
Anthracycline Therapy Modifies Immune Checkpoint Signaling in the Heart

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. DOX Treated Mice Show a Profound Effect on Cardiac Function
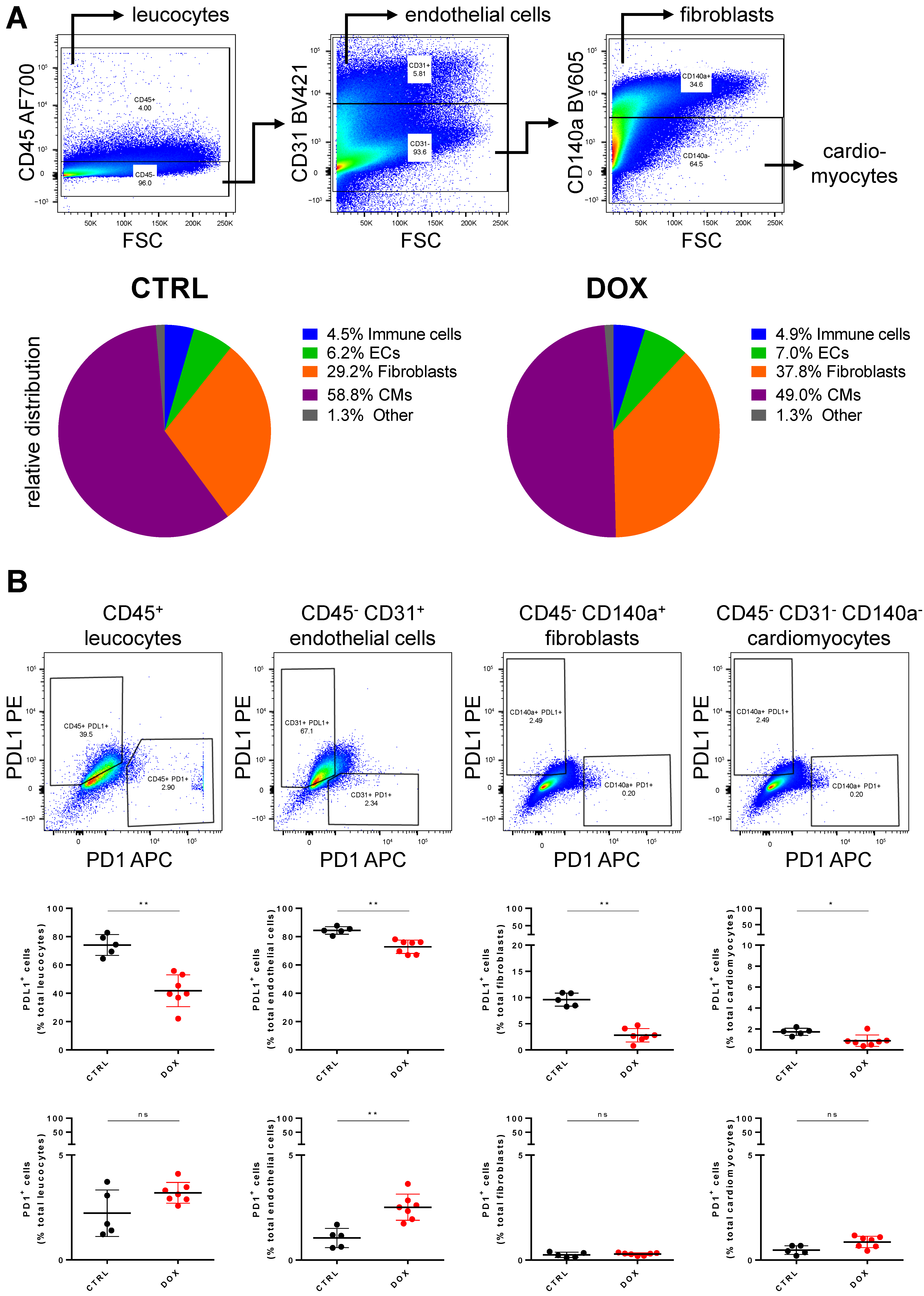
2.2. PDL1 Expression following Anthracycline Treatment
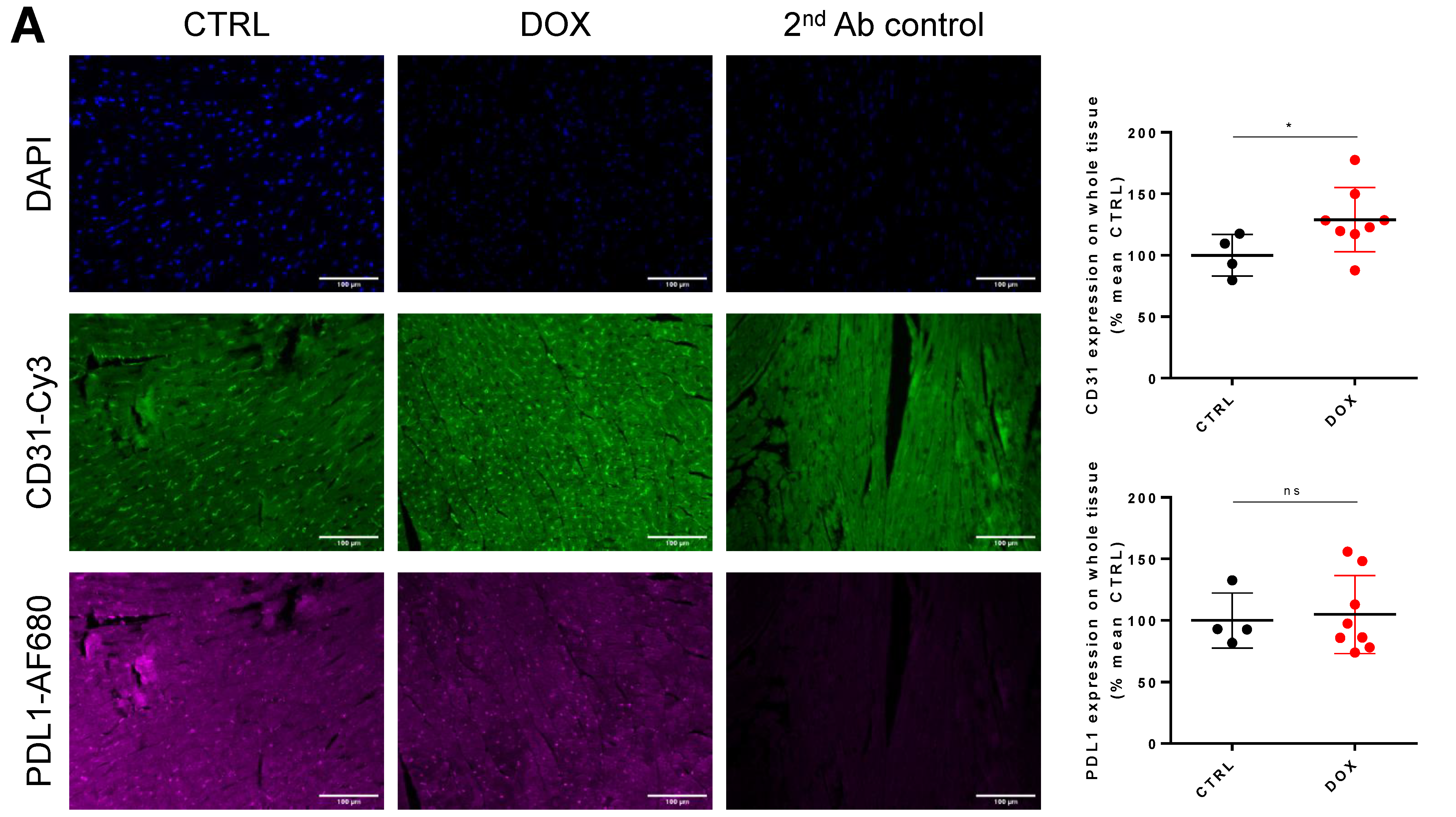
2.3. DOX Treatment Does Not Increase Overall Tissue CD31 Expression
3. Discussion
4. Materials and Methods
4.1. Animals and Ethical Statement
4.2. DOX Treatment Model and Experiment Setup
4.3. Cardiac Function and Blood Analysis
4.4. Flow Cytometry
4.5. Immuno-Histology
4.6. Western Blot
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol. 2015, 12, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early Detection of Anthracycline Cardiotoxicity and Improvement with Heart Failure Therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; Lamantia, G.; Colombo, N.; Civelli, M.; De Giacomi, G.; Rubino, M.; Veglia, F.; Fiorentini, C.; Cipolla, C.M. Anthracycline-Induced Cardiomyopathy: Clinical Relevance and Response to Pharmacologic Therapy. J. Am. Coll. Cardiol. 2010, 55, 213–220. [Google Scholar] [CrossRef]
- Huang, J.; Wu, R.; Chen, L.; Yang, Z.; Yan, D.; Li, M. Understanding Anthracycline Cardiotoxicity from Mitochondrial Aspect. Front. Pharmacol. 2022, 13, 811406. [Google Scholar] [CrossRef]
- Li, M.; Russo, M.; Pirozzi, F.; Tocchetti, C.G.; Ghigo, A. Autophagy and cancer therapy cardiotoxicity: From molecular mechanisms to therapeutic opportunities. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118493. [Google Scholar] [CrossRef]
- Kim, D.J.; Jang, J.H.; Ham, S.Y.; Choi, S.H.; Park, S.S.; Jeong, S.Y.; Kim, B.C.; Jeon, D.Y.; Lee, B.J.; Ko, B.K.; et al. Doxorubicin inhibits PD-L1 expression by enhancing TTP-mediated decay of PD-L1 mRNA in cancer cells. Biochem. Biophys. Res. Commun. 2020, 522, 402–407. [Google Scholar] [CrossRef]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.B.; Blohmer, J.U.; Grischke, E.-M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; Rezai, M.; et al. Randomized phase II neoadjuvant study (GeparNuevo) to investigate the addition of durvalumab to a taxane-anthracycline containing chemotherapy in triple negative breast cancer (TNBC). J. Clin. Oncol. 2018, 36, 104. [Google Scholar] [CrossRef]
