

Next Generation of Ovarian Cancer Detection Using Aptamers
 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Results
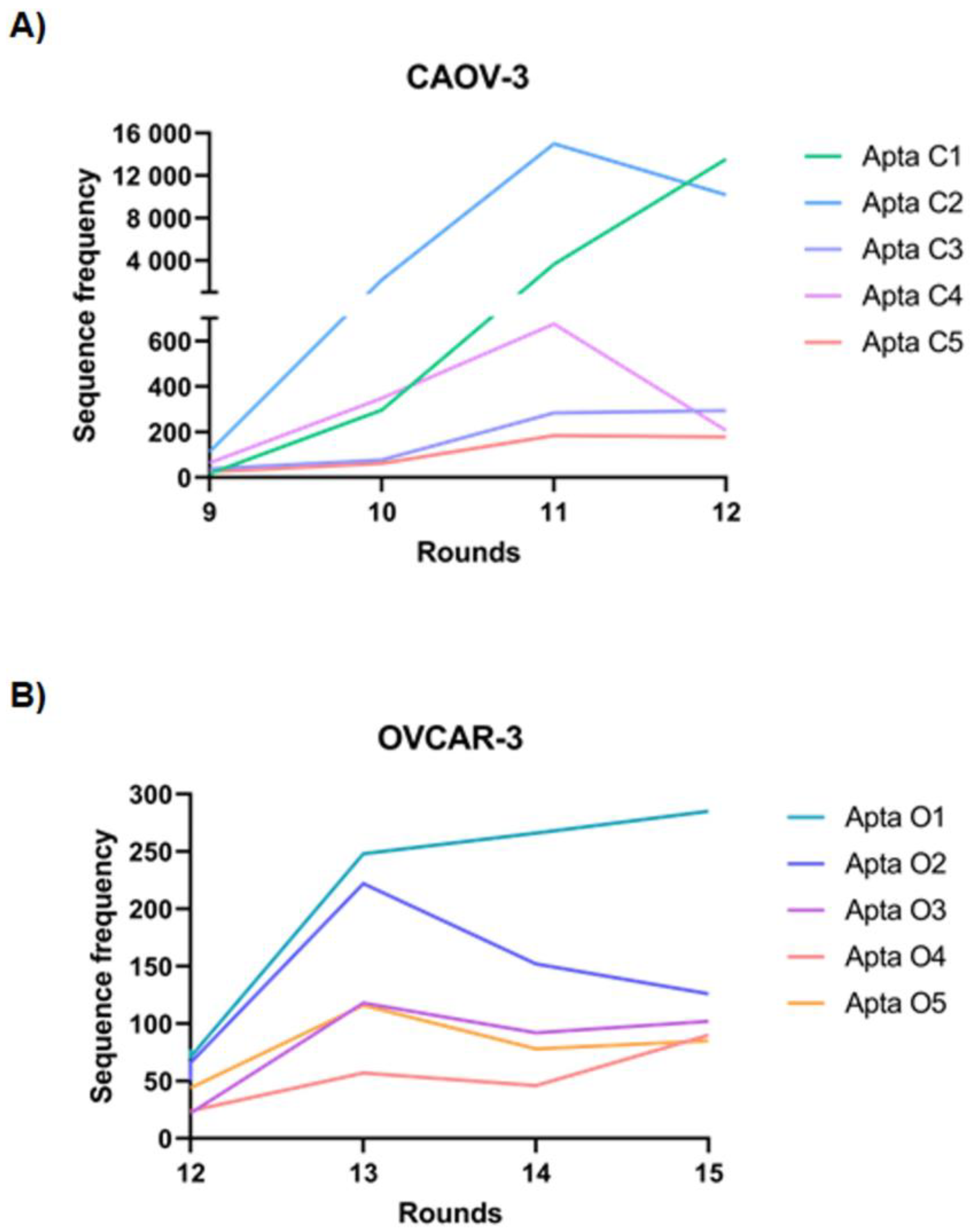
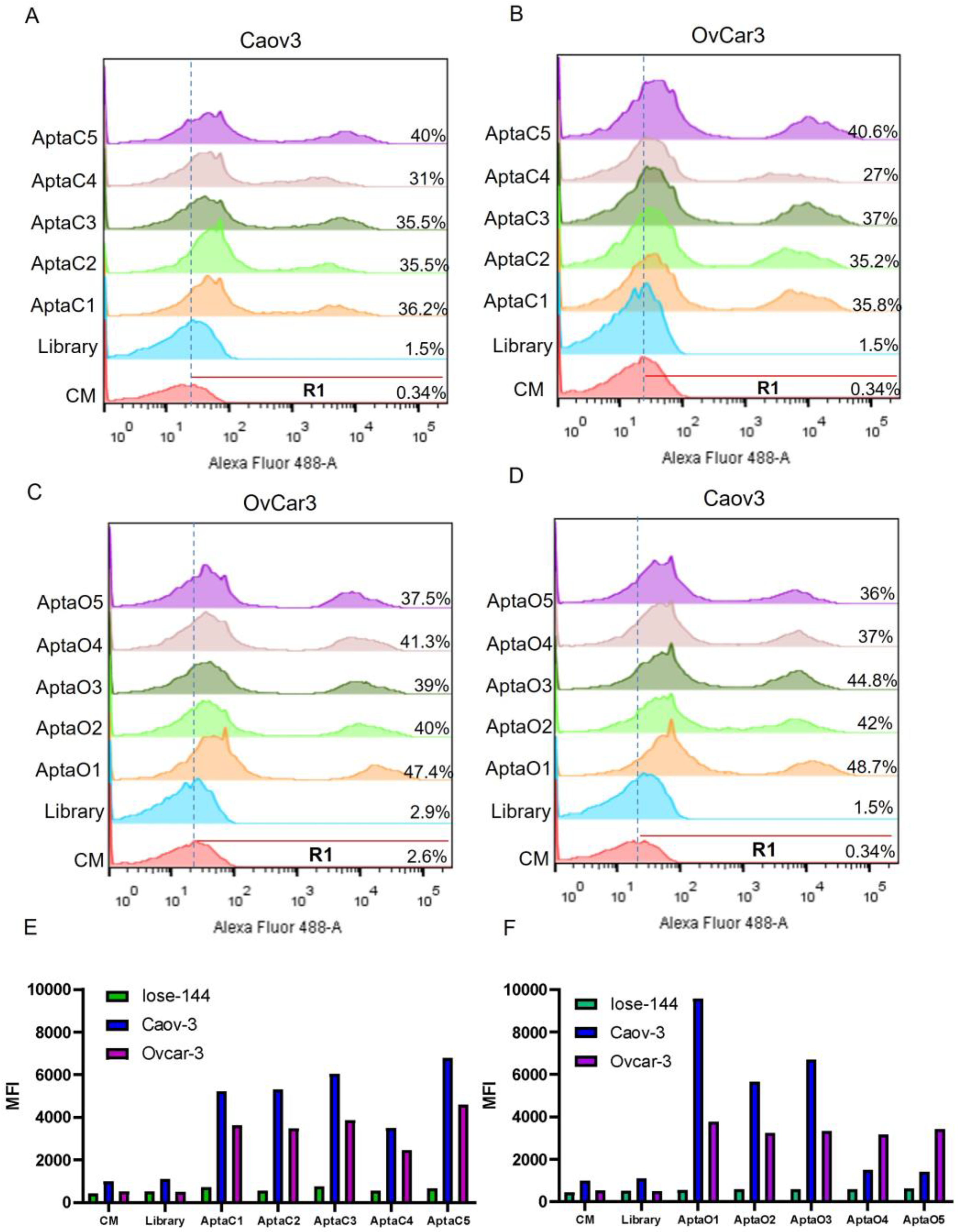
2.1. Selection and Identification of Tumor-Specific Aptamers for the Caov-3 and OvCar-3 Cell Lines
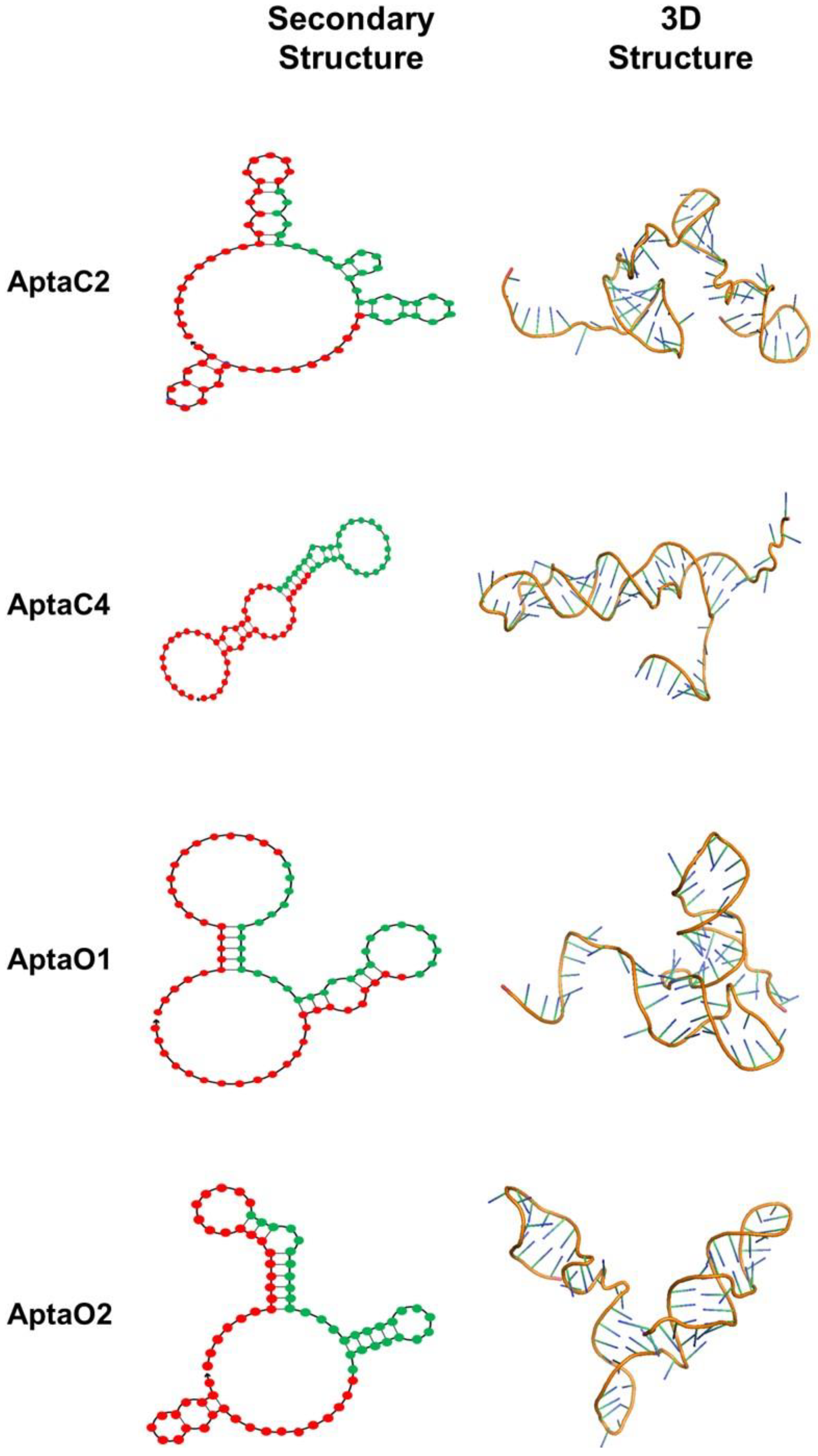
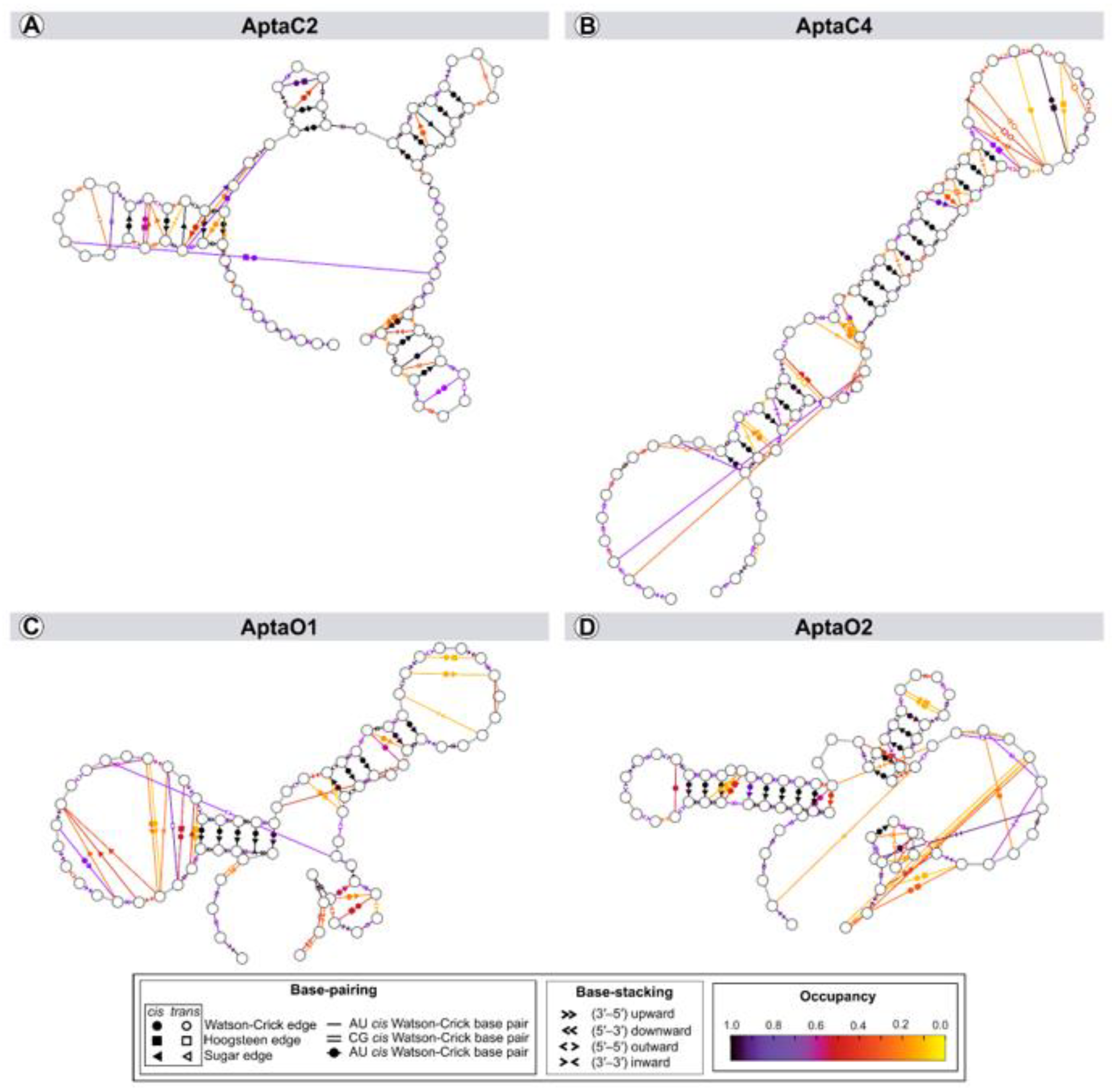
2.2. Structural Characterization of Selected Aptamers for Caov-3 and OvCar-3 Cells
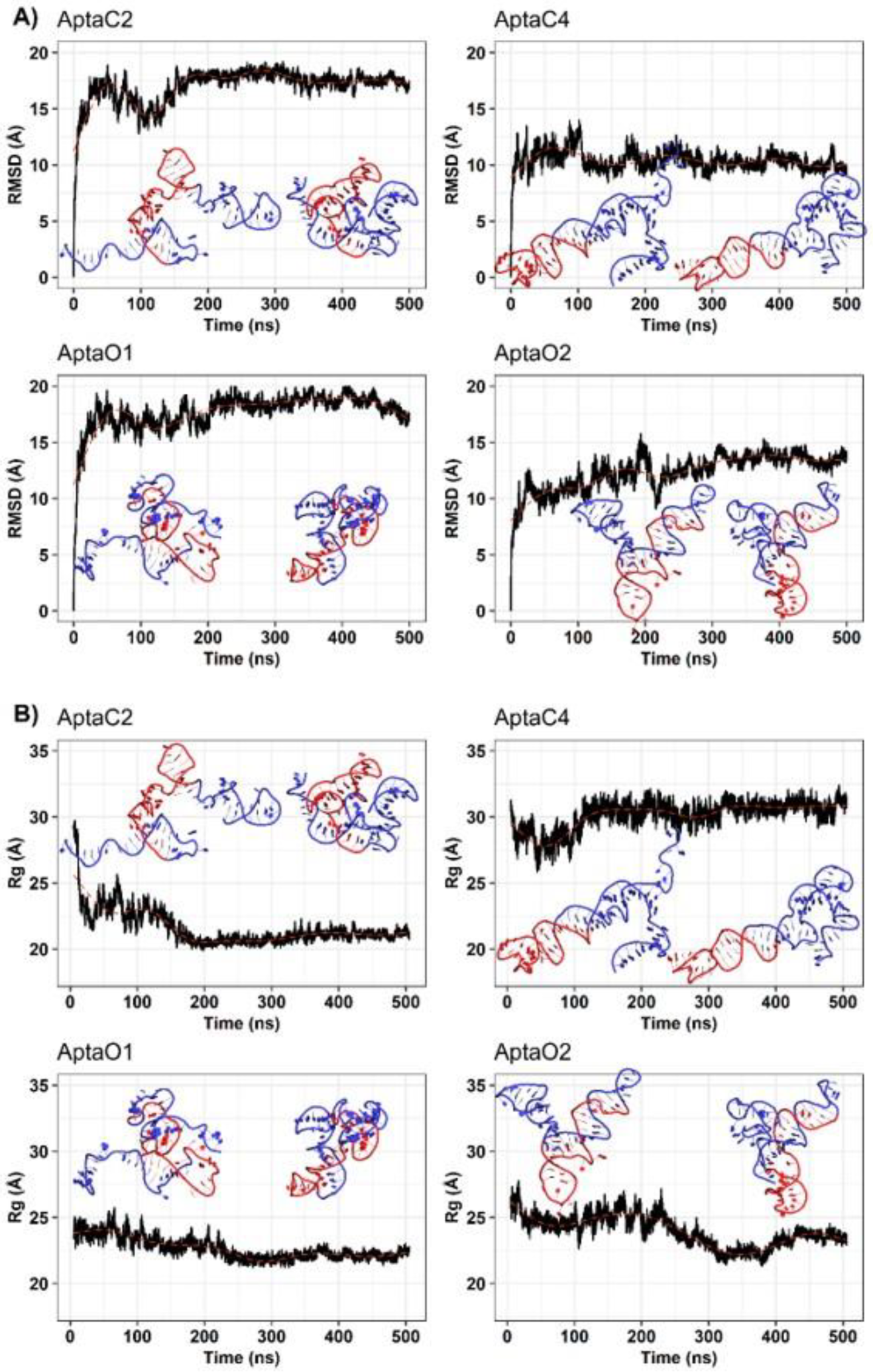
2.3. Characterization of the Three-Dimensional Structure Stability of Selected Aptamers for CaoV-3 and OvCar-3 Cells by Molecular Dynamics
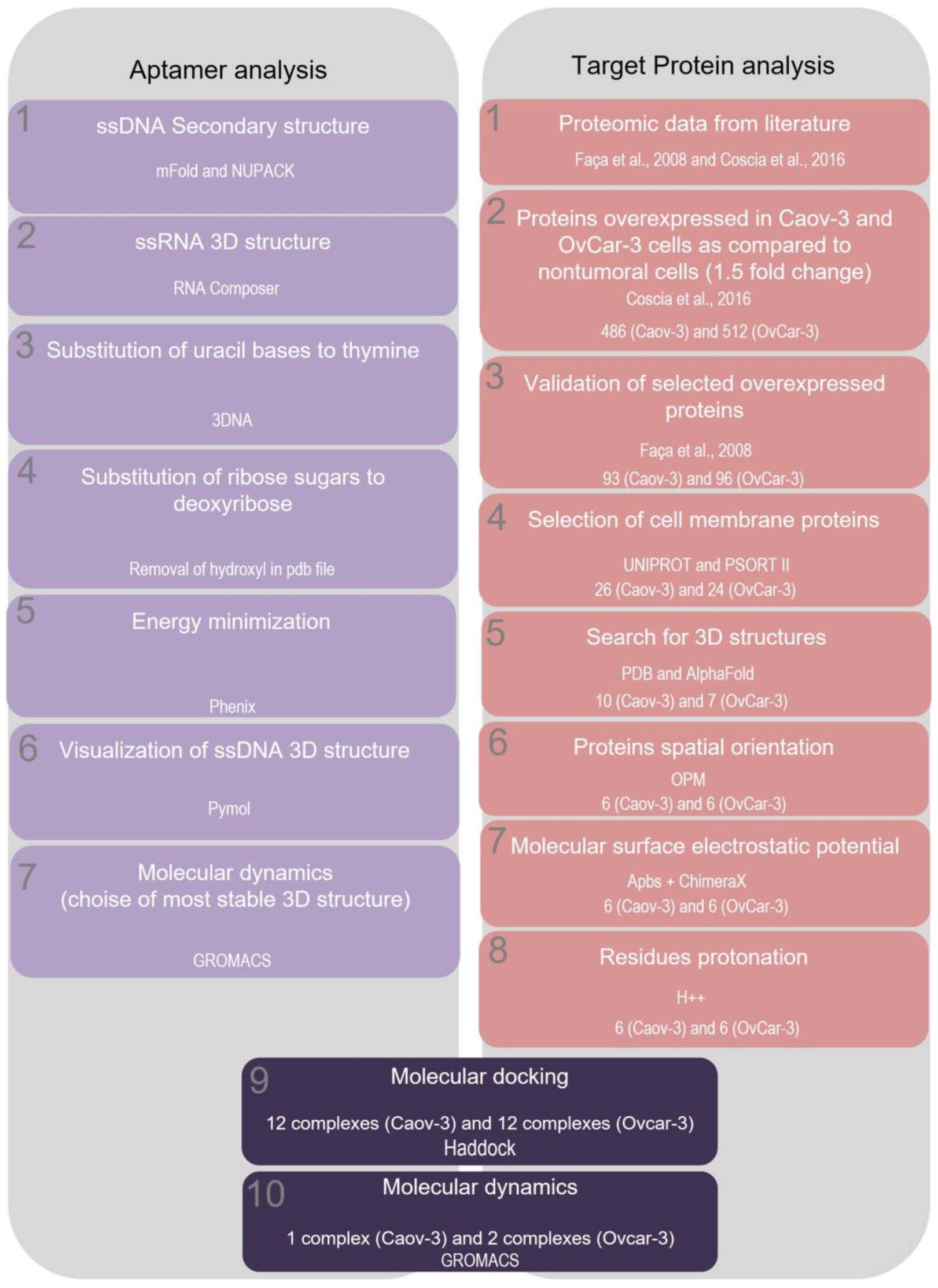
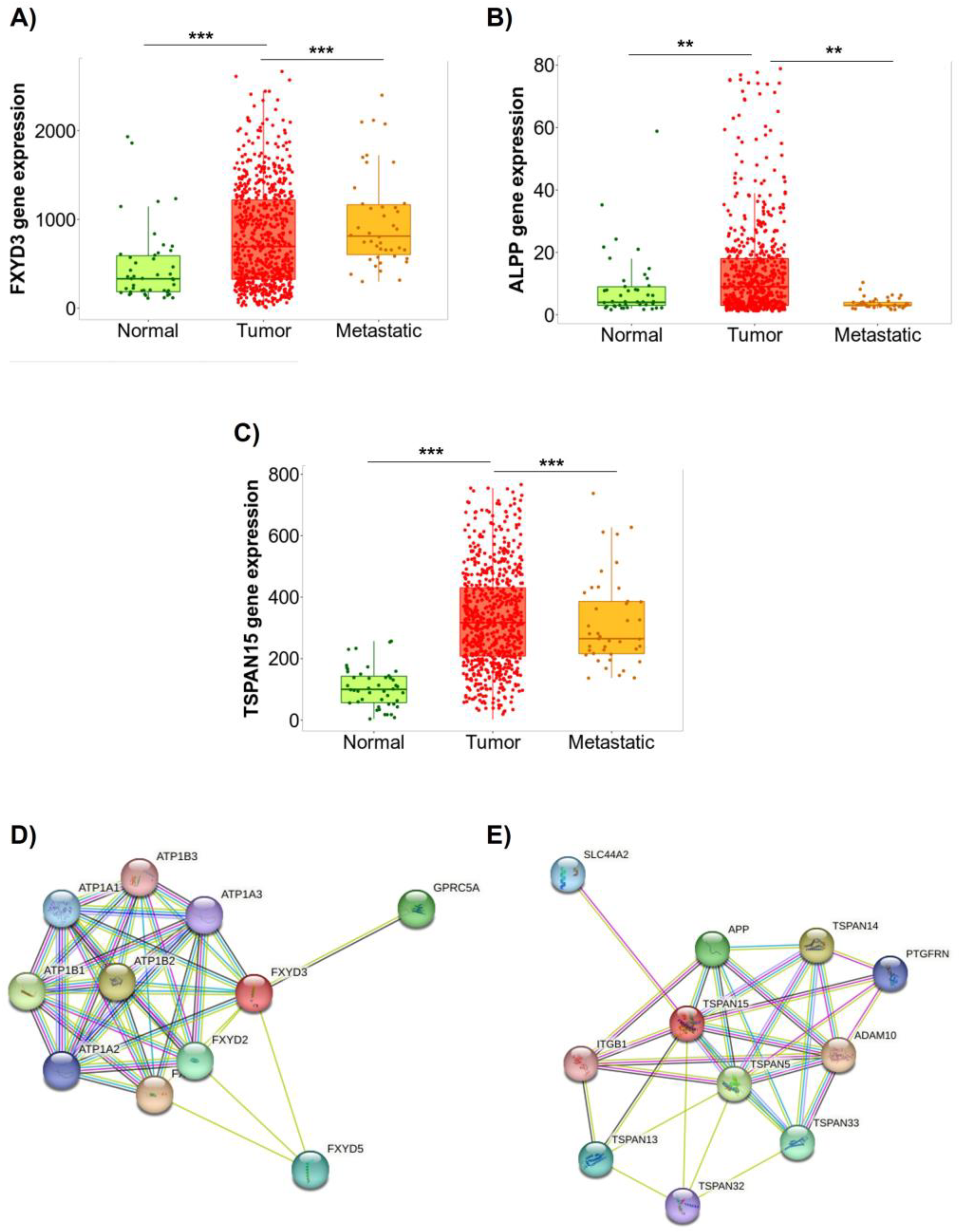
2.4. Selection of Potential Aptamer Targets in Caov-3 and OvCar-3 Cell Lines
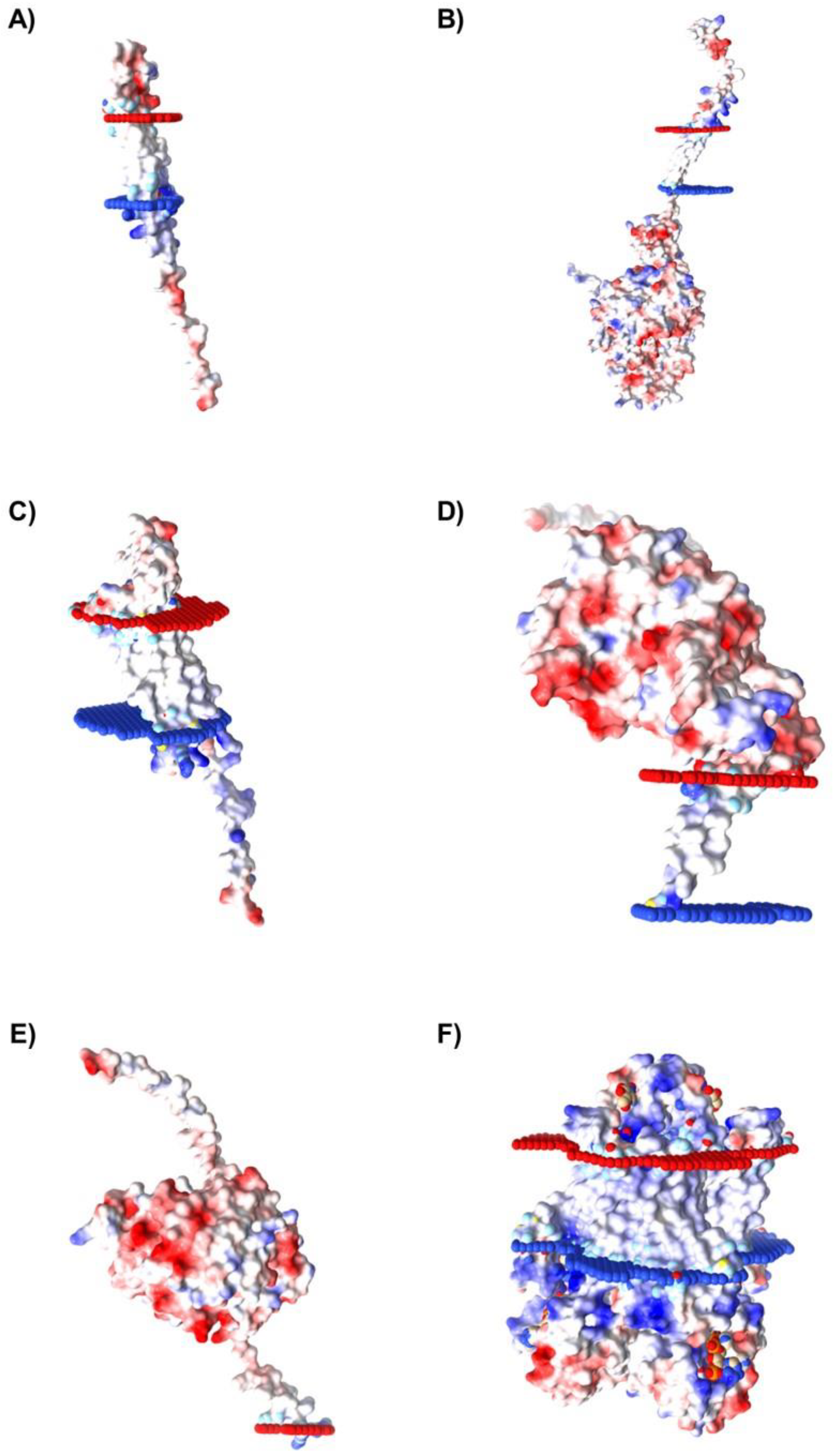
2.5. Characterization of Spatial Orientation and Electrostatic Potential of Membrane Proteins Selected for Caov-3 and OvCar-3 Cells
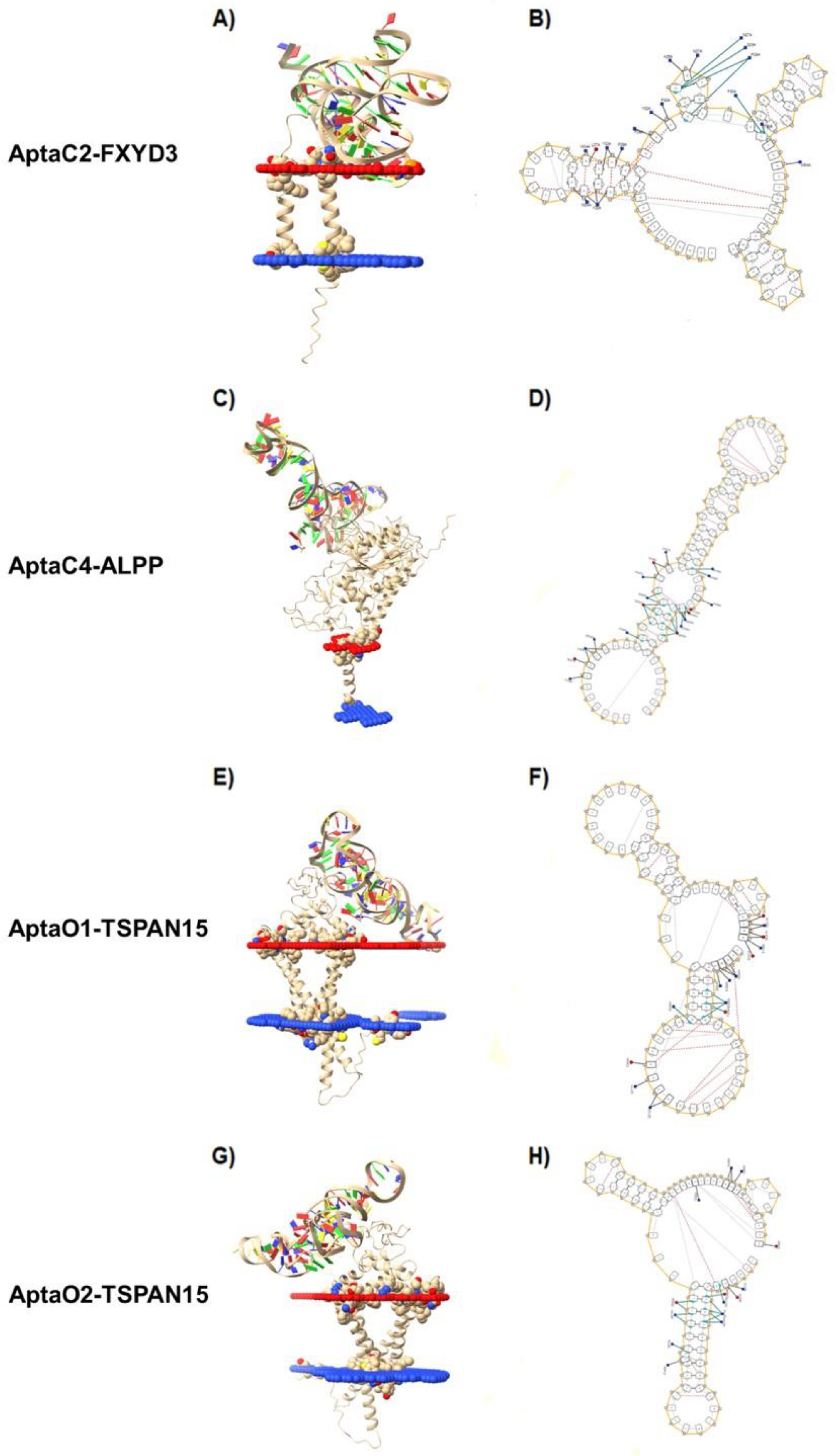
2.6. Characterization of Protein-Aptamer Complexes by Molecular Docking
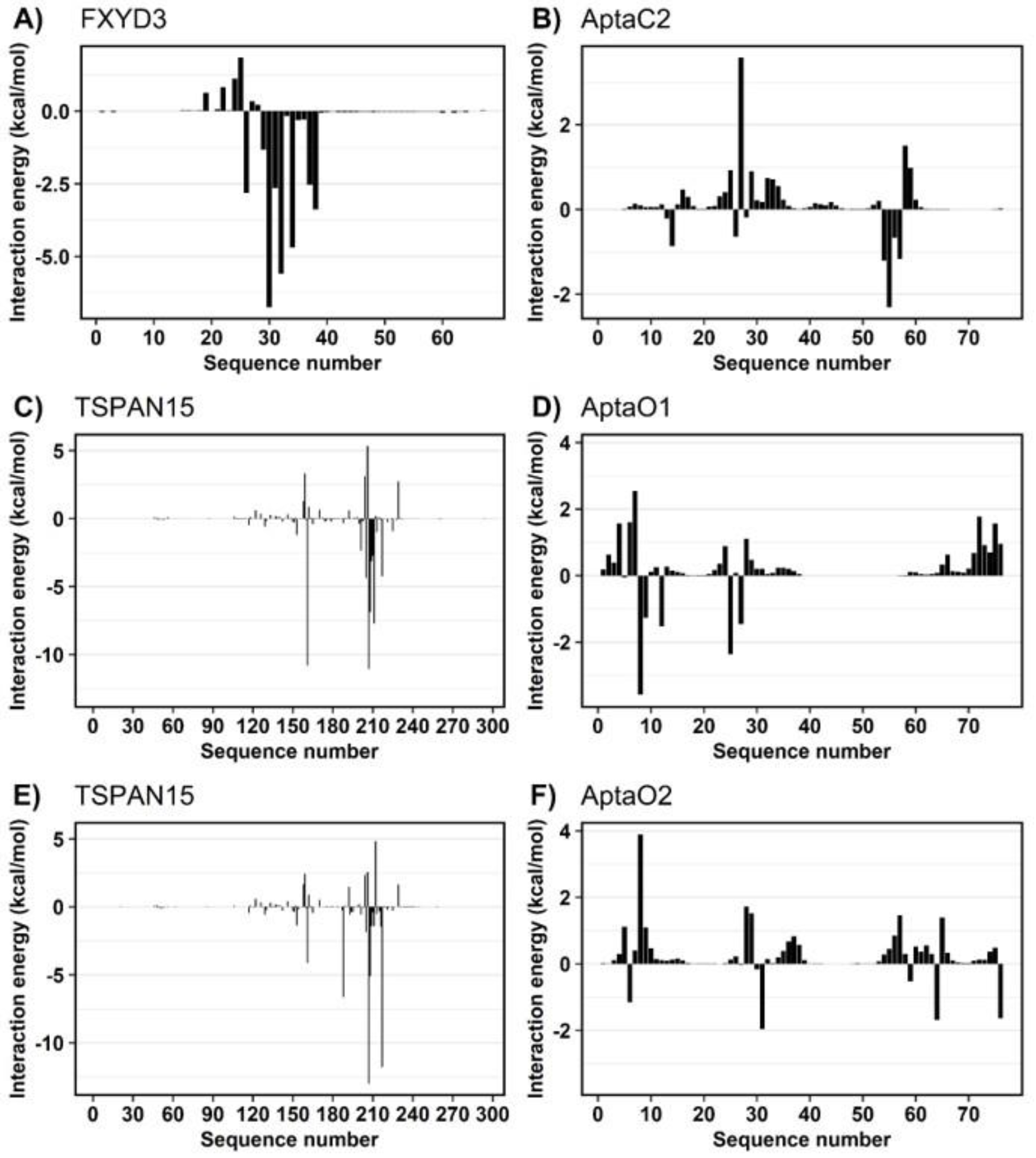
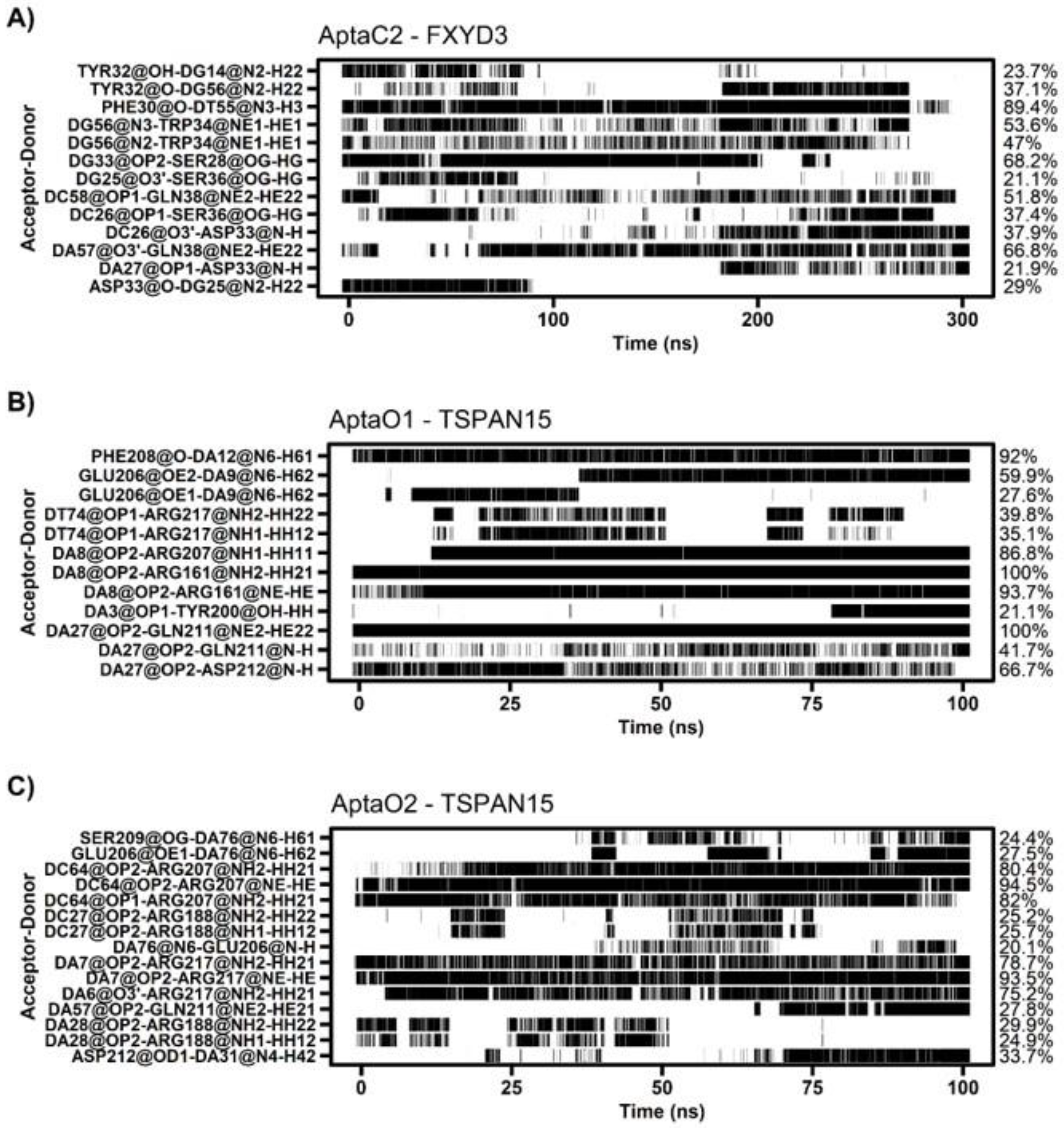
2.7. Characterization of Protein-Aptamer Complex Stability by Molecular Dynamics
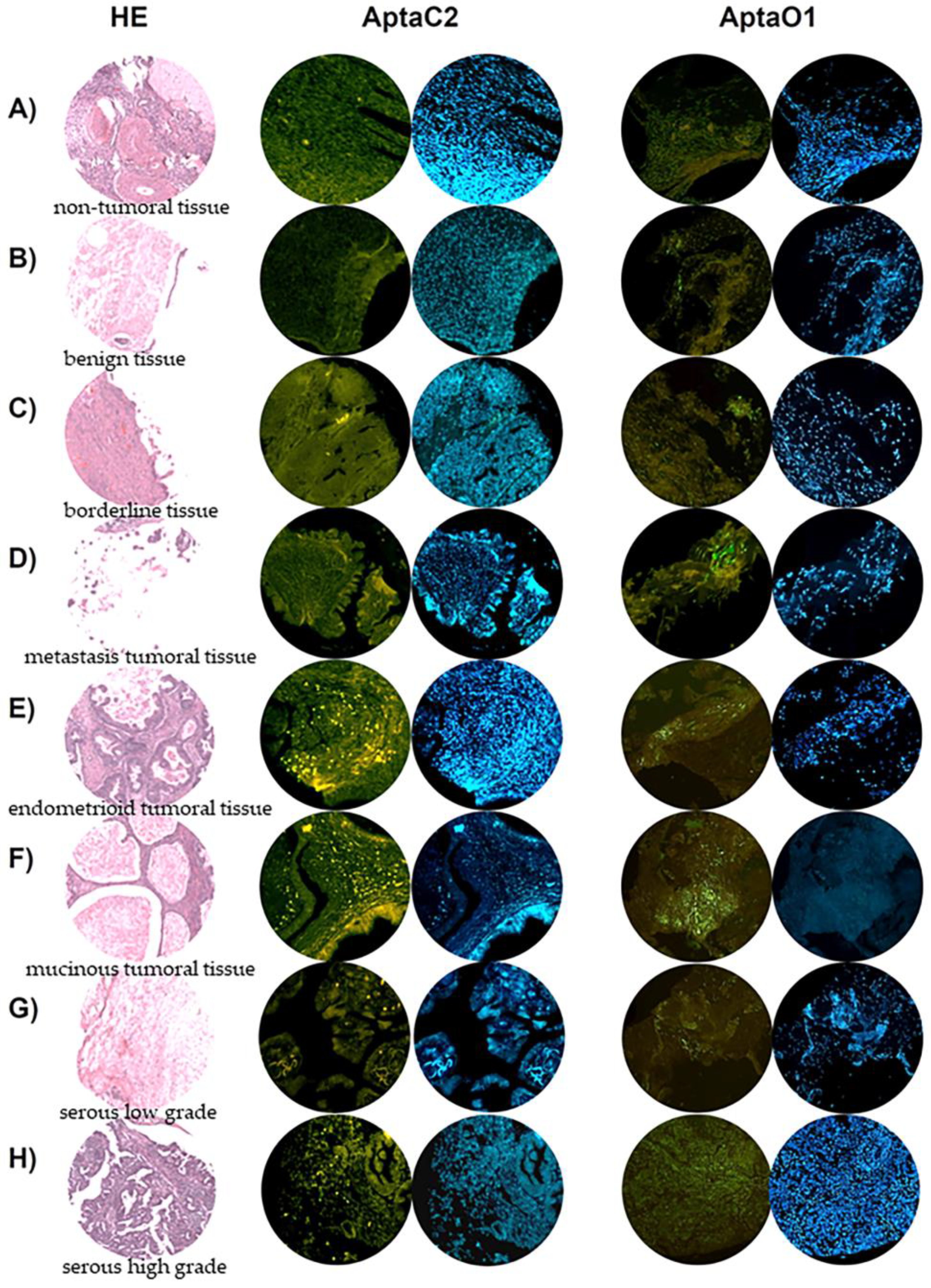
2.8. AptaC2 and AptaO1 Detect Human Ovarian Tumors from Different Subtypes
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell-SELEX
4.3. Identification and Analysis of Selected Aptamer Sequences for Caov-3 and OvCar-3 Cells
4.4. Evaluation of Specificity of the Selected Aptamers
