
Chitinase Signature in the Plasticity of Neurodegenerative Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
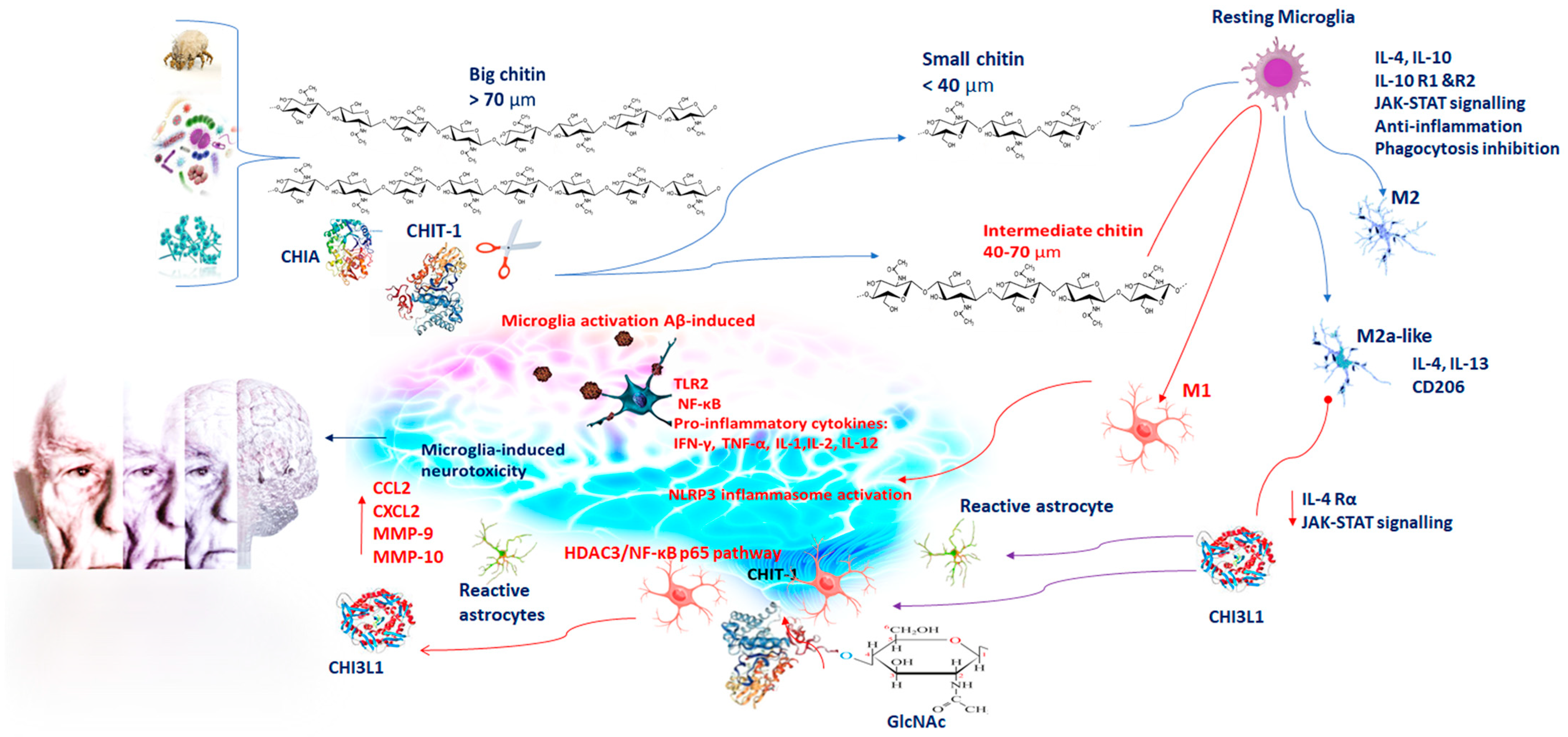
2. Chitin and Chitinase Induction in Alzheimer’s Disease (AD)
3. Chitinases and Activated Microglia in Neuroinflammation
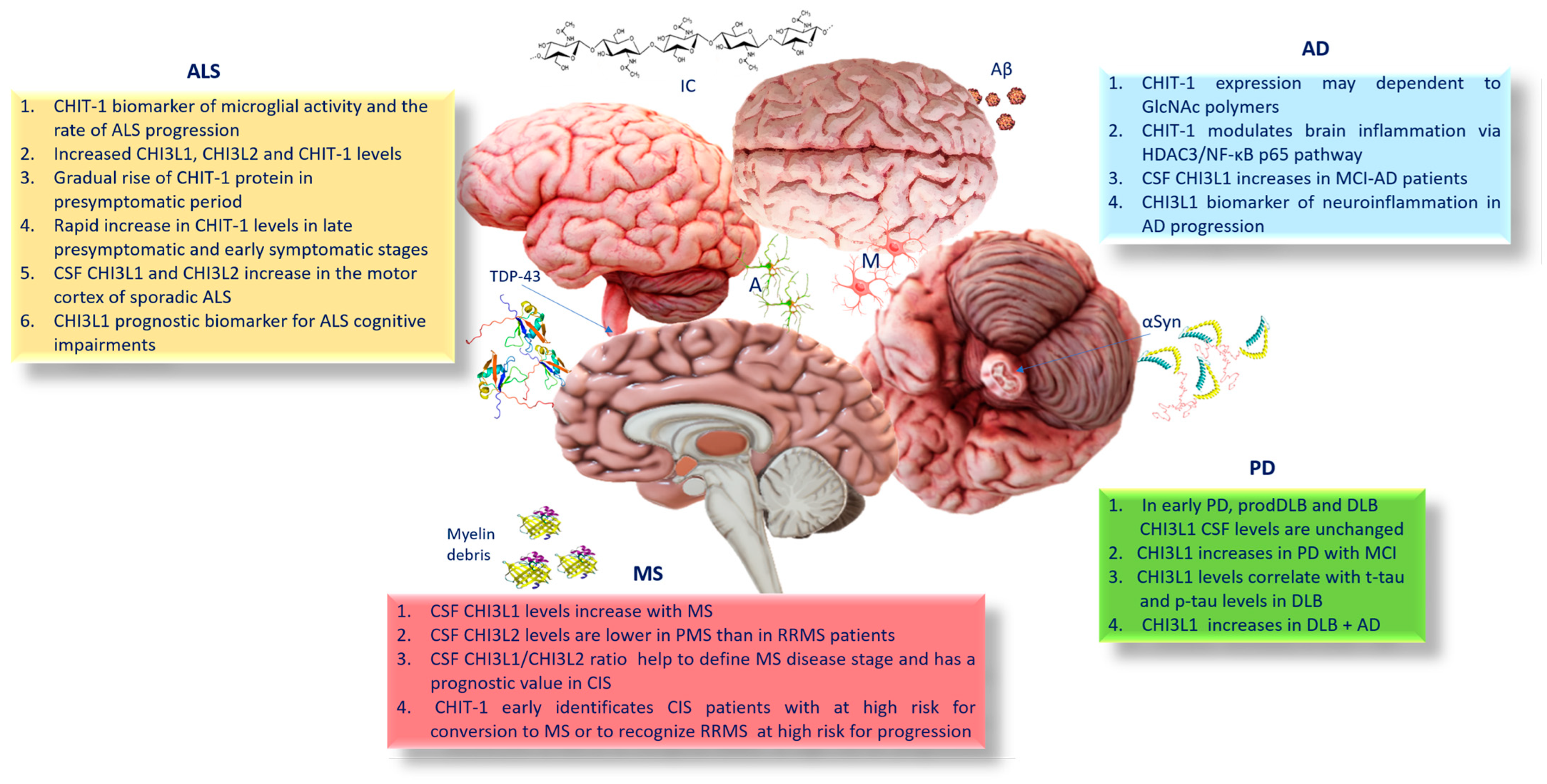
4. CHIT-1 and AD
5. CHI3L1 and AD
6. Chitinases, Parkinson’s Disease (PD), and Dementia with Lewy Bodies (DLB)
7. Chitinases and Amyotrophic Lateral Sclerosis (ALS)
8. Chitinases and Multiple Sclerosis (MS)
9. Conclusions
10. Future Prospects
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, 028035. [Google Scholar] [CrossRef]
- Dharshini, S.A.P.; Jemimah, S.; Taguchi, Y.H.; Gromiha, M.M. Exploring Common Therapeutic Targets for Neurodegenerative Disorders Using Transcriptome Study. Front. Genet. 2021, 12, 639160. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Pathology of Neurodegenerative Diseases. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, L. Chitotriosidase: The yin and yang. Cell. Mol. Life Sci. 2006, 63, 3018–3029. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Distefano, G.; Zorena, K.; Malaguarnera, L. Chitinases and immunity: Ancestral molecules with new functions. Immunobiology 2016, 221, 399–411. [Google Scholar] [CrossRef]
- Di Rosa, M.; De Gregorio, C.; Malaguarnera, G.; Tuttobene, M.; Biazzo, F.; Malaguarnera, L. Evaluation of AMCase and CHIT-1 expression in monocyte macrophages lineage. Mol. Cell. Biochem. 2013, 374, 73–80. [Google Scholar] [CrossRef]
- Zhao, T.; Su, Z.; Li, Y.; Zhang, X.; You, Q. Chitinase-3 like-protein-1 function and its role in diseases. Signal Transduct. Target Ther. 2020, 5, 201. [Google Scholar] [CrossRef]
- Di Rosa, M.; Malaguarnera, L. Chitotriosidase: A New Inflammatory Marker in Diabetic Complications. Pathobiology 2016, 83, 211–219. [Google Scholar] [CrossRef]
- Di Rosa, M.; Malaguarnera, L. Chitinase 3 Like-1: An Emerging Molecule Involved in Diabetes and Diabetic Complications. Pathobiology 2016, 83, 228–242. [Google Scholar] [CrossRef]
- Di Rosa, M.; Dell’Ombra, N.; Zambito, A.M.; Malaguarnera, M.; Nicoletti, F.; Malaguarnera, L. Chitotriosidase and inflammatory mediator levels in Alzheimer’s disease and cerebrovascular dementia. Eur. J. Neurosci. 2006, 23, 2648–2656. [Google Scholar] [CrossRef]
- Steinacker, P.; Verde, F.; Fang, L.; Feneberg, E.; Oeckl, P.; Roeber, S.; Anderl-Straub, S.; Danek, A.; Diehl-Schmid, J.; Fassbender, K.; et al. Chitotriosidase (CHIT1) is increased in microglia and macrophages in spinal cordof amyotrophic lateral sclerosis and cerebrospinal fluid levels correlate with disease severity and progression. J. Neurol. Neurosurg. Psychiatry 2018, 89, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Pinteac, R.; Montalban, X.; Comabella, M. Chitinases and chitinase-like proteins as biomarkers in neurologic disorders. Neurol. Neuroimmunol. Neuroinflamm. 2020, 8, e921. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Gray, E.; Bampton, A.; Raciborska, D.; Talbot, K.; Turner, M.R. CSF chitinase proteins in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Petržalka, M.; Meluzínová, E.; Libertínová, J.; Mojžišová, H.; Hanzalová, J.; Ročková, P.; Elišák, M.; Kmetonyová, S.; Šanda, J.; Sobek, O.; et al. IL-2; IL-6 and chitinase 3-like 2 might predict early relapse activity in multiple sclerosis. PLoS ONE 2022, 17, e0270607. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Farhat, S.M.; Ahmed, T. Neuroprotective and Neurotoxic Implications of α7 Nicotinic Acetylcholine Receptor and Aβ Interaction: Therapeutic Options in Alzheimer’s Disease. Curr. Drug Targets 2017, 18, 1537–1544. [Google Scholar] [CrossRef]
