
A Machine Learning Model to Predict Survival and Therapeutic Responses in Multiple Myeloma

 , and
, and 
Abstract
1. Introduction
2. Results
2.1. Subject Selection and Baseline Patient Characteristics
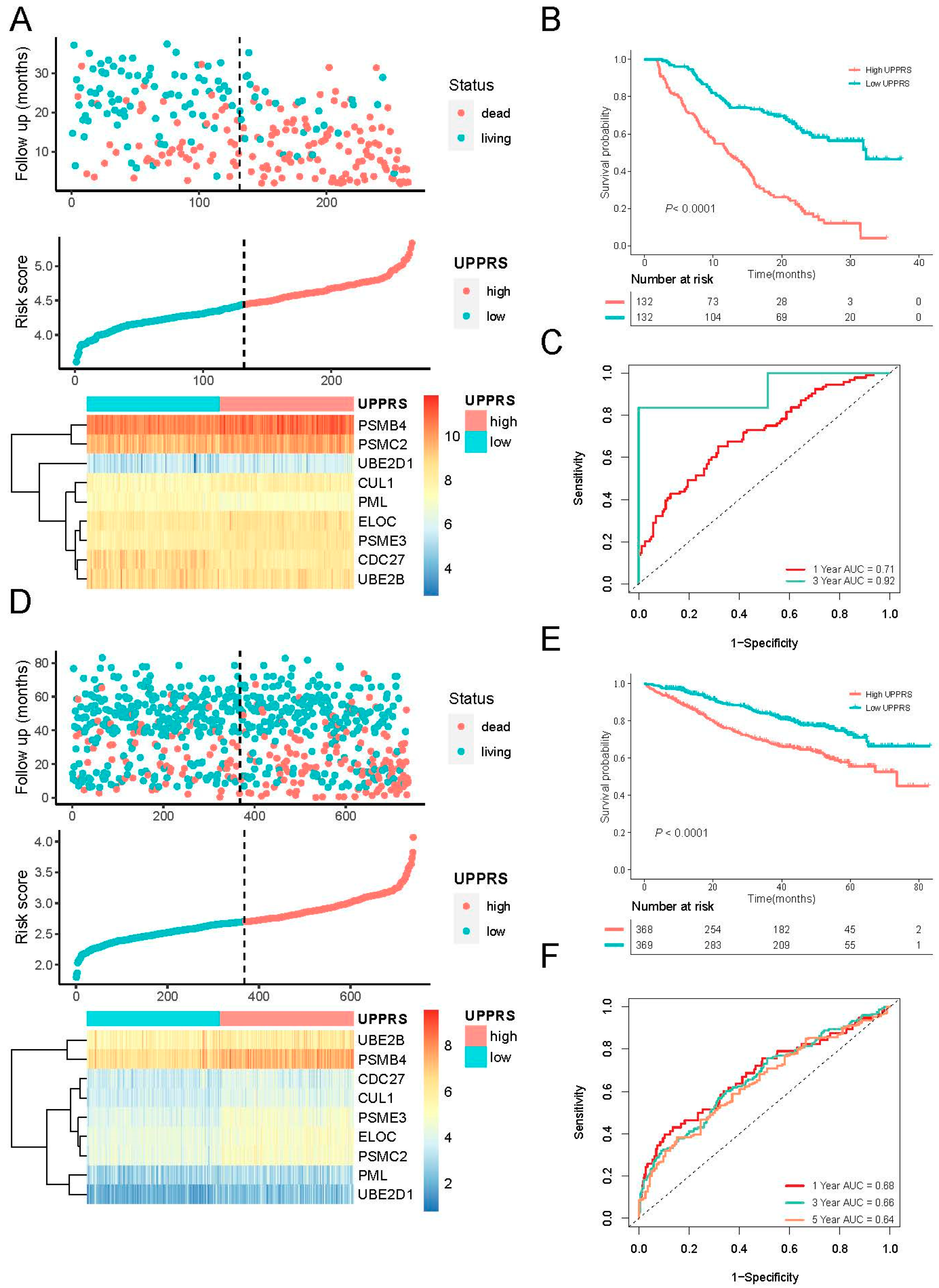
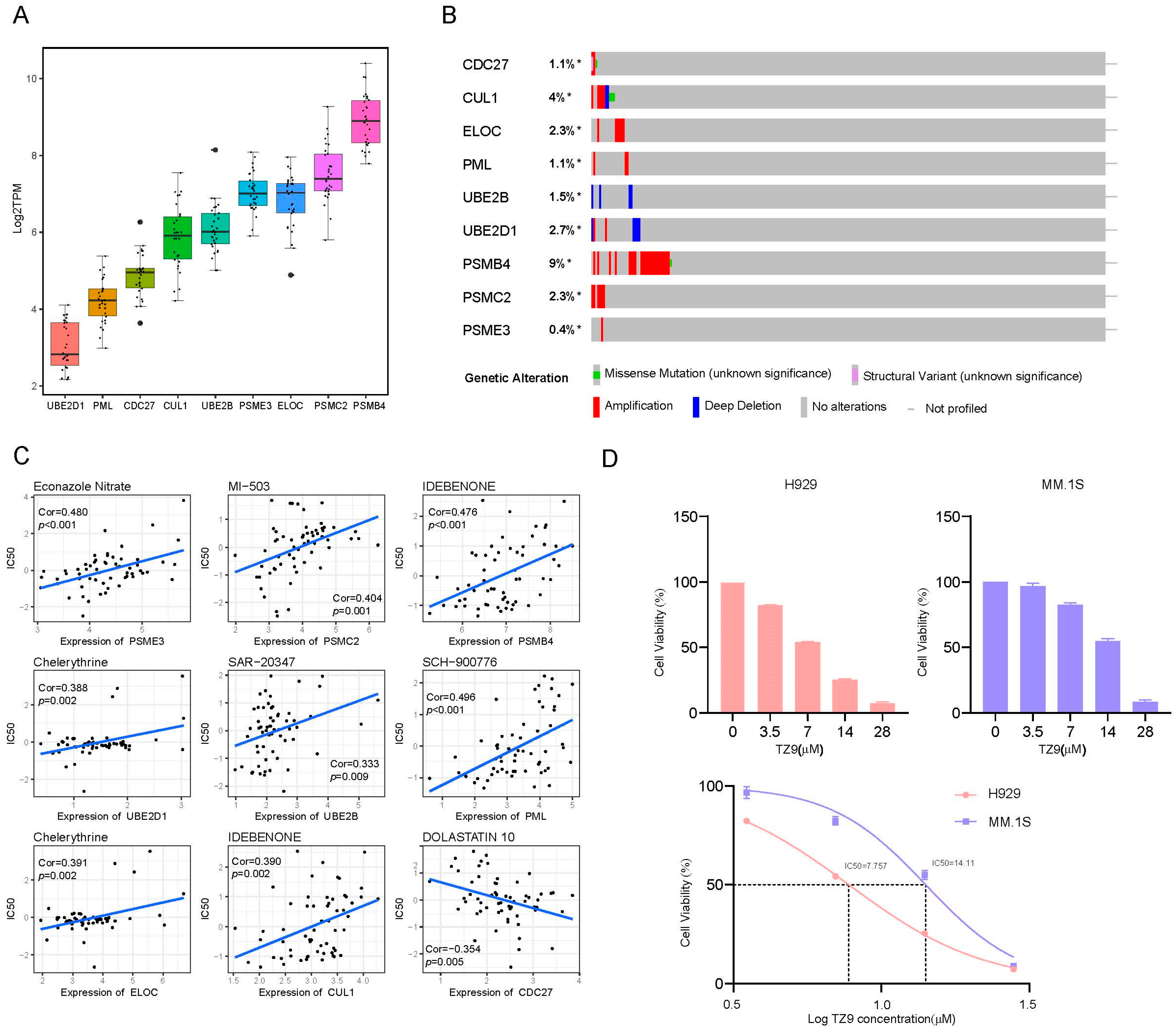
2.2. Construction of a Prognostic Gene Signature
2.3. Evaluation of UPPRS in MM Cohorts
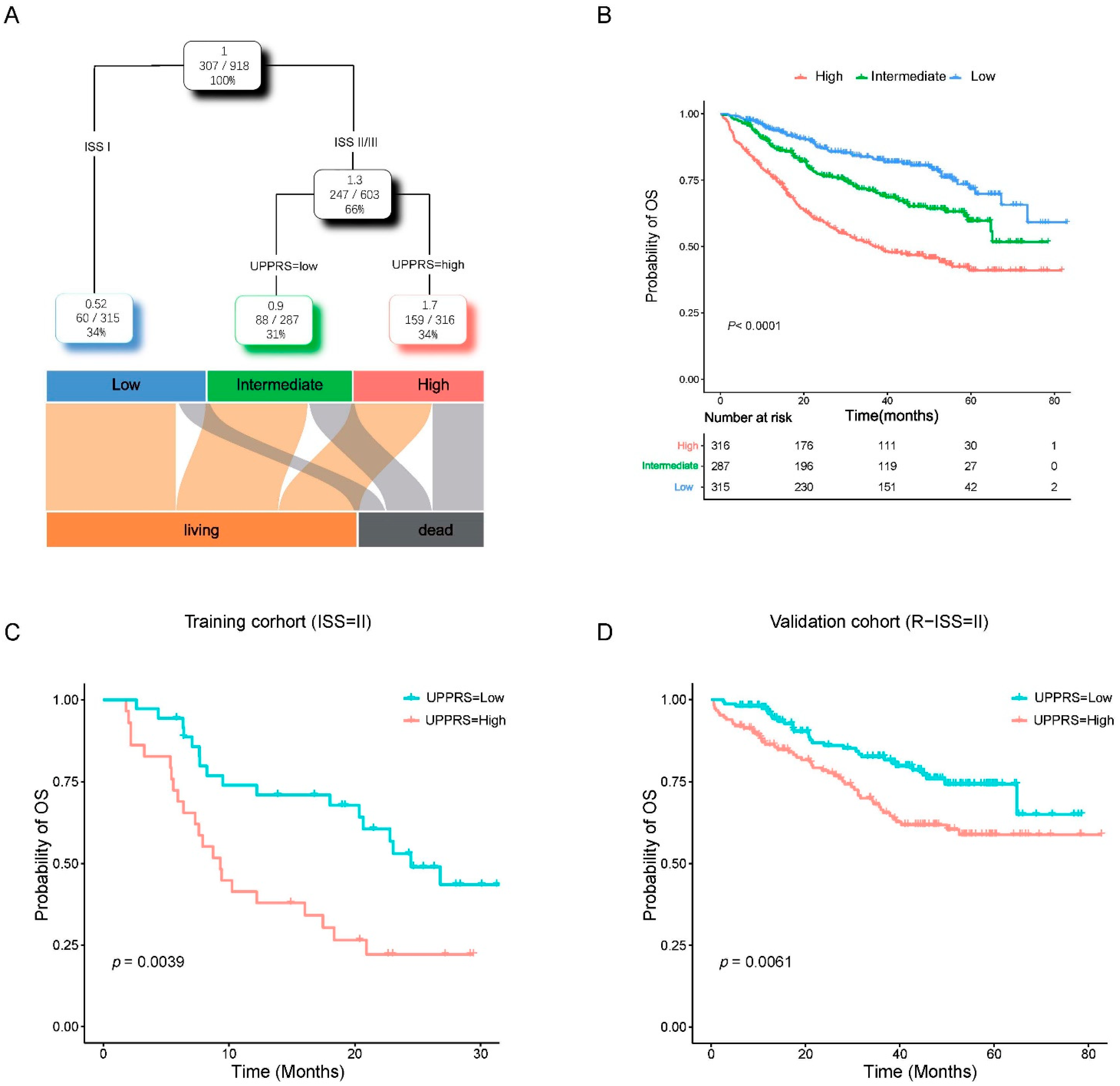
2.4. Clinicopathological Features with UPPRS
2.5. UPPRS and Patients’ Response to PIs
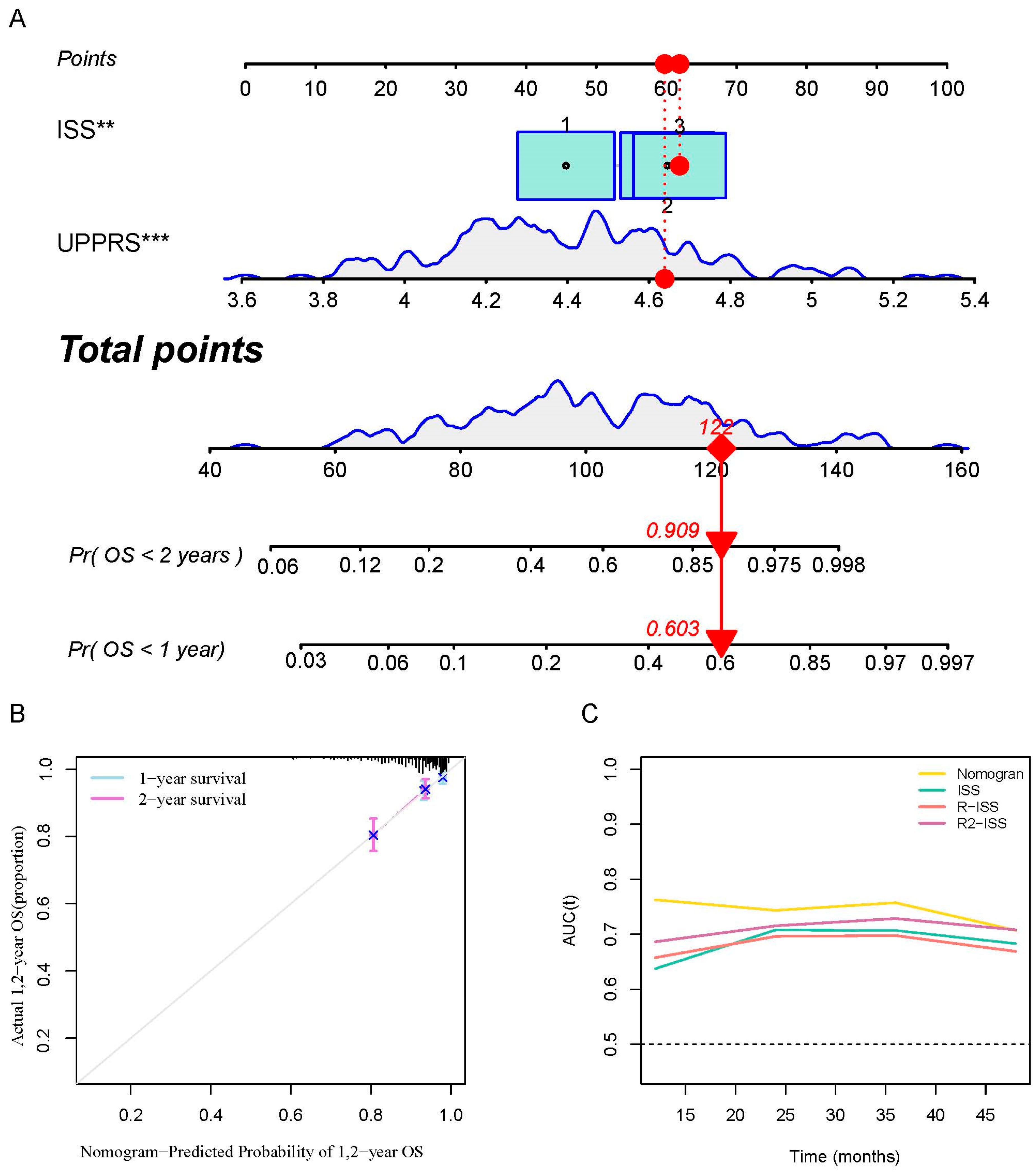
2.6. A prognostic Model Combined UPPRS and Other Clinical Factors
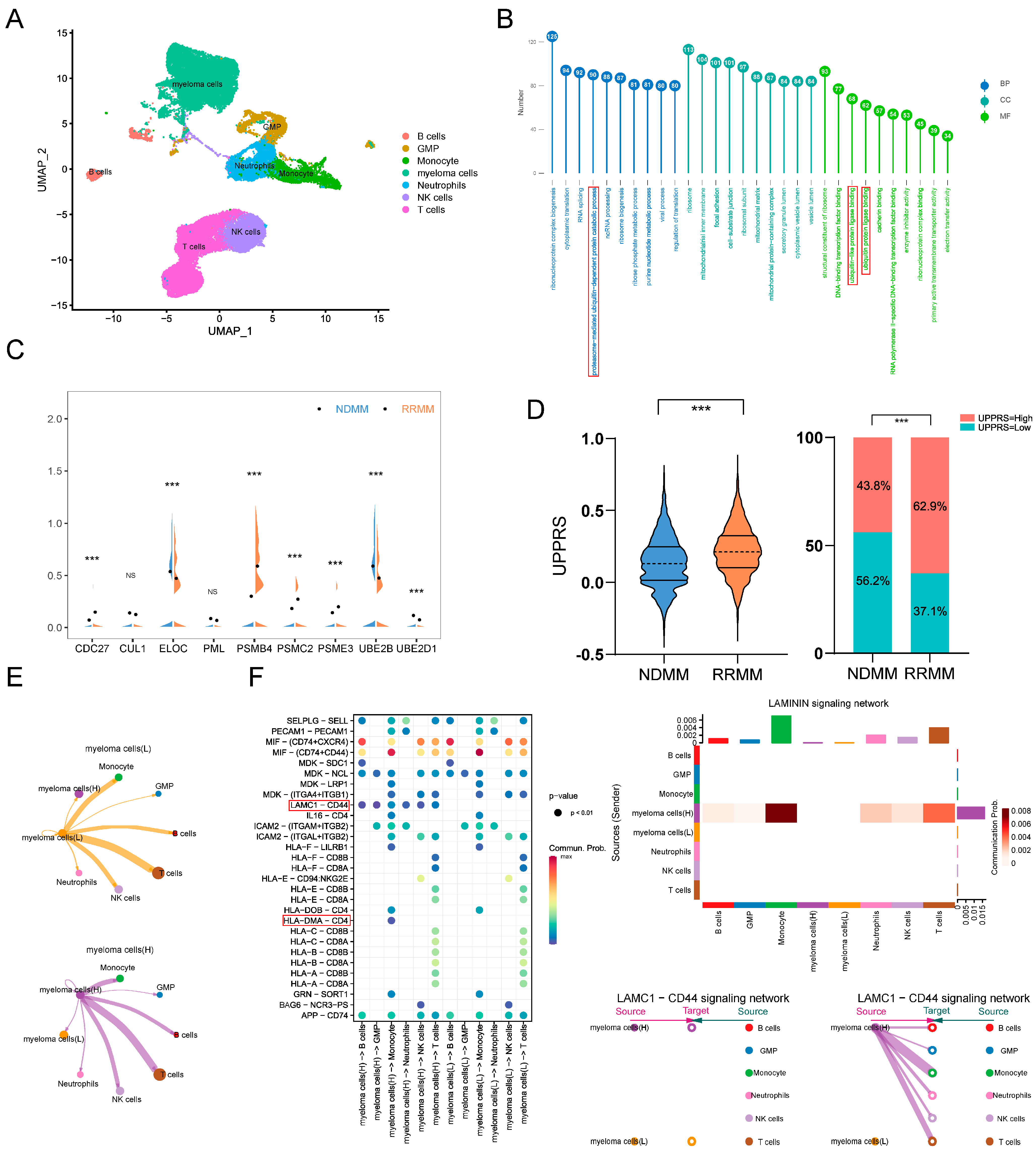
2.7. UPPRS Analysis in Single-Cell Level
2.8. External Validation in Cell Lines
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Constructing of the Prognostic Gene Signatures
4.3. Constructing of the Prognostic Nomogram
4.4. Single-Cell Samples and Data Processing
4.5. Cell Culture and Reagents
4.6. Cell Viability Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Greipp, P.R.; Miguel, J.S.; Durie, B.G.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Avet-Loiseau, H.; Kyle, R.A.; et al. International Staging System for Multiple Myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Hulin, C.; Campion, L.; Rodon, P.; Marit, G.; Attal, M.; Royer, B.; Dib, M.; Voillat, L.; Bouscary, D.; et al. Chromosomal Abnormalities Are Major Prognostic Factors in Elderly Patients with Multiple Myeloma: The Intergroupe Francophone du Myélome Experience. J. Clin. Oncol. 