4.5. Three-Dimensional Structure Characterization of the Identified Aptamers
4.6. Identification of Target Proteins Potentially Recognized by Tumor-Specific Aptamers
4.7. Three-Dimensional Structure of the Selected Target Molecules
4.8. Identification of Transmembrane Domains in the Selected Proteins
4.9. Evaluation of the Electrostatic Potential of Selected Proteins
4.10. Molecular Docking
4.11. Analysis of Differentially Expressed Gene in Ovarian Cancer
4.12. Molecular Dynamics
4.13. Tissue Microarray
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Women’s Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Coburn, S.B.; Bray, F.; Sherman, M.E.; Trabert, B. International patterns and trends in ovarian cancer incidence, overall and by histologic subtype. Int. J. Cancer 2017, 140, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Stavnes, H.T.; Holth, A.; Chen, X.; Yang, Y.; Shih, I.-M.; Wang, T.-L. Gene expression signatures differentiate ovarian/peritoneal serous carcinoma from breast carcinoma in effusions. J. Cell. Mol. Med. 2011, 15, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Mrochem, J.; Sodowski, K.; Deja, R.; Walaszek-Gruszka, A.; Wojcieszek, A.; Kołosza, Z.; Chmura, A.; Czernik, E.; Masłyk, B.; Bartnik, W.; et al. Evaluation of selected serum protein markers as early detectors of ovarian cancer. Ginekol. Pol. 2008, 79, 271–275. [Google Scholar]
- Meden, H.; Fattahi-Meibodi, A. CA 125 in benign gynecological conditions. Int. J. Biol. Mark. 1998, 13, 231–237. [Google Scholar] [CrossRef]