- Castellani, R.J.; Siedlak, S.L.; Fortino, A.E.; Perry, G.; Ghetti, B.; Smith, M.A. Chitin- like polysaccharides in Alzheimer’s disease brains. Curr. Alzheimer Res. 2005, 2, 419–423. [Google Scholar] [CrossRef]
- Castellani, R.J.; Perry, G.; Smith, M.A. The role of novel chitin-like polysaccharides in Alzheimer disease. Neurotox. Res. 2007, 12, 269–274. [Google Scholar] [CrossRef]
- Lee, C.G.; Da Silva, C.A.; Cruz, C.S.D.; Ahangari, F.; Ma, B.; Kang, M.J.; He, C.H.; Takyar, S.; Elias, J.A. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Ann. Rev. Physiol. 2011, 73, 479–501. [Google Scholar] [CrossRef]
- Da Silva, C.A.; Chalouni, C.; Williams, A.; Hartl, D.; Lee, C.G.; Elias, J.A. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. J. Immunol. 2009, 182, 3573–3582. [Google Scholar] [CrossRef]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef]
- Wills-Karp, M.; Nathan, A.; Page, K.; Karp, C.L. New insights into innate immune mechanisms underlying allergenicity. Mucosal. Immunol. 2010, 3, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Turano, E.; Busetto, G.; Marconi, S.; Guzzo, F.; Farinazzo, A.; Commisso, M.; Bistaffa, E.; Angiari, S.; Musumeci, S.; Sotgiu, S.; et al. Neurotoxicity and synaptic plasticity impairment of N-acetylglucosamine polymers: Implications for Alzheimer’s disease. Neurobiol. Aging 2015, 36, 1780–1791. [Google Scholar] [CrossRef]
- Saitgareeva, A.R.; Bulygin, K.V.; Gareev, I.F.; Beylerli, O.A.; Akhmadeeva, L.R. The role of microglia in the development of neurodegeneration. Neurol. Sci. 2020, 41, 3609–3615. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Villar-Piqué, A.; Schmitz, M.; Hermann, P.; Goebel, S.; Bunck, T.; Varges, D.; Ferrer, I.; Riggert, J.; Llorens, F.; Zerr, I. Plasma YKL-40 in the spectrum of neurodegenerative dementia. J. Neuroinflamm. 2019, 16, 145. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Pepe, G.; Calderazzi, G.; De Maglie, M.; Villa, A.M.; Vegeto, E. Heterogeneous induction of microglia M2a phenotype by central administration of interleukin-4. J. Neuroinflamm. 2014, 11, 211. [Google Scholar] [CrossRef]
- Russo, C.; Valle, M.S.; Russo, A.; Malaguarnera, L. The Interplay between Ghrelin and Microglia in Neuroinflammation: Implications for Obesity and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 13432. [Google Scholar] [CrossRef]
- Malaguarnera, L.; Di Rosa, M.; Zambito, A.M.; dell’Ombra, N.; Nicoletti, F.; Malaguarnera, M. Chitotriosidase gene expression in Kupffer cells from patients with non-alcoholic fatty liver disease. Gut 2006, 55, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Watabe-Rudolph, M.; Song, Z.; Lausser, L.; Schnack, C.; Begus-Nahrmann, Y.; Scheithauer, M.O.; Rettinger, G.; Otto, M.; Tumani, H.; Thal, D.R.; et al. Chitinase enzyme activity in CSF is a powerful biomarker of Alzheimer disease. Neurology 2012, 78, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Rosén, C.; Andersson, C.H.; Andreasson, U.; Molinuevo, J.L.; Bjerke, M.; Rami, L.; Lladó, A.; Blennow, K.; Zetterberg, H. Increased Levels of Chitotriosidase and YKL-40 in Cerebrospinal Fluid from Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Dis. Extra 2014, 4, 297–304. [Google Scholar] [CrossRef]
- Casal, J.A.; Robles, A.; Tutor, J.C. Serum markers of monocyte/macrophage activation in patients with Alzheimer’s disease and other types of dementia. Clin. Biochem. 2003, 36, 553–556. [Google Scholar] [CrossRef]