2013, 31, 2806–2809. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Fonseca, R.; Ketterling, R.P.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Knudson, R.A.; et al. Trisomies in multiple myeloma: Impact on survival in patients with high-risk cytogenetics. Blood 2012, 119, 2100–2105. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- D’Agostino, M.; Cairns, D.A.; Lahuerta, J.J.; Wester, R.; Bertsch, U.; Waage, A.; Zamagni, E.; Mateos, M.-V.; Dall’Olio, D.; van de Donk, N.W.; et al. Second Revision of the International Staging System (R2-ISS) for Overall Survival in Multiple Myeloma: A European Myeloma Network (EMN) Report within the HARMONY Project. J. Clin. Oncol. 2022, 40, 3406–3418. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Yoon, D.H.; Lee, J.B.; Kim, S.-Y.; Moon, J.H.; Do, Y.R.; Lee, J.H.; Park, Y.; Lee, H.S.; Eom, H.S.; et al. Comprehensive evaluation of the revised international staging system in multiple myeloma patients treated with novel agents as a primary therapy. Am. J. Hematol. 2017, 92, 1280–1286. [Google Scholar] [CrossRef]
- Jung, S.-H.; Kim, K.; Kim, J.S.; Kim, S.J.; Cheong, J.-W.; Kim, S.-J.; Ahn, J.-S.; Ahn, S.-Y.; Yang, D.-H.; Kim, H.-J.; et al. A prognostic scoring system for patients with multiple myeloma classified as stage II with the Revised International Staging System. Br. J. Haematol. 2018, 181, 707–710. [Google Scholar] [CrossRef]