- Pollack, S.M.; Redman, M.W.; Baker, K.K.; Wagner, M.J.; Schroeder, B.A.; Loggers, E.T.; Trieselmann, K.; Copeland, V.C.; Zhang, S.; Black, G.; et al. Assessment of Doxorubicin and Pembrolizumab in Patients with Advanced Anthracycline-Naive Sarcoma: A Phase 1/2 Nonrandomized Clinical Trial. JAMA Oncol. 2020, 6, 1778–1782. [Google Scholar] [CrossRef]
- Sadighi, S.; Sharifian, R.; Kazemimanesh, M.; Muhammadnejad, A.; Amanpour, S.; Muhammadnejad, S. Downregulation of immune checkpoints by doxorubicin and carboplatin-containing neoadjuvant regimens in breast cancer: A new insight into the mechanism of induction of antitumor immunity. Ann. Oncol. 2019, 30, iii12. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Wang, J.N.; Ma, S.Y.; Cai, J.; Su, N.; Huang, H.Q.; Li, Z.M.; Xia, Z.J.; Huang, H.; Liu, P.P.; et al. Combination of PD-1 inhibitor with GVD (gemcitabine, vinorelbine, liposomal doxorubicin) versus GVD regimen as second-line therapy for relapsed/refractory classical Hodgkin lymphoma. Br. J. Haematol. 2022, 196, 127–135. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Michel, L.; Rassaf, T.; Totzeck, M. Cardiotoxicity from immune checkpoint inhibitors. Int. J. Cardiol. Heart Vasc. 2019, 25, 100420. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.; Totzeck, M.; Lehmann, L.; Finke, D. Emerging role of immune checkpoint inhibitors and their relevance for the cardiovascular system. Herz 2020, 45, 645–651. [Google Scholar] [CrossRef]
- Michel, L.; Helfrich, I.; Hendgen-Cotta, U.B.; Mincu, R.-I.; Korste, S.; Mrotzek, S.M.; Spomer, A.; Odersky, A.; Rischpler, C.; Herrmann, K.; et al. Targeting early stages of cardiotoxicity from anti-PD1 immune checkpoint inhibitor therapy. Eur. Heart J. 2021, 43, 316–329. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Hall, P.E.; Schmid, P. Emerging strategies for TNBC with early clinical data: New chemoimmunotherapy strategies. Breast Cancer Res. Treat. 2022, 193, 21–35. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef]