- Mattsson, N.; Tabatabaei, S.; Johansson, P.; Hansson, O.; Andreasson, U.; Månsson, J.E.; Johansson, J.O.; Olsson, B.; Wallin, A.; Svensson, J.; et al. Cerebrospinal fluid microglial markers in Alzheimer’s disease: Elevated chitotriosidase activity but lack of diagnostic utility. Neuromolecular. Med. 2011, 13, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, W.; Wu, L.; Yang, W.; Lü, Y. Chitotriosidase attenuates brain inflammation via HDAC3/NF-κB pathway in D-galactose and aluminum-induced rat model with cognitive impairments. Neurosci. Res. 2021, 172, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Johansen, J.S. Studies on serum YKL-40 as a biomarker in diseases with inflammation; tissue remodelling; fibroses and cancer. Dan. Med. Bull. 2006, 53, 172–209. [Google Scholar]
- Rehli, M.; Krause, S.W.; Andreesen, R. Molecular characterization of the gene for human cartilage gp-39 (CHI3L1); a member of the chitinase protein family and marker for late stages of macrophage differentiation. Genomics 1997, 43, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Tibullo, D.; Vecchio, M.; Nunnari, G.; Saccone, S.; Di Raimondo, F.; Malaguarnera, L. Determination of chitinases family during osteoclastogenesis. Bone 2014, 61, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Libreros, S.; Garcia-Areas, R.; Shibata, Y.; Carrio, R.; Torroella-Kouri, M.; Iragavarapu-Charyulu, V. Induction of proinflammatory mediators by CHI3L1 is reduced by chitin treatment: Decreased tumor metastasis in a breast cancer model. Int. J. Cancer 2012, 131, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, H.W.; Suk, K. Plasma level of chitinase 3-like 1 protein increases in patients with early Alzheimer’s disease. J. Neurol. 2011, 258, 2181–2185. [Google Scholar] [CrossRef]
- Rehli, M.; Niller, H.H.; Ammon, C.; Langmann, S.; Schwarzfischer, L.; Andreesen, R.; Krause, S.W. Transcriptional regulation of CHI3L1; a marker gene for late stages of macrophage differentiation. J. Biol. Chem. 2003, 278, 44058–44067. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature; definitions; and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte biomarkers in Alzheimer’s disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef]
- Bellaver, B.; Ferrari-Souza, J.P.; da Ros, L.U.; Carter, S.F.; Rodriguez-Vieitez, E.; Nordberg, A.; Pellerin, L.; Rosa-Neto, P.; Leffa, D.T.; Zimmer, E.R. Astrocyte biomarkers in Alzheimer disease: A systematic review and meta-analysis. Neurology 2021, 96, e2944–e2955. [Google Scholar] [CrossRef]
- Antonell, A.; Mansilla, A.; Rami, L.; Llado, A.; Iranzo, A.; Olives, J.; Balasa, M.; Sánchez-Valle, R.; Molinuevo, J.L. Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 901–908. [Google Scholar] [CrossRef]
- Gispert, J.D.; Monté, G.C.; Falcon, C.; Tucholka, A.; Rojas, S.; Sánchez-Valle, R.; Antonell, A.; Lladó, A.; Rami, L.; Molinuevo, J.L. CSF YL40 and pTau181 are related to different cerebral morphometric patterns in early AD. Neurobiol. Aging 2016, 38, 47–55. [Google Scholar] [CrossRef]
- Janelidze, S.; Hertze, J.; Zetterberg, H.; Landqvist Waldö, M.; Santillo, A.; Blennow, K.; Hansson, O. Cerebrospinal fluid neurogranin and YKL-40 as biomarkers of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2015, 3, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Kester, M.I.; Teunissen, C.E.; Sutphen, C.; Herries, E.M.; Ladenson, J.H.; Xiong, C.; Scheltens, P.; van der Flier, W.M.; Morris, J.C.; Holtzman, D.M.; et al. Cerebrospinal fluid VILIP-1 and YKL-40, candidate biomarkers to diagnose, predict and monitor Alzheimer’s