- Topalak, O.; Saygili, U.; Soyturk, M.; Karaca, N.; Batur, Y.; Uslu, T.; Erten, O. Serum, pleural effusion, and ascites CA-125 levels in ovarian cancer and nonovarian benign and malignant diseases: A comparative study. Gynecol. Oncol. 2002, 85, 108–113. [Google Scholar] [CrossRef]
- Germer, C.T.; Mann, B. Robotik: Innovation in der minimal-invasiven Chirurgie [Robotics: Innovation in minimally invasive surgery]. Chirurgie 2013, 84, 633–634. [Google Scholar] [CrossRef]
- Faria, M.; Ulrich, H. The use of synthetic oligonucleotides as protein inhibitors and anticode drugs in cancer therapy: Accomplishments and limitations. Curr. Cancer Drug Targets 2002, 2, 355–368. [Google Scholar] [CrossRef]
- Mairal, T.; Özalp, V.C.; Lozano, S.P.; Mir, M.; Katakis, I.; O’Sullivan, C.K. Aptamers: Molecular tools for analytical applications. Anal. Bioanal. Chem. 2008, 390, 989–1007. [Google Scholar] [CrossRef]
- Que-Gewirth, N.S.; Sullenger, B.A. Gene therapy progress and prospects: RNA aptamers. Gene Ther. 2007, 14, 283–291. [Google Scholar] [CrossRef]
- Ohuchi, S. Cell-SELEX Technology. BioRes. Open Access 2012, 1, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Bonvin, A.M. 3D-DART: A DNA structure modelling server. Nucleic Acids Res. 2009, 37, W235–W239. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; Pastor, F.; Villanueva, H.; Soldevilla, M.M.; Hervas-Stubbs, S.; Oyarzabal, J. In Silico Aptamer Docking Studies: From a Retrospective Validation to a Prospective Case Study-TIM3 Aptamers Binding. Mol. Ther. Nucleic Acids 2016, 5, e376. [Google Scholar] [CrossRef]