- Kastritis, E.; Terpos, E.; Roussou, M.; Gavriatopoulou, M.; Migkou, M.; Eleutherakis-Papaiakovou, E.; Fotiou, D.; Ziogas, D.; Panagiotidis, I.; Kafantari, E.; et al. Evaluation of the Revised International Staging System in an independent cohort of unselected patients with multiple myeloma. Haematologica 2017, 102, 593–599. [Google Scholar] [CrossRef]
- Chen, H.; Zhou, N.; Hu, X.; Wang, D.; Wei, W.; Peng, R.; Chen, X.; Shi, H.; Wu, L.; Yu, W.; et al. The applicability of the Second Revision of the International Staging System for patients with multiple myeloma receiving immunomodulatory drugs or proteasome inhibitor-based regimens as induction treatment: A real-world analysis. Hematol. Oncol. 2023, 41, 139–146. [Google Scholar] [CrossRef]
- Tan, J.L.C.; Wellard, C.; Moore, E.M.; Mollee, P.; Rajagopal, R.; Quach, H.; Harrison, S.J.; McDonald, E.J.; Ho, P.J.; Prince, H.M.; et al. The second revision of the International Staging System (R2-ISS) stratifies progression-free and overall survival in multiple myeloma: Real world data results in an Australian and New Zealand Population. Br. J. Haematol. 2023, 200, e17–e21. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Dispenzieri, A.; Chim, C.S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.S.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef]
