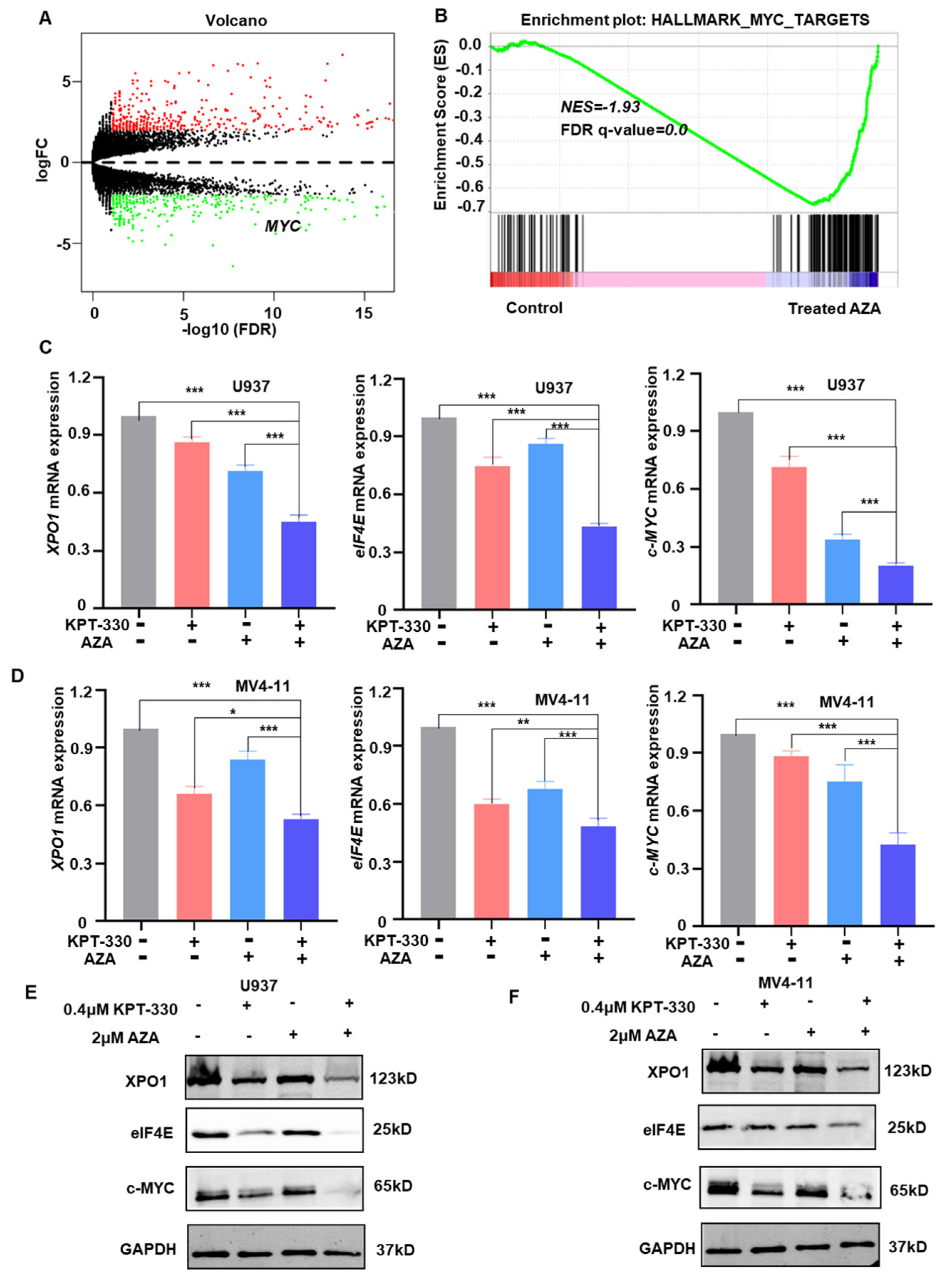
2.3. Transcriptome Analysis to Identify the Key Genes and Pathway Responsible for the Synergistic Effect
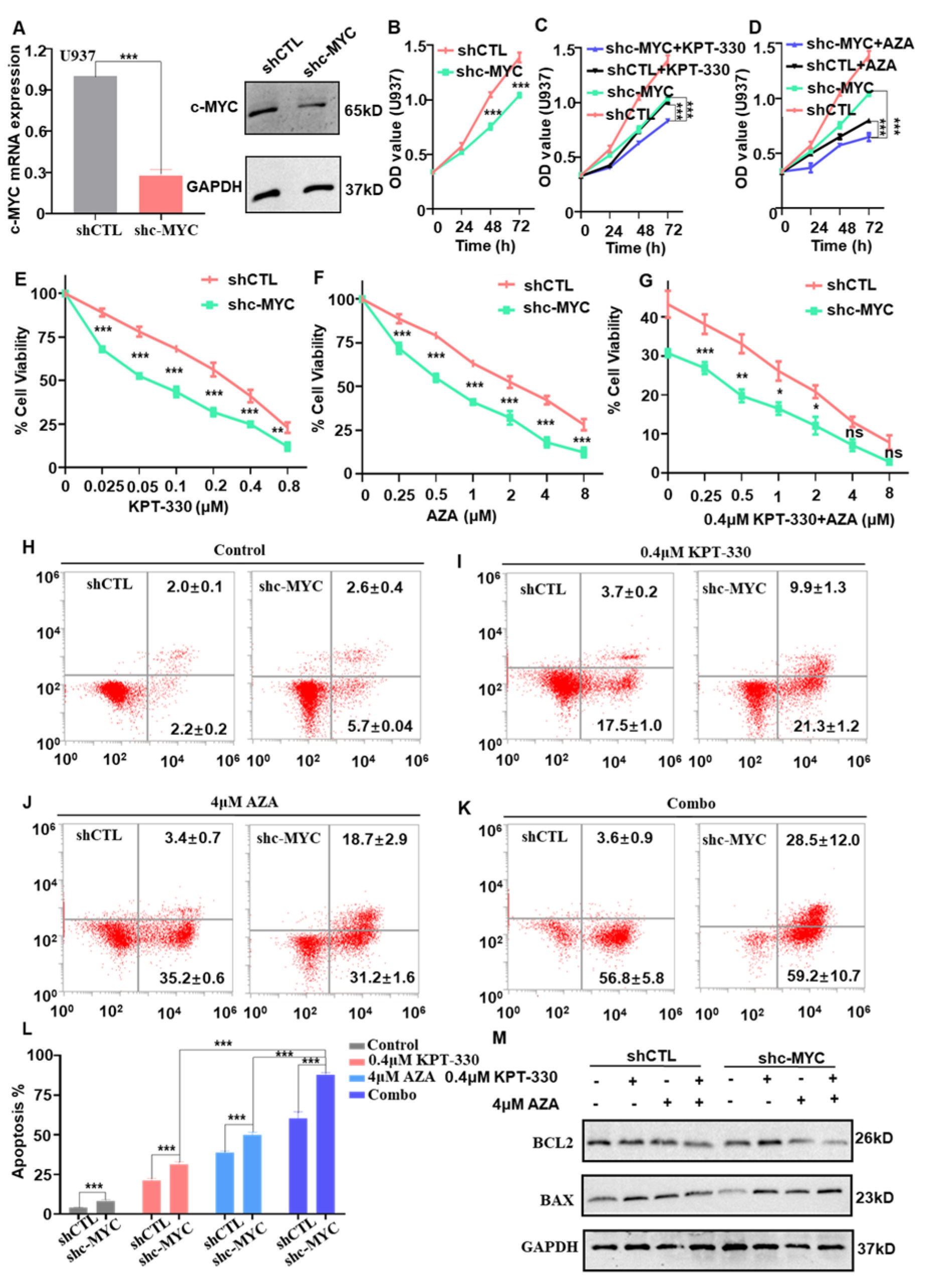
2.4. c-MYC-Dependence on the Combination-Mediated Proliferation Arrest and Apoptosis in U937 Cells
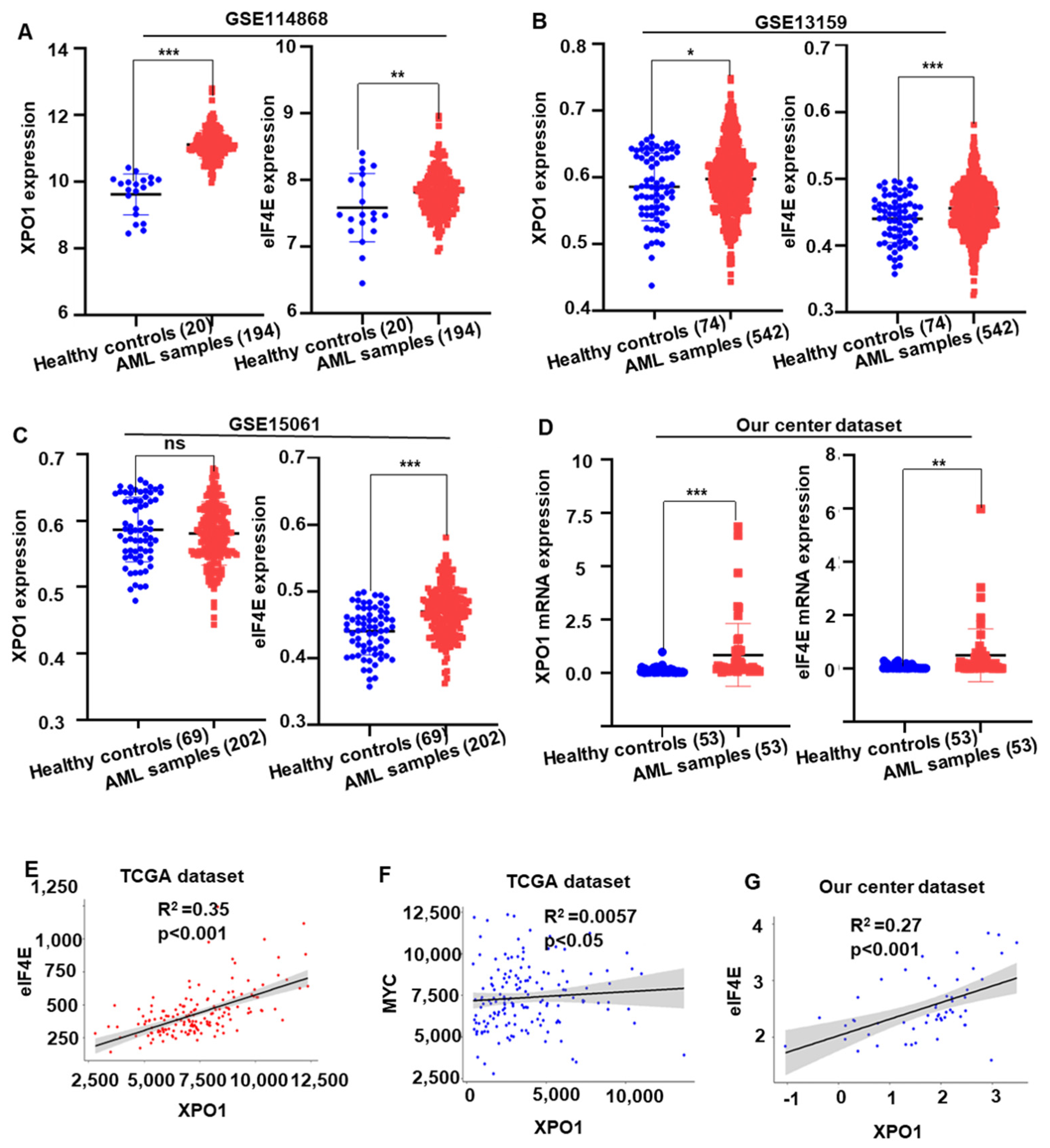
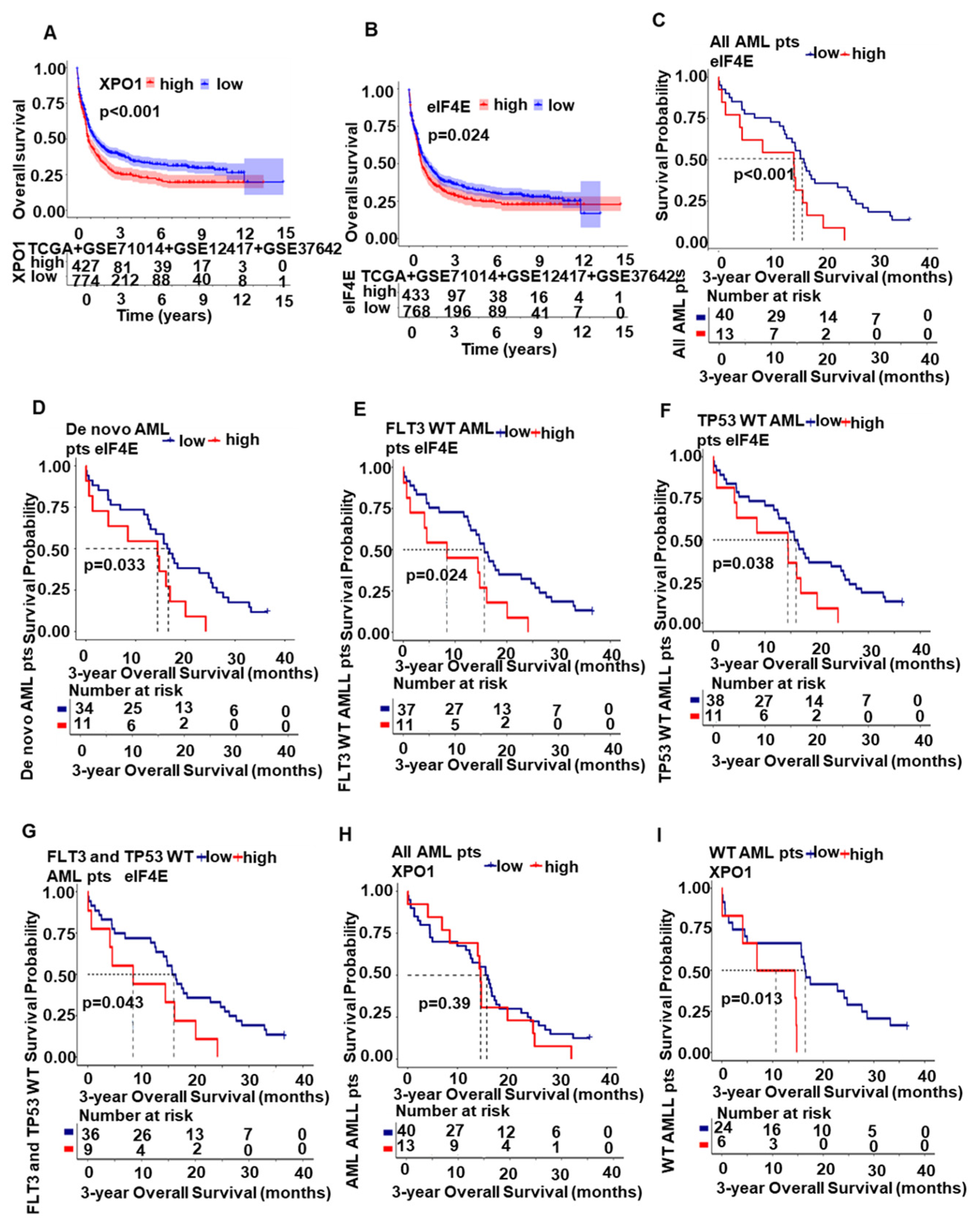
2.5. XPO1/eIF4E Was Up-Regulated in AML Patients and Expression Was Associated with a Worse Prognosis
2.6. Synergistic Effect of KPT-330 with AZA on Cell Growth Arrest in Primary Cells from the AML Patients
2.7. Synergistic Effect of KPT-330 with AZA on Cell Growth Arrest in Primary Cells from the AML Patients
3. Discussion
4. Materials and Methods
4.1. Samples from AML Patients
4.2. Cell Lines
4.3. Reagents
4.4. Cell Proliferation Assay
4.5. Apoptosis Assay by Flow Cytometry
4.6. Cell Cycle Test
4.7. RNA-Seq Analysis
4.8. Real-Time Quantitative PCR (RT-qPCR)
4.9. Western Blot
4.10. c-MYC shRNA Knockdown
4.11. Bioinformatics Analysis of Public Databases
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Estey, E.H. Acute myeloid leukemia: 2019 update on risk-stratification and management. Am. J. Hematol. 2018, 93, 1267–1291. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, Q.; Huang, Y.; Wei, Y.; Zi, J.; Zhao, L.; Cai, Z.; Lu, X.; Xiao, R.; Zhang, Y.; et al. High efficacy of Azacitidine plus HAG in acute myeloid leukemia: An open-label, single-arm, multi-center, phase 2 study. Blood Cancer J. 2022, 12, 145. [Google Scholar] [CrossRef]
- Gu, S.; Hou, Y.; Dovat, K.; Dovat, S.; Song, C.; Ge, Z. Synergistic effect of HDAC inhibitor Chidamide with Cladribine on cell cycle arrest and apoptosis by targeting HDAC2/c-Myc/RCC1 axis in acute myeloid leukemia. Exp. Hematol. Oncol. 2023, 12, 23. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Büchner, T.; Berdel, W.E.; Haferlach, C.; Haferlach, T.; Schnittger, S.; Müller-Tidow, C.; Braess, J.; Spiekermann, K.; Kienast, J.; Staib, P.; et al. Age-related risk profile and chemotherapy dose response in acute myeloid leukemia: A study by the German Acute Myeloid Leukemia Cooperative Group. J. Clin. Oncol. 2009, 27, 61–69. [Google Scholar] [CrossRef]