disease in a memory clinic cohort. Alzheimers Res. Ther. 2015, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Hertze, J.; Lautner, R.; Zetterberg, H.; Nägga, K.; Höglund, K.; Basun, H.; Annas, P.; Lannfelt, L.; Andreasen, N.; et al. Microglial markers are elevated in the prodromal phase of Alzheimer’s disease and vascular dementia. J. Alzheimers Dis. 2013, 33, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Sutphen, C.L.; Jasielec, M.S.; Shah, A.R.; Macy, E.M.; Xiong, C.; Vlassenko, A.G.; Benzinger, T.L.; Stoops, E.E.; Vanderstichele, H.M.; Brix, B.; et al. Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA Neurol. 2015, 72, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, D.; Vilaplana, E.; Pegueroles, J.; Montal, V.; Sánchez-Juan, P.; González-Suárez, A.; Pozueta, A.; Rodríguez-Rodríguez, E.; Bartrés-Faz, D.; Vidal-Piñeiro, D.; et al. Relationship between cortical thickness and cerebrospinal fluid YKL-40in predementia stages of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2018–2023. [Google Scholar] [CrossRef]
- Dichev, V.; Kazakova, M.; Sarafian, V. YKL-40 e enolasi neurone-specifica nella neurodegenerazione e neuroinfiammazione. Rev. Neurosci. 2020, 31, 539–553. [Google Scholar] [CrossRef]
- Kim, M.N.; Lee, K.E.; Hong, J.Y.; Heo, W.I.; Kim, K.W.; Kim, K.E.; Sohn, M.H. Involvement of the MAPK and PI3K pathways in chitinase 3-like 1-regulated hyperoxia-induced airway epithelial cell death. Biochem. Biophys. Res. Commun. 2012, 421, 790–796. [Google Scholar] [CrossRef]
- Im, J.H.; Yeo, I.J.; Park, P.H.; Choi, D.Y.; Han, S.B.; Yun, J.; Hong, J.T. Deletion of Chitinase-3-like 1 accelerates stroke development through enhancement of Neuroinflammation by STAT6-dependent M2 microglial inactivation in Chitinase-3-like 1 knockout mice. Exp. Neurol. 2020, 323, 113082. [Google Scholar] [CrossRef]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol. Psychiatry 2010, 68, 903–912. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Querol-Vilaseca, M.; Colom-Cadena, M.; Pegueroles, J.; San Martín-Paniello, C.; Clarimon, J.; Belbin, O.; Fortea, J.; Lleó, A. YKL-40 (Chitinase 3-like I) is expressed in a subset of astrocytes in Alzheimer’s disease and other tauopathies. J. Neuroinflamm. 2017, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Ferrari-Souza, J.P.; Ferreira, P.C.L.; Bellaver, B.; Tissot, C.; Wang, Y.-T.; Leffa, D.T.; Brum, W.S.; Benedet, A.L.; Ashton, N.J.; De Bastiani, M.A.; et al. Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer’s disease. Mol. Psychiatry 2022, 27, 4781–4789. [Google Scholar] [CrossRef] [PubMed]
- McLarnon, J.G. Chemokine Interleukin-8 (IL-8) in Alzheimer’s and Other Neurodegenerative Diseases. J. Alzheimers Dis. Parkinsonism 2016, 6, 273. [Google Scholar] [CrossRef]
- Zhang, K.; Tian, L.; Liu, L.; Feng, Y.; Dong, Y.-B.; Li, B.; Shang, D.-S.; Fang, W.-G.; Cao, Y.-P.; Chen, Y.-H. CXCL1 contributes to β-amyloid-induced transendothelial migration of monocytes in Alzheimer’s disease. PLoS ONE 2013, 8, e72744. [Google Scholar] [CrossRef]
- Duits, F.H.; Hernandez-Guillamon, M.; Montaner, J.; Goos, J.D.; Montañola, A.; Wattjes, M.P.; Barkhof, F.; Scheltens, P.; Teunissen, C.E.; van der Flier, W.M. Matrix Metalloproteinases in Alzheimer’s Disease and Concurrent Cerebral Microbleeds. J. Alzheimers Dis. 2015, 48, 711–720. [Google Scholar] [CrossRef]
- Bolós, M.; Llorens-Martín, M.; Perea, J.R.; Jurado-Arjona, J.; Rábano, A.; Hernández, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.J. Levels of mTOR and its downstream targets 4E-BP1; eEF2; and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef]