- Calame, K.L.; Lin, K.-I.; Tunyaplin, C. Regulatory Mechanisms that Determine the Development and Function of Plasma Cells. Annu. Rev. Immunol. 2003, 21, 205–230. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Patil, C.; Walter, P. Intracellular signaling from the endoplasmic reticulum to the nucleus: The unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 2001, 13, 349–355. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Workman, P.; Davies, F.E. A stressful life (or death): Combinatorial proteotoxic approaches to cancer-selective therapeutic vulnerability. Oncotarget 2011, 2, 277–280. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A Phase 2 Study of Bortezomib in Relapsed, Refractory Myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef]
- Ahn, J.-S.; Jung, S.-H.; Yang, D.-H.; Bae, S.-Y.; Kim, Y.-K.; Kim, H.-J.; Lee, J.-J. Patterns of Relapse or Progression After Bortezomib-Based Salvage Therapy in Patients With Relapsed/Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2014, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, M.T.; Giraldo, P.; Corradini, P.; Teixeira, A.; Dimopoulos, M.; Blau, I.W.; Drach, J.; Angermund, R.; Allietta, N.; Broer, E.; et al. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. Br. J. Haematol. 2013, 160, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, G.; Mitsiades, C.; Bryant, B.; Zhan, F.; Chng, W.J.; Roels, S.; Koenig, E.; Fergus, A.; Huang, Y.; Richardson, P.; et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2007, 109, 3177–3188. [Google Scholar] [CrossRef] [PubMed]