- Fernandez, H.F.; Sun, Z.; Yao, X.; Litzow, M.R.; Luger, S.M.; Paietta, E.M.; Racevskis, J.; Dewald, G.W.; Ketterling, R.P.; Bennett, J.M.; et al. dose intensification in acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1249–1259. [Google Scholar] [CrossRef]
- Deneberg, S. Epigenetics in myeloid malignancies. Methods Mol. Biol. 2012, 863, 119–137. [Google Scholar]
- Kroeger, H.; Jelinek, J.; Estécio, M.R.; He, R.; Kondo, K.; Chung, W.; Zhang, L.; Shen, L.; Kantarjian, H.M.; Bueso-Ramos, C.E.; et al. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood 2008, 112, 1366–1373. [Google Scholar] [CrossRef]
- Hackanson, B.; Daskalakis, M. Decitabine. Recent Results Cancer Res. 2014, 201, 269–297. [Google Scholar] [PubMed]
- El Fakih, R.; Komrokji, R.; Shaheen, M.; Almohareb, F.; Rasheed, W.; Hassanein, M. Azacitidine Use for Myeloid Neoplasms. Clin. Lymphoma Myeloma Leuk. 2018, 18, e147–e155. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Almeida, A.; Giagounidis, A.; Platzbecker, U.; Garcia, R.; Voso, M.T.; Larsen, S.R.; Valcarcel, D.; Silverman, L.R.; Skikne, B.; et al. Design and rationale of the QUAZAR Lower-Risk MDS (AZA-MDS-003) trial: A randomized phase 3 study of CC-486 (oral azacitidine) plus best supportive care vs placebo plus best supportive care in patients with IPSS lower-risk myelodysplastic syndromes and poor prognosis due to red blood cell transfusion-dependent anemia and thrombocytopenia. BMC Hematol. 2016, 16, 12. [Google Scholar]