- Murphy, G.M., Jr.; Zhao, F.; Yang, L.; Cordell, B. Expression of macrophage colony-stimulating factor receptor is increased in the AbetaPP(V717F) transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 2000, 157, 895–904. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Z.; Wang, Q.; Lv, X.; Cheng, Z.; Wu, Y.; Tang, F.; Shen, Y.; Gao, F. Identification of hub proteins in cerebrospinal fluid as potential biomarkers of Alzheimer’s disease by integrated bioinformatics. J. Neurol. 2023, 270, 1487–1500, Epub ahead of print. [Google Scholar] [CrossRef]
- Alarcón, T.A.; Presti-Silva, S.M.; Simões, A.P.T.; Ribeiro, F.M.; Pires, R.G.W. Molecular mechanisms underlying the neuroprotection of environmental enrichment in Parkinson’s disease. Neural Regen Res. 2023, 18, 1450–1456. [Google Scholar]
- Sezgin, M.; Bilgic, B.; Tinaz, S.; Emre, M. Parkinson’s Disease Dementia and Lewy Body Disease. Semin. Neurol. 2019, 39, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Heywood, W.E.; Galimberti, D.; Bliss, E.; Sirka, E.; Paterson, R.W.; Magdalinou, N.K.; Carecchio, M.; Reid, E.; Heslegrave, A.; Fenoglio, C.; et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high- throughput multiplexed targeted proteomic assay. Mol. Neurodegener. 2015, 10, 64. [Google Scholar] [CrossRef]
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein; tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- Surendranathan, A.; Rowe, J.B.; O’Brien, J.T. Neuroinflammation in Lewy body dementia. Parkinsonism Relat. Disord. 2015, 21, 1398–1406. [Google Scholar] [CrossRef]
- Brück, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef]
- Lv, Q.K.; Tao, K.X.; Wang, X.B.; Yao, X.Y.; Pang, M.Z.; Liu, J.Y.; Wang, F.; Liu, C.F. Role of α-synuclein in microglia: Autophagy and phagocytosis balance neuroinflammation in Parkinson’s disease. Inflamm. Res. 2023, 72, 443–462, Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Bartl, M.; Dakna, M.; Galasko, D.; Hutten, S.J.; Foroud, T.; Quan, M.; Marek, K.; Siderowf, A.; Franz, J.; Trenkwalder, C.; et al. Parkinson’s Progression Markers Initiative. Biomarkers of neurodegeneration and glial activation validated in Alzheimer’s disease assessed in longitudinal cerebrospinal fluid samples of Parkinson’s disease. PLoS ONE 2021, 16, e0257372. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, D.S.; Fixemer, S.; Heurtaux, T.; Jeannelle, F.; Frauenknecht, K.B.M.; Mittelbronn, M. The Multifaceted Neurotoxicity of Astrocytes in Ageing and Age-Related Neurodegenerative Diseases: A Translational Perspective. Front. Physiol. 2022, 13, 814889. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends. Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Jellinger, K.A.; Wenning, G.K.; Stefanova, N. Glial dysfunction in the pathogenesis of α-synucleinopathies: Emerging concepts. Acta Neuropathol. 2011, 121, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Pelech, S.; Giasson, B.I.; Maguire, J.; Zhang, H.; McGeer, E.G.; McGeer, P.L. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol. Aging 2008, 29, 739–752. [Google Scholar] [CrossRef]
- Dettmer, U.; Selkoe, D.; Bartels, T. New insights into cellular α-synuclein homeostasis in health and disease. Curr. Opin. Neurobiol. 2016, 36, 15–22. [Google Scholar] [CrossRef]
- Lu, S.Z.; Guo, Y.S.; Liang, P.Z.; Zhang, S.Z.; Yin, S.; Yin, Y.Q.; Wang, X.M.; Ding, F.; Gu, X.S.; Zhou, J.W. Suppression of astrocytic autophagy by αB-crystallin contributes to α-synuclein inclusion formation. Transl. Neurodegener. 2019, 8, 3. [Google Scholar] [CrossRef]