- DepMap, Broad (2021): DepMap 21Q3 Public. Figshare. Dataset. Available online: https://doi.org/10.6084/m9.figshare.15160110.v2 (accessed on 2 November 2021).
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A Web-Based Suite of Genomic and Pharmacologic Tools to Explore Transcript and Drug Patterns in the NCI-60 Cell Line Set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef]
- Gulla, A.; Anderson, K.C. Multiple myeloma: The revolution of current therapy and a glance into future. Haematologica 2020, 105, 2358–2367. [Google Scholar] [CrossRef]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef]
- Wirth, M.; Schick, M.; Keller, U.; Krönke, J. Ubiquitination and Ubiquitin-Like Modifications in Multiple Myeloma: Biology and Therapy. Cancers 2020, 12, 3764. [Google Scholar] [CrossRef]
- Malek, E.; Abdel-Malek, M.A.; Jagannathan, S.; Vad, N.; Karns, R.; Jegga, A.G.; Broyl, A.; van Duin, M.; Sonneveld, P.; Cottini, F.; et al. Pharmacogenomics and chemical library screens reveal a novel SCF(SKP2) inhibitor that overcomes Bortezomib resistance in multiple myeloma. Leukemia 2017, 31, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.C.; Zada, M.; Wang, S.-Y.; Bornstein, C.; David, E.; Moshe, A.; Li, B.; Shlomi-Loubaton, S.; Gatt, M.E.; Gur, C.; et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 2021, 27, 491–503. [Google Scholar] [CrossRef]