- Yati, S.; Silathapanasakul, A.; Thakaeng, C.; Chanasakulniyom, M.; Songtawee, N.; Porntadavity, S.; Pothacharoen, P.; Pruksakorn, D.; Kongtawelert, P.; Yenchitsomanus, P.T.; et al. Extracellular Vesicle-Mediated IL-1 Signaling in Response to Doxorubicin Activates PD-L1 Expression in Osteosarcoma Models. Cells 2022, 11, 1042. [Google Scholar] [CrossRef]
- Moon, Y.; Shim, M.K.; Choi, J.; Yang, S.; Kim, J.; Yun, W.S.; Cho, H.; Park, J.Y.; Kim, Y.; Seong, J.K.; et al. Anti-PD-L1 peptide-conjugated prodrug nanoparticles for targeted cancer immunotherapy combining PD-L1 blockade with immunogenic cell death. Theranostics 2022, 12, 1999–2014. [Google Scholar] [CrossRef]
- Hu, Q.; Yao, J.; Wang, X.; Wang, Y.; Fu, X.; Ma, J.; Lin, H.; Xu, J.; Shen, L.; Yu, X. Combinational Chemoimmunotherapy for Breast Cancer by Codelivery of Doxorubicin and PD-L1 siRNA Using a PAMAM-Incorporated Liposomal Nanoplatform. ACS Appl. Mater. Interfaces 2022, 14, 8782–8792. [Google Scholar] [CrossRef]
- Cheong, A.; McGrath, S.; Robinson, T.; Maliki, R.; Spurling, A.; Lock, P.; Rephaeli, A.; Nudelman, A.; Parker, B.S.; Pepe, S.; et al. A switch in mechanism of action prevents doxorubicin-mediated cardiac damage. Biochem. Pharmacol. 2021, 185, 114410. [Google Scholar] [CrossRef]
- Hou, K.; Shen, J.; Yan, J.; Zhai, C.; Zhang, J.; Pan, J.A.; Zhang, Y.; Jiang, Y.; Wang, Y.; Lin, R.Z.; et al. Loss of TRIM21 alleviates cardiotoxicity by suppressing ferroptosis induced by the chemotherapeutic agent doxorubicin. EBioMedicine 2021, 69, 103456. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Wang, B.; Chi, W.; Li, Z.; Zhang, M.; Shen, Y.; Liu, X.; Lu, Y.; Liu, Y. Dihydromyricetin protects against Doxorubicin-induced cardiotoxicity through activation of AMPK/mTOR pathway. Phytomedicine 2022, 99, 154027. [Google Scholar] [CrossRef]
- Kuno, A.; Hosoda, R.; Tsukamoto, M.; Sato, T.; Sakuragi, H.; Ajima, N.; Saga, Y.; Tada, K.; Taniguchi, Y.; Iwahara, N.; et al. SIRT1 in the cardiomyocyte counteracts doxorubicin-induced cardiotoxicity via regulating histone H2AX. Cardiovasc. Res. 2022, 118, 3360–3373. [Google Scholar] [CrossRef]