- Morenas-Rodríguez, E.; Alcolea, D.; Suárez-Calvet, M.; Muñoz-Llahuna, L.; Vilaplana, E.; Sala, I.; Subirana, A.; Querol-Vilaseca, M.; Carmona-Iragui, M.; Illán-Gala, I.; et al. Different pattern of CSF glial markers between dementia with Lewy bodies and Alzheimer’s disease. Sci. Rep. 2019, 9, 7803. [Google Scholar] [CrossRef]
- Wennström, M.; Surova, Y.; Hall, S.; Nilsson, C.; Minthon, L.; Hansson, O.; Nielsen, H.M. The Inflammatory Marker YKL-40 Is Elevated in Cerebrospinal Fluid from Patients with Alzheimer’s but Not Parkinson’s Disease or Dementia with Lewy Bodies. PLoS ONE 2015, 10, e0135458. [Google Scholar] [CrossRef]
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell. Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef]
- Wattmo, C.; Blennow, K.; Hansson, O. Cerebro-spinal fluid biomarker levels: Phosphorylated tau (T) and total tau (N) as markers for rate of progression in Alzheimer’s disease. BMC Neurol. 2020, 20, 10. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O.; Kiernan, M.C.; Chiò, A.; Rix-Brooks, B.; van den Berg, L.H. Amyotrophic lateral sclerosis: Moving towards a new classification system. Lancet Neurol. 2016, 15, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- Burrell, J.R.; Halliday, G.M.; Kril, J.J.; Ittner, L.M.; Götz, J.; Kiernan, M.C.; Hodges, J.R. The frontotemporal dementia-motor neuron disease continuum. Lancet 2016, 388, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.; Heverin, M.; McLaughlin, R.L.; Hardiman, O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019, 76, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes; environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psichiatria 2020, 92, 86–95. [Google Scholar] [CrossRef]
- Tan, R.H.; Ke, Y.D.; Ittner, L.M.; Halliday, G.M. ALS/FTLD: Experimental models and reality. Acta Neuropathol. 2017, 133, 177–196. [Google Scholar] [CrossRef]
- Swarup, V.; Phaneuf, D.; Bareil, C.; Robertson, J.; Rouleau, G.A.; Kriz, J.; Julien, J.P. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 2011, 134 Pt 9, 2610–2626. [Google Scholar] [CrossRef]
- Huang, C.; Zhou, H.; Tong, J.; Chen, H.; Liu, Y.J.; Wang, D.; Wei, X.; Xia, X.G. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011, 7, e1002011. [Google Scholar] [CrossRef]
- Uchida, A.; Sasaguri, H.; Kimura, N.; Tajiri, M.; Ohkubo, T.; Ono, F.; Sakaue, F.; Kanai, K.; Hirai, T.; Sano, T.; et al. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain 2012, 135 Pt 3, 833–846. [Google Scholar] [CrossRef]
- Bilican, B.; Serio, A.; Barmada, S.J.; Nishimura, A.L.; Sullivan, G.J.; Carrasco, M.; Phatnani, H.P.; Puddifoot, C.A.; Story, D.; Fletcher, J.; et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. USA 2012, 109, 5803–5808. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Wu, L.S.; Cheng, W.C.; Shen, C.K. Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J. Biol. Chem. 2012, 287, 27335–27344. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.; Huang, C.; Tong, J.; Qiu, G.; Huang, B.; Wu, Q.; Li, F.; Xu, Z.; Bowser, R.; Xia, X.-G.; et al. Reactive astrocytes secrete lcn2 to promote neuron death. Proc. Natl. Acad. Sci. USA 2013, 110, 4069–4074. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of amyotrophic lateral sclerosis: Seeking therapeutic targets in the era of gene therapy. J. Hum. Genet. 2023, 68, 131–152. [Google Scholar] [CrossRef]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.-G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Gille, B.; De Schaepdryver, M.; Dedeene, L.; Goossens, J.; Claeys, K.; Bosch, L.V.D.; Tournoy, J.; Van Damme, P.; Poesen, K. Inflammatory markers in cerebrospinal fluid: Independent prognostic biomarkers in amyotrophic lateral sclerosis? J. Neurol. Neurosurg. Psychiatry 2019, 90, 1338–1346. [Google Scholar] [CrossRef]