- Pérez-Galán, P.; Mora-Jensen, H.; Weniger, M.A.; Shaffer, A.L., 3rd; Rizzatti, E.G.; Chapman, C.M.; Mo, C.C.; Stennett, L.S.; Rader, C.; Liu, P.; et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood 2011, 117, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Jung, S.-H.; Jo, J.-C.; Lee, Y.J.; Yoon, S.E.; Park, S.-S.; Kim, D.Y.; Shin, H.-J.; Mun, Y.-C.; Yi, J.H.; et al. Development of a new risk stratification system for patients with newly diagnosed multiple myeloma using R-ISS and 18F-FDG PET/CT. Blood Cancer J. 2021, 11, 190. [Google Scholar] [CrossRef]
- Hervé, A.-L.; Florence, M.; Philippe, M.; Michel, A.; Thierry, F.; Kenneth, A.; Jean-Luc, H.; Nikhil, M.; Stéphane, M. Molecular Heterogeneity of Multiple Myeloma: Pathogenesis, Prognosis, and Therapeutic Implications. J. Clin. Oncol. 2011, 29, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Tirier, S.M.; Mallm, J.-P.; Steiger, S.; Poos, A.M.; Awwad, M.H.S.; Giesen, N.; Casiraghi, N.; Susak, H.; Bauer, K.; Baumann, A.; et al. Subclone-specific microenvironmental impact and drug response in refractory multiple myeloma revealed by single-cell transcriptomics. Nat. Commun. 2021, 12, 6960. [Google Scholar] [CrossRef]