- Jeddi, I.; Saiz, L. Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Faça, V.M.; Ventura, A.P.; Fitzgibbon, M.P.; Pereira-Faça, S.R.; Pitteri, S.J.; Green, A.E.; Ireton, R.C.; Zhang, Q.; Wang, H.; O’Briant, K.C.; et al. Proteomic Analysis of Ovarian Cancer Cells Reveals Dynamic Processes of Protein Secretion and Shedding of Extra-Cellular Domains. PLoS ONE 2008, 3, e2425. [Google Scholar] [CrossRef]
- Memorial Sloan Kettering Cancer Center (MSKCC). Available online: https://www.mskcc.org/researchadvantage/support/technology/tangible-material/caov-3-human-ovarian-cell-line (accessed on 16 October 2022).
- Hamilton, T.C.; Young, R.C.; McKoy, W.M.; Grotzinger, K.R.; Green, J.A.; Chu, E.W.; Whang-Peng, J.; Rogan, A.M.; Green, W.R.; Ozols, R.F. Characterization of a human ovarian carcinoma cell line (NIH:OVCAR-3) with androgen and estrogen receptors. Cancer Res. 1983, 43, 5379–5389. [Google Scholar]
- Li, P.; Maines-Bandiera, S.L.; Collins, C.; Gray, J.W.; Auersperg, N. Immortalization of human ovarian epithelial cells (OSE) by overexpression of the ZNF217 gene. Cancer Res. 2004, 64, 414. [Google Scholar]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Buglak, A.A.; Samokhvalov, A.V.; Zherdev, A.V.; Dzantiev, B.B. Methods and Applications of In Silico Aptamer Design and Modeling. Int. J. Mol. Sci. 2020, 21, 8420. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, L.; Zhang, B.-T.; Lu, A.; Wang, Y.; Yu, Y.; Zhang, G. Artificial Intelligence in Aptamer–Target Binding Prediction. Int. J. Mol. Sci. 2021, 22, 3605. [Google Scholar] [CrossRef] [PubMed]