- Costa, J.; Gromicho, M.; Pronto-Laborinhom, A.; Almeida, C.; Gomes, R.A.; Guerreiro, A.C.L.; Oliva, A.; Pinto, S.; de Carvalho, M. Cerebrospinal Fluid Chitinases as Biomarker for Amyotrophic Lateral Sclerosis. Diagnostics 2021, 11, 1210. [Google Scholar] [CrossRef]
- Gray, E.; Thompson, A.G.; Wuu, J.; Pelt, J.; Talbot, K.; Benatar, M.; Turner, M.R. CSF chitinases before and after symptom onset in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Feneberg, E.; Halbgebauer, S.; Witzel, S.; Verde, F.; Oeckl, P.; Van Damme, P.; Gaur, N.; Gray, E.; Grosskreutz, J.; et al. Chitotriosidase as biomarker for early stage amyotrophic lateral sclerosis: A multicenter study. Amyotroph. Lateral. Scler. Frontotemporal. Degener. 2021, 22, 276–286. [Google Scholar] [CrossRef]
- Oeckl, P.; Weydt, P.; Steinacker, P.; Anderl-Straub, S.; Nordin, F.; E Volk, A.; Diehl-Schmid, J.; Andersen, P.M.; Kornhuber, J.; Danek, A.; et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J. Neurol. Neurosurg. Psychiatry 2019, 90, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.; An, J.; Kovalik, T.; Gendron, T.; Petrucelli, L.; Bowser, R. Cross-sectional and longitudinal measures of chitinase proteins in amyotrophic lateral sclerosis and expression of CHI3L1 in activated astrocytes. J. Neurol. Neurosurg. Psychiatry 2020, 91, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Hol, E.M.; Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Bellenberg, B.; Gisevius, B.; Hirschberg, S.; Sankowski, R.; Prinz, M.; Gold, R.; Lukas, C.; Haghikia, A. Chitinase 3-like 1 and neurofilament light chain in CSF and CNS atrophy in MS. Neurol. Neuroimmunol. Neuroinflamm. 2020, 8, e906. [Google Scholar] [CrossRef]
- Gaur, N.; Perner, C.; Witte, O.W.; Grosskreutz, J. The Chitinases as Biomarkers for Amyotrophic Lateral Sclerosis: Signals from the CNS and Beyond. Front. Neurol. 2020, 11, 377. [Google Scholar] [CrossRef]
- Hendrickx, D.A.E.; van Eden, C.G.; Schuurman, K.G.; Hamann, J.; Huitinga, I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J. Neuroimmunol. 2017, 309, 12–22. [Google Scholar] [CrossRef]
- Cossburn, M.D.; Harding, K.; Ingram, G.; El-Shanawany, T.; Heaps, A.; Pickersgill, T.P.; Jolles, S.; Robertson, N.P. Clinical relevance of differential lymphocyte recovery after alemtuzumab therapy for multiple sclerosis. Neurology 2013, 80, 55–61. [Google Scholar] [CrossRef]
- Ziemssen, T.; Medin, J.; Couto, C.A.; Mitchell, C.R. Multiple sclerosis in the real world: A systematic review of fingolimod as a case study. Autoimmun. Rev. 2017, 16, 355–376. [Google Scholar] [CrossRef]
- Højsgaard Chow, H.; Schreiber, K.; Magyari, M.; Ammitzbøll, C.; Börnsen, L.; Romme Christensen, J.; Ratzer, R.; Soelberg Sørensen, P.; Sellebjerg, F. Progressive multiple sclerosis; cognitive function; and quality of life. Brain Behav. 2018, 8, e00875. [Google Scholar] [CrossRef]
- Cashion, J.M.; Young, K.M.; Sutherland, B.A. How does neurovascular unit dysfunction contribute to multiple sclerosis? Neurobiol. Dis. 2023, 178, 106028. [Google Scholar] [CrossRef]
- Degelman, M.L.; Herman, K.M. Smoking and multiple sclerosis: A systematic review and meta-analysis using the Bradford Hill criteria for causation. Mult. Scler. Relat. Disord. 2017, 17, 207–216. [Google Scholar] [CrossRef]