- Edlin, R.; Connock, M.; Tubeuf, S.; Round, J.; Fry-Smith, A.; Hyde, C.; Greenheld, W. Azacitidine for the treatment of myelodysplastic syndrome, chronic myelomonocytic leukaemia and acute myeloid leukaemia. Health Technol. Assess 2010, 14 (Suppl. S1), 69–74. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Saliba, A.N.; John, A.J.; Kaufmann, S.H. Resistance to venetoclax and hypomethylating agents in acute myeloid leukemia. Cancer Drug Resist. 2021, 4, 125–142. [Google Scholar] [CrossRef]
- Robert, G.; Auberger, P. Azacitidine resistance caused by LAMP2 deficiency: A therapeutic window for the use of autophagy inhibitors in MDS/AML patients? Autophagy 2019, 15, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Leisch, M.; Greil, R.; Pleyer, L. IDO in MDS/AML disease progression and its role in resistance to azacitidine: A potential new drug target? Br. J. Haematol. 2020, 190, 314–317. [Google Scholar] [CrossRef]
- Gargantilla, M.; López-Fernández, J.; Camarasa, M.J.; Persoons, L.; Daelemans, D.; Priego, E.M.; Pérez-Pérez, M.J. Inhibition of XPO-1 Mediated Nuclear Export through the Michael-Acceptor Character of Chalcones. Pharmaceuticals 2021, 14, 1131. [Google Scholar] [CrossRef]
- Fukuda, M.; Asano, S.; Nakamura, T.; Adachi, M.; Yoshida, M.; Yanagida, M.; Nishida, E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 1997, 390, 308–311. [Google Scholar] [CrossRef]
- Mendonca, J.; Sharma, A.; Kim, H.S.; Hammers, H.; Meeker, A.; De Marzo, A.; Carducci, M.; Kauffman, M.; Shacham, S.; Kachhap, S. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget 2014, 5, 6102–6112. [Google Scholar] [CrossRef]
- Etchin, J.; Montero, J.; Berezovskaya, A.; Le, B.T.; Kentsis, A.; Christie, A.L.; Conway, A.S.; Chen, W.C.; Reed, C.; Mansour, M.R.; et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice. Leukemia 2016, 30, 190–199. [Google Scholar] [CrossRef]