- D’haeseleer, P. What are DNA sequence motifs? Nat. Biotechnol. 2006, 24, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, J.N.; Steenberg, C.D.; Bois, J.S.; Wolfe, B.R.; Pierce, M.B.; Khan, A.R.; Dirks, R.M.; Pierce, N.A. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 2011, 32, 170–173. [Google Scholar] [CrossRef]
- Sakamoto, T.; Ennifar, E.; Nakamura, Y. Thermodynamic study of aptamers binding to their target proteins. Biochimie 2018, 145, 91–97. [Google Scholar] [CrossRef]
- Lai, Y.T.; De Stefano, J.J. DNA aptamers to human immunodeficiency virus reverse transcriptase selected by a primer-free SELEX method: Characterization and comparison with other aptamers. Nucleic Acid Ther. 2012, 22, 162–176. [Google Scholar] [CrossRef]
- von Heijne, G. Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 2006, 7, 909–918. [Google Scholar] [CrossRef]
- PSORT II Localization Features. Available online: https://wolfpsort.hgc.jp/WoLFPSORTdoc/almDescription.html (accessed on 3 August 2022).
- Tsuchiya, Y.; Kinoshita, K.; Nakamura, H. Structure-based prediction of DNA-binding sites on proteins Using the empirical preference of electrostatic potential and the shape of molecular surfaces. Proteins 2004, 55, 885–894. [Google Scholar] [CrossRef]
- Jones, S.; Shanahan, H.P.; Berman, H.M.; Thornton, J.M. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 2003, 31, 7189–7198. [Google Scholar] [CrossRef]