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A.; et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Sanders, M.A.; Brahemi, G.; Nangia-Makker, P.; Balan, V.; Morelli, M.; Kothayer, H.; Westwell, A.D.; Shekhar, M.P. Novel Inhibitors of Rad6 Ubiquitin Conjugating Enzyme: Design, Synthesis, Identification, and Functional Characterization. Mol. Cancer Ther. 2013, 12, 373–383. [Google Scholar] [CrossRef]
- Zhang, Z.; Kattan, M.W. Drawing Nomograms with R: Applications to categorical outcome and survival data. Ann. Transl. Med. 2017, 5, 211. [Google Scholar] [CrossRef] [PubMed]
- Blanche, P.; Dartigues, J.F.; Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 2013, 32, 5381–5397. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Strobl, C.; Malley, J.; Tutz, G. An introduction to recursive partitioning: Rationale, application, and characteristics of classification and regression trees, bagging, and random forests. Psychol. Methods 2009, 14, 323–348. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Training Cohort GSE9782 | Validation Cohort CoMMpass |
|---|---|---|
| N = 264 | N = 737 | |
| Age, years(median, range) | 61 (27–86) | 63(27–93) |
| <65 | 177 (67%) | 407 (55%) |
| ≥65 | 87 (33%) | 330 (45%) |
| Sex | ||
| Male | 159 (60%) | 438 (59%) |
| Female | 105 (40%) | 299 (41%) |
| Hemoglobin (g/L) | ||
| ≥100 | - | 454 (62%) |
| <100 | - | 283 (38%) |
| ALB (g/L) | ||
| ≥35 | 167 (63%) | 440 (60%) |
| <35 | 83 (31%) | 297 (40%) |
| β2-Mg (mg/L) | ||