- Gandhi, U.H.; Senapedis, W.; Baloglu, E.; Unger, T.J.; Chari, A.; Vogl, D.; Cornell, R.F. Clinical Implications of Targeting XPO1-mediated Nuclear Export in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Lapalombella, R.; Sun, Q.; Williams, K.; Tangeman, L.; Jha, S.; Zhong, Y.; Goettl, V.; Mahoney, E.; Berglund, C.; Gupta, S.; et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 2012, 120, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Aboukameel, A.; Bao, B.; Sarkar, F.H.; Philip, P.A.; Kauffman, M.; Shacham, S.; Mohammad, R.M. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology 2013, 144, 447–456. [Google Scholar] [CrossRef]
- Soung, Y.H.; Kashyap, T.; Nguyen, T.; Yadav, G.; Chang, H.; Landesman, Y.; Chung, J. Selective Inhibitors of Nuclear Export (SINE) compounds block proliferation and migration of triple negative breast cancer cells by restoring expression of ARRDC3. Oncotarget 2017, 8, 52935–52947. [Google Scholar] [CrossRef]
- Etchin, J.; Sun, Q.; Kentsis, A.; Farmer, A.; Zhang, Z.C.; Sanda, T.; Mansour, M.R.; Barcelo, C.; McCauley, D.; Kauffman, M.; et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia 2013, 27, 66–74. [Google Scholar] [CrossRef]
- Garzon, R.; Savona, M.; Baz, R.; Andreeff, M.; Gabrail, N.; Gutierrez, M.; Savoie, L.; Mau-Sorensen, P.M.; Wagner-Johnston, N.; Yee, K.; et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood 2017, 129, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- Sweet, K.; Bhatnagar, B.; Döhner, H.; Donnellan, W.; Frankfurt, O.; Heuser, M.; Kota, V.; Liu, H.; Raffoux, E.; Roboz, G.J.; et al. A 2:1 randomized, open-label, phase II study of selinexor vs. physician’s choice in older patients with relapsed or refractory acute myeloid leukemia. Leuk. Lymphoma 2021, 62, 3192–3203. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, B.; Zhao, Q.; Mims, A.S.; Vasu, S.; Behbehani, G.K.; Larkin, K.; Blachly, J.S.; Blum, W.; Klisovic, R.B.; Ruppert, A.S.; et al. Selinexor in combination with decitabine in patients with acute myeloid leukemia: Results from a phase 1 study. Leuk. Lymphoma 2020, 61, 387–396. [Google Scholar] [CrossRef]