- Morrison, B.W.; Moorman, J.R.; Kowdley, G.C.; Kobayashi, Y.M.; Jones, L.R.; Leder, P. Mat-8, a novel phospholemman-like protein expressed in human breast tumors, induces a chloride conductance in Xenopus oocytes. J. Biol. Chem. 1995, 270, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Mukaisho, K.; Sugihara, H.; Hattori, T.; Asano, S. Down-regulation of FXYD3 is induced by transforming growth factor-β signaling via ZEB1/δEF1 in human mammary epithelial cells. Biol. Pharm. Bull. 2011, 34, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pang, S.T.; Kasper, K.A.; Luan, C.; Wondergem, B.; Lin, F.; Chuang, C.K.; Teh, B.T.; Yang, X.J. FXYD3: A Promising Biomarker for Urothelial Carcinoma. Biomark Insights 2011, 6, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, X.; Xu, S.; Ge, J.; Liu, J.; Li, L.; Fang, G.; Meng, Y.; Zhang, H.; Sun, X. Expression and clinical significance of FXYD3 in endometrial cancer. Oncol. Lett. 2014, 8, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Lai, L.; Lian, W.; Tu, X.; Zhou, J.; Dong, P.; Su, D.; Wang, X.; Cao, X.; Chen, Y.; et al. SOX9/FXYD3/Src Axis Is Critical for ER+ Breast Cancer Stem Cell Function. Mol. Cancer Res. 2019, 17, 238–249. [Google Scholar] [CrossRef]
- Jian-Hua, T.; Xiao-Jie, Z.; Yi-Dan, W.; Wen-Kai, L.; Wang-Jiao, H.; Cheng-Hong, W.; Gui-Yuan, L. Effect of glycosylphosphatidylinositol specific phospholipase D gene expression levels on complement mediated killing of leukemic cells in patients with chronic myeloid leukemia. Clin. Chim. Acta 2005, 359, 115–124. [Google Scholar] [CrossRef]