- Huang, J.; Kockum, I.; Stridh, P. Trends in the environmental risks associated with earlier onset in multiple sclerosis. Mult. Scler. Relat. Disord. 2022, 68, 104250. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Münz, C.; Cohen, J.I.; Ascherio, A. Epstein-Barr virus as a leading cause of multiple sclerosis: Mechanisms and implications. Nat. Rev. Neurol. 2023, 19, 160–171. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated with Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Baxter, P.S.; Dando, O.; Emelianova, K.; He, X.; McKay, S.; Hardingham, G.E.; Qiu, J. Microglial identity and inflammatory responses are controlled by the combined effects of neurons and astrocytes. Cell. Rep. 2021, 34, 108882. [Google Scholar] [CrossRef]
- Bonneh-Barkay, D.; Wang, G.; Starkey, A.; Hamilton, R.L.; Wiley, C.A. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J. Neuroinflamm. 2010, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Fernández, M.; Martin, R.; Rivera-Vallvé, S.; Borràs, E.; Chiva, C.; Julià, E.; Rovira, A.; Cantó, E.; Alvarez-Cermeño, J.C.; et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain 2010, 133 Pt 4, 1082–1093. [Google Scholar] [CrossRef]
- Hinsinger, G.; Galéotti, N.; Nabholz, N.; Urbach, S.; Rigau, V.; Demattei, C.; Lehmann, S.; Camu, W.; Labauge, P.; Castelnovo, G.; et al. Chitinase 3-like proteins as diagnostic and prognostic biomarkers of multiple sclerosis. Mult. Scler. 2015, 21, 1251–1261. [Google Scholar] [CrossRef]
- De Fino, C.; Lucchini, M.; Lucchetti, D.; Nociti, V.; Losavio, F.; Bianco, A.; Colella, F.; Ricciardi-Tenore, C.; Sgambato, A.; Mirabella, M. The predictive value of CSF multiple assay in multiple sclerosis: A single center experience. Mult. Scler. Relat. Disord. 2019, 35, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Samaha, D.Y.; Zeitoun, Y.A.; Fouad, N.T.; Abdel Aziz, A.A.; Saad, M.A. Oligoclonal band versus chitinase-3-like protein-1 in CSF of newly diagnosed relapsing remitting multiple sclerosis. Egypt J. Immunol. 2023, 30, 42–48. [Google Scholar] [CrossRef]
- Oldoni, E.; Smets, I.; Mallants, K.; Vandebergh, M.; Van Horebeek, L.; Poesen, K.; Dupont, P.; Dubois, B.; Goris, A. CHIT1 at Diagnosis Reflects Long-Term Multiple Sclerosis Disease Activity. Ann. Neurol. 2020, 87, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Novakova, L.; Axelsson, M.; Khademi, M.; Zetterberg, H.; Blennow, K.; Malmeström, C.; Piehl, F.; Olsson, T.; Lycke, J. Cerebrospinal fluid biomarkers as a measure of disease activity and treatment efficacy in relapsing-remitting multiple sclerosis. J. Neurochem. 2017, 141, 296–304. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, C.; Valle, M.S.; Casabona, A.; Malaguarnera, L. Chitinase Signature in the Plasticity of Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 6301. https://doi.org/10.3390/ijms24076301
Russo C, Valle MS, Casabona A, Malaguarnera L. Chitinase Signature in the Plasticity of Neurodegenerative Diseases. International Journal of Molecular Sciences. 2023; 24(7):6301. https://doi.org/10.3390/ijms24076301
Chicago/Turabian StyleRusso, Cristina, Maria Stella Valle, Antonino Casabona, and Lucia Malaguarnera. 2023. "Chitinase Signature in the Plasticity of Neurodegenerative Diseases" International Journal of Molecular Sciences 24, no. 7: 6301. https://doi.org/10.3390/ijms24076301
APA StyleRusso, C., Valle, M. S., Casabona, A., & Malaguarnera, L. (2023). Chitinase Signature in the Plasticity of Neurodegenerative Diseases. International Journal of Molecular Sciences, 24(7), 6301. https://doi.org/10.3390/ijms24076301