- Martínez Sánchez, M.P.; Megías-Vericat, J.E.; Rodríguez-Veiga, R.; Vives, S.; Bergua, J.M.; Torrent, A.; Suárez-Varela, S.; Boluda, B.; Martínez-López, J.; Cano-Ferri, I.; et al. A phase I trial of selinexor plus FLAG-Ida for the treatment of refractory/relapsed adult acute myeloid leukemia patients. Ann. Hematol. 2021, 100, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Abboud, R.; Chendamarai, E.; Rettig, M.P.; Trinkaus, K.M.; Riedell, P.A.; Abboud, C.N.; Ghobadi, A.; Pusic, I.; Stockerl-Goldstein, K.; Schroeder, M.A.; et al. Selinexor combined with cladribine, cytarabine, and filgrastim in relapsed or refractory acute myeloid leukemia. Haematologica 2020, 105, e404–e407. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.E.; Rahimi, S.; Zarandi, B.; Chegeni, R.; Safa, M. MYC: A multipurpose oncogene with prognostic and therapeutic implications in blood malignancies. J. Hematol. Oncol. 2021, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.A.; Xie, W.; Li, X.; Huang, S. MYC-mediated synthetic lethality for treatment of hematological malignancies. Curr. Cancer Drug Targets 2015, 15, 53–70. [Google Scholar] [CrossRef]
- Yang, C.; Gu, Y.; Ge, Z.; Song, C. Targeting EZH2 Promotes Chemosensitivity of BCL-2 Inhibitor through Suppressing PI3K and c-KIT Signaling in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2022, 23, 11393. [Google Scholar] [CrossRef]
- Li, J.; Huang, Y.; Hou, Y.; Gu, Y.; Song, C.; Ge, Z. High efficacy of azacitidine combined with homoharringtonine, idarubicin, and cytarabine in newly diagnosed patients with AML: A single arm, phase 2 trial. Front. Oncol. 2022, 12, 1069246. [Google Scholar] [CrossRef]
- Šimoničová, K.; Janotka, Ľ.; Kavcová, H.; Sulová, Z.; Breier, A.; Messingerova, L. Different mechanisms of drug resistance to hypomethylating agents in the treatment of myelodysplastic syndromes and acute myeloid leukemia. Drug Resist. Updates 2022, 61, 100805. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Das, M. Venetoclax with decitabine or azacitidine for AML. Lancet Oncol. 2018, 19, e672. [Google Scholar] [CrossRef]