- Dua, P.; Kang, H.S.; Hong, S.M.; Tsao, M.S.; Kim, S.; Lee, D.K. Alkaline phosphatase ALPPL-2 is a novel pancreatic carcinoma-associated protein. Cancer Res. 2013, 73, 1934–1945. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Z.; Li, L.; Qin, Y.R.; Liu, H.; Jiang, C.; Zeng, T.T.; Li, M.Q.; Xie, D.; Li, Y.; et al. TSPAN15 interacts with BTRC to promote oesophageal squamous cell carcinoma metastasis via activating NF-κB signaling. Nat. Commun. 2018, 9, 1423. [Google Scholar] [CrossRef]
- Hiroshima, K.; Shiiba, M.; Oka, N.; Hayashi, F.; Ishida, S.; Fukushima, R.; Koike, K.; Iyoda, M.; Nakashima, D.; Tanzawa, H.; et al. Tspan15 plays a crucial role in metastasis in oral squamous cell carcinoma. Exp. Cell Res. 2019, 384, 111622. [Google Scholar] [CrossRef]
- Sidahmed-Adrar, N.; Ottavi, J.F.; Benzoubir, N.; Ait Saadi, T.; Bou Saleh, M.; Mauduit, P.; Guettier, C.; Desterke, C.; Le Naour, F. Tspan15 Is a New Stemness-Related Marker in Hepatocellular Carcinoma. Proteomics 2019, 19, e1900025. [Google Scholar] [CrossRef]
- Huang, R.; Sun, H.; Lin, R.; Zhang, J.; Yin, H.; Xian, S.; Li, M.; Wang, S.; Li, Z.; Qiao, Y.; et al. The role of tetraspanins pan-cancer. iScience 2022, 25, 104777. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.S. Desenvolvimento de Aptâmeros Como Agentes Antitumorais. Ph.D. Thesis, Programa de Biologia Celular e Molecular-Instituto Oswaldo Cruz/Fiocruz, Rio de Janeiro, Brazil, 2018. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 September 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Seqtk. Available online: https://github.com/lh3/seqtk (accessed on 28 September 2022).