| <5.5 | 136 (52%) | 503 (68%) |
| ≥5.5 | 68 (26%) | 169 (23%) |
| LDH (U/L) | ||
| <250 | - | 496 (67%) |
| ≥250 | - | 119 (16%) |
| ISS | ||
| stage-I | 69(26%) | 246 (33%) |
| stage-II | 65(25%) | 261 (35%) |
| stage-III | 68(26%) | 209 (28%) |
| Cytogenetics | ||
| del (17p) | - | 70 (9.5%) |
| t (4; 14) | - | 69 (9.4%) |
| t (4; 16) | - | 23 (3.1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, L.; Xu, B.; Xu, J.; Li, J.; Jiang, J.; Ren, Y.; Liu, P. A Machine Learning Model to Predict Survival and Therapeutic Responses in Multiple Myeloma. Int. J. Mol. Sci. 2023, 24, 6683. https://doi.org/10.3390/ijms24076683
Ren L, Xu B, Xu J, Li J, Jiang J, Ren Y, Liu P. A Machine Learning Model to Predict Survival and Therapeutic Responses in Multiple Myeloma. International Journal of Molecular Sciences. 2023; 24(7):6683. https://doi.org/10.3390/ijms24076683
Chicago/Turabian StyleRen, Liang, Bei Xu, Jiadai Xu, Jing Li, Jifeng Jiang, Yuhong Ren, and Peng Liu. 2023. "A Machine Learning Model to Predict Survival and Therapeutic Responses in Multiple Myeloma" International Journal of Molecular Sciences 24, no. 7: 6683. https://doi.org/10.3390/ijms24076683
APA StyleRen, L., Xu, B., Xu, J., Li, J., Jiang, J., Ren, Y., & Liu, P. (2023). A Machine Learning Model to Predict Survival and Therapeutic Responses in Multiple Myeloma. International Journal of Molecular Sciences, 24(7), 6683. https://doi.org/10.3390/ijms24076683