- Azizi, A.; Ediriwickrema, A.; Dutta, R.; Patel, S.A.; Shomali, W.; Medeiros, B.; Iberri, D.; Gotlib, J.; Mannis, G.; Greenberg, P.; et al. Venetoclax and hypomethylating agent therapy in high risk myelodysplastic syndromes: A retrospective evaluation of a real-world experience. Leuk. Lymphoma 2020, 61, 2700–2707. [Google Scholar] [CrossRef]
- Chaudhry, G.E.; Akim, A.M.; Sung, Y.Y.; Muhammad, T.S.T. Cancer and Apoptosis. Methods Mol. Biol. 2022, 2543, 191–210. [Google Scholar]
- Tang, R.; Cheng, A.; Guirales, F.; Yeh, W.; Tirado, C.A. C-MYC Amplification in AML. J. Assoc. Genet. Technol. 2021, 47, 202–212. [Google Scholar] [PubMed]
- Alitalo, K.; Saksela, K.; Winqvist, R.; Alitalo, R.; Keski-Oja, J.; Laiho, M.; Ilvonen, M.; Knuutila, S.; de la Chapelle, A. Acute myelogenous leukaemia with c-myc amplification and double minute chromosomes. Lancet 1985, 2, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Sharma, S.; Qi, J.; Shah, B.; Devaraj, S.G.; Leveque, C.; Portier, B.P.; Iyer, S.; Bradner, J.E.; Bhalla, K.N. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther. 2014, 13, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Zhang, W.N.; Chen, B.; Xi, W.D.; Lu, Y.; Huang, J.Y.; Wang, Y.Y.; Long, J.; Wu, S.F.; Zhang, Y.X.; et al. Homoharringtonine deregulates MYC transcriptional expression by directly binding NF-κB repressing factor. Proc. Natl. Acad. Sci. USA 2019, 116, 2220–2225. [Google Scholar] [CrossRef] [PubMed]
- Handschuh, L.; Kaźmierczak, M.; Milewski, M.C.; Góralski, M.; Łuczak, M.; Wojtaszewska, M.; Uszczyńska-Ratajczak, B.; Lewandowski, K.; Komarnicki, M.; Figlerowicz, M. Gene expression profiling of acute myeloid leukemia samples from adult patients with AML-M1 and -M2 through boutique microarrays, real-time PCR and droplet digital PCR. Int. J. Oncol. 2018, 52, 656–678. [Google Scholar] [CrossRef]