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Klein, P.; Kanehisa, M.; Delsi, C. The detection and classification of membrane spanning proteins. Biochem. Biophys. Acta 1985, 815, 468–476. [Google Scholar] [CrossRef]
- Hartmann, E.; Rapoport, T.A.; Lodish, H.F. Predicting the orientation of eukaryotic membrane-spanning proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 5786–5790. [Google Scholar] [CrossRef]
- Nakai, K.; Horton, P. PSORT: A program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 1999, 24, 34–36. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Fraczkiewicz, R.; Braun, W. Exact and Efficient Analytical Calculation of the Accessible Surface Areas and Their Gradients for Macromolecules. J. Comput. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; Mcgee, T.D.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; et al. Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery. J. Chem. Inf. Model. 2020, 620, 5595–5623. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Bottaro, S.; Bussi, G.; Pinamonti, G.; Reiber, S.; Boomsma, W.; Lindorff-Larsen, K. Barnaba: Software for Analysis of Nucleic Acid Structures and Trajectories. Rna 2019, 25, 219–231. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AptaC2 | AptaC4 | |||||||
|---|---|---|---|---|---|---|---|---|
| Cluster | HADDOCK Score | Cluster Size | RMSD | Cluster | HADDOCK Score | Cluster Size | RMSD | |
| ALPP | Cluster 5 | −21.9 +/− 30.2 | 5 | 0.7 +/− 0.4 | Cluster 3 | −27.6 +/− 9.6 | 7 | 1.0 +/− 0.9 |
| CLDN7 | Cluster 2 | −2.7 +/− 17.9 | 20 | 0.8 +/− 0.5 | Cluster 11 | 25.7 +/− 12.5 | 4 | 23.6 +/− 0.5 |
| FXYD3 | Cluster 3 | −40.7 +/− 10.5 | 11 | 1.4 +/− 0.8 | Cluster 6 | 33.9 +/− 12.1 | 12 | 1.4 +/− 0.9 |
| ITGB2 | Cluster 4 | −24.9 +/− 2.6 | 17 | 10.9 +/− 0.2 | Cluster 5 | 21.8 +/− 6.3 | 9 | 22.9 +/− 0.2 |
| SMPDL3B | Cluster 12 | −18.8 +/− 7.3 | 5 | 18.6 +/− 0.1 | Cluster 4 | 15.0 +/− 5.5 | 11 | 13.2 +/− 0.8 |
| STEAP4 | Cluster 9 | −49.4 +/− 20.1 | 7 | 9.4 +/− 0.1 | Cluster 4 | −10.8 +/− 9.3 | 12 | 2.2 +/− 1.8 |
| AptaO1 | AptaO2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Cluster | HADDOCK Score | Cluster Size | RMSD | Cluster | HADDOCK Score | Cluster Size | RMSD | |
| CD47 | Cluster 10 | 5.2 +/− 12.8 | 8 | 5.5 +/− 0.1 | Cluster 11 | −8.9 +/− 20.8 | 5 | 3.2 +/− 2.3 |
| CLDN6 | Cluster 9 | 4.3 +/− 2.9 | 6 | 19.2 +/− 0.3 | Cluster 2 | 14.9 +/− 5.7 | 12 | 15.6 +/− 0.2 |
| CLDN7 | Cluster 1 | 2.3 +/− 9.7 | 13 | 14.9 +/− 0.2 | Cluster 1 | 3.6 +/− 9.2 | 14 | 1.5 +/− 1.1 |
| EPHA1 | Cluster 1 | 8.5 +/− 4.1 | 26 | 5.9 +/− 0.1 | Cluster 4 | 18.9 +/− 3.4 | 9 | 15.5 +/− 0.2 |
| TSPAN15 | Cluster 4 | −36.8 +/− 6.9 | 13 | 13.9 +/− 0.3 | Cluster 1 | −11.5 +/− 17.7 | 26 | 25.5 +/− 0.1 |
| UPK1B | Cluster 11 | 6.0 +/− 17.4 | 6 | 1.1 +/− 0.9 | Cluster 2 | 19.1 +/− 7.1 | 14 | 2.1 +/− 1.2 |
| Items | AptaC2—FXYD3 | AptaO1—TSPAN15 | AptaO2—TSPAN15 |
|---|---|---|---|
| ΔEvdw | −75.93 ± 0.31 | −80.99 ± 0.53 | −80.46 ± 0.41 |
| ΔEele | 1579.04 ± 6.56 | 1627.33 ± 11.74 | 1498.08 ± 7.76 |
| ΔGegb | −1510.61 ± 6.32 | −1563.69 ± 11.16 | −1422.75 ± 7.51 |
| ΔGesurf | −8.36 ± 0.03 | −10.03 ± 0.06 | −10.13 ± 0.04 |
| ΔGele+egb a | 68.43 ± 6.44 | 63.64 ± 11.45 | 75.33 ± 7.63 |
| ΔGbind b | −15.86 ± 0.39 | −27.39 ± 0.50 | −15.27 ± 0.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abreu, R.d.S.; Antunes, D.; Moreira, A.d.S.; Passetti, F.; Mendonça, J.B.; de Araújo, N.S.; Sassaro, T.F.; Alberto, A.V.P.; Carrossini, N.; Fernandes, P.V.; et al. Next Generation of Ovarian Cancer Detection Using Aptamers. Int. J. Mol. Sci. 2023, 24, 6315. https://doi.org/10.3390/ijms24076315
Abreu RdS, Antunes D, Moreira AdS, Passetti F, Mendonça JB, de Araújo NS, Sassaro TF, Alberto AVP, Carrossini N, Fernandes PV, et al. Next Generation of Ovarian Cancer Detection Using Aptamers. International Journal of Molecular Sciences. 2023; 24(7):6315. https://doi.org/10.3390/ijms24076315
Chicago/Turabian StyleAbreu, Rayane da Silva, Deborah Antunes, Aline dos Santos Moreira, Fabio Passetti, Julia Badaró Mendonça, Natássia Silva de Araújo, Tayanne Felippe Sassaro, Anael Viana Pinto Alberto, Nina Carrossini, Priscila Valverde Fernandes, and et al. 2023. "Next Generation of Ovarian Cancer Detection Using Aptamers" International Journal of Molecular Sciences 24, no. 7: 6315. https://doi.org/10.3390/ijms24076315
APA StyleAbreu, R. d. S., Antunes, D., Moreira, A. d. S., Passetti, F., Mendonça, J. B., de Araújo, N. S., Sassaro, T. F., Alberto, A. V. P., Carrossini, N., Fernandes, P. V., Costa, M. A., Guimarães, A. C. R., Degrave, W. M. S., & Waghabi, M. C. (2023). Next Generation of Ovarian Cancer Detection Using Aptamers. International Journal of Molecular Sciences, 24(7), 6315. https://doi.org/10.3390/ijms24076315