- Reagan, W.J.; York, M.; Berridge, B.; Schultze, E.; Walker, D.; Pettit, S. Comparison of cardiac troponin I and T, including the evaluation of an ultrasensitive assay, as indicators of doxorubicin-induced cardiotoxicity. Toxicol. Pathol. 2013, 41, 1146–1158. [Google Scholar] [CrossRef]
- Ray, M.R.; Lakshmi, C.; Deb, C.; Ray, C.; Lahiri, T. Modulatory Effect of Dopamine on Doxorubicin-Induced Myelosuppression. Comp. Haematol. Int. 2001, 10, 212–220. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, C.; Kong, C.Y.; Song, P.; Wu, H.M.; Xu, S.C.; Yuan, Y.P.; Deng, W.; Ma, Z.G.; Tang, Q.Z. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020, 27, 540–555. [Google Scholar] [CrossRef]
- Catanzaro, M.P.; Weiner, A.; Kaminaris, A.; Li, C.; Cai, F.; Zhao, F.; Kobayashi, S.; Kobayashi, T.; Huang, Y.; Sesaki, H.; et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. 2019, 33, 11096–11108. [Google Scholar] [CrossRef]
- Zhang, D.X.; Ma, D.Y.; Yao, Z.Q.; Fu, C.Y.; Shi, Y.X.; Wang, Q.L.; Tang, Q.Q. ERK1/2/p53 and NF-kappaB dependent-PUMA activation involves in doxorubicin-induced cardiomyocyte apoptosis. Eur. Rev. Med. Pharm. Sci. 2016, 20, 2435–2442. [Google Scholar]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Favreau-Lessard, A.J.; Blaszyk, H.; Jones, M.A.; Sawyer, D.B.; Pinz, I.M. Systemic and cardiac susceptibility of immune compromised mice to doxorubicin. Cardiooncology 2019, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Wang, H.; Li, C.; Fang, J.Y.; Xu, J. Cancer Cell-Intrinsic PD-1 and Implications in Combinatorial Immunotherapy. Front. Immunol. 2018, 9, 1774. [Google Scholar] [CrossRef]
- Baban, B.; Liu, J.Y.; Qin, X.; Weintraub, N.L.; Mozaffari, M.S. Upregulation of Programmed Death-1 and Its Ligand in Cardiac Injury Models: Interaction with GADD153. PLoS ONE 2015, 10, e0124059. [Google Scholar] [CrossRef]
- Naba, N.M.; Tolay, N.; Erman, B.; Sayi Yazgan, A. Doxorubicin inhibits miR-140 expression and upregulates PD-L1 expression in HCT116 cells, opposite to its effects on MDA-MB-231 cells. Turk. J. Biol. 2020, 44, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.C.; Spalding, P.B.; Gutierrez, J.C.; Balkan, W.; Namias, N.; Koniaris, L.G.; Zimmers, T.A. Body surface area prediction in normal, hypermuscular, and