- Delgado, M.D.; Albajar, M.; Gomez-Casares, M.T.; Batlle, A.; León, J. MYC oncogene in myeloid neoplasias. Clin. Transl. Oncol. 2013, 15, 87–94. [Google Scholar] [CrossRef]
- Ohanian, M.; Rozovski, U.; Kanagal-Shamanna, R.; Abruzzo, L.V.; Loghavi, S.; Kadia, T.; Futreal, A.; Bhalla, K.; Zuo, Z.; Huh, Y.O.; et al. MYC protein expression is an important prognostic factor in acute myeloid leukemia. Leuk. Lymphoma 2019, 60, 37–48. [Google Scholar] [CrossRef]
- Ci, W.; Polo, J.M.; Cerchietti, L.; Shaknovich, R.; Wang, L.; Yang, S.N.; Ye, K.; Farinha, P.; Horsman, D.E.; Gascoyne, R.D.; et al. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood 2009, 113, 5536–5548. [Google Scholar] [CrossRef]
- Deng, C.; Lipstein, M.R.; Scotto, L.; Jirau Serrano, X.O.; Mangone, M.A.; Li, S.; Vendome, J.; Hao, Y.; Xu, X.; Deng, S.X.; et al. Silencing c-Myc translation as a therapeutic strategy through targeting PI3Kδ and CK1ε in hematological malignancies. Blood 2017, 129, 88–99. [Google Scholar] [CrossRef]
- Carabet, L.A.; Rennie, P.S.; Cherkasov, A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int. J. Mol. Sci. 2018, 20, 120. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Yu, X.; Na, C.; Santhanam, R.; Shacham, S.; Kauffman, M.; Walker, A.; Klisovic, R.; Blum, W.; Caligiuri, M.; et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 2012, 120, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhou, H. Phospho-eIF4E: A New Target for Acute Myeloid Leukemia. Curr. Protein Pept. Sci. 2021, 22, 328–335. [Google Scholar] [CrossRef]
- Kojima, K.; Kornblau, S.M.; Ruvolo, V.; Dilip, A.; Duvvuri, S.; Davis, R.E.; Zhang, M.; Wang, Z.; Coombes, K.R.; Zhang, N.; et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 2013, 121, 4166–4174. [Google Scholar] [CrossRef]
- Zhao, L.; Luo, B.; Wang, L.; Chen, W.; Jiang, M.; Zhang, N. Pan-cancer analysis reveals the roles of XPO1 in predicting prognosis and tumorigenesis. Transl. Cancer Res. 2021, 10, 4664–4679. [Google Scholar] [CrossRef]
- Marullo, R.; Revuelta, M.; Zamponi, N.; Culjkovic, B.; Yang, S.N.; Ahn, H.; Inghirami, G.; Borden, K.L.; Cerchietti, L. Exportin-1 Connects Dynamic Transcription and Translation of Genotoxic Stress Genes in Diffuse Large B-Cell Lymphoma Patients. Blood 2017, 130, 312. [Google Scholar]
- Zi, J.; Han, Q.; Gu, S.; McGrath, M.; Kane, S.; Song, C.; Ge, Z. Targeting NAT10 Induces Apoptosis Associated with Enhancing Endoplasmic Reticulum Stress in Acute Myeloid Leukemia Cells. Front. Oncol. 2020, 10, 598107. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, H.; Hou, Y.; Li, J.; Song, C.; Ge, Z. Azacitidine Is Synergistically Lethal with XPO1 Inhibitor Selinexor in Acute Myeloid Leukemia by Targeting XPO1/eIF4E/c-MYC Signaling. Int. J. Mol. Sci. 2023, 24, 6816. https://doi.org/10.3390/ijms24076816
Long H, Hou Y, Li J, Song C, Ge Z. Azacitidine Is Synergistically Lethal with XPO1 Inhibitor Selinexor in Acute Myeloid Leukemia by Targeting XPO1/eIF4E/c-MYC Signaling. International Journal of Molecular Sciences. 2023; 24(7):6816. https://doi.org/10.3390/ijms24076816
Chicago/Turabian StyleLong, Huideng, Yue Hou, Jun Li, Chunhua Song, and Zheng Ge. 2023. "Azacitidine Is Synergistically Lethal with XPO1 Inhibitor Selinexor in Acute Myeloid Leukemia by Targeting XPO1/eIF4E/c-MYC Signaling" International Journal of Molecular Sciences 24, no. 7: 6816. https://doi.org/10.3390/ijms24076816
APA StyleLong, H., Hou, Y., Li, J., Song, C., & Ge, Z. (2023). Azacitidine Is Synergistically Lethal with XPO1 Inhibitor Selinexor in Acute Myeloid Leukemia by Targeting XPO1/eIF4E/c-MYC Signaling. International Journal of Molecular Sciences, 24(7), 6816. https://doi.org/10.3390/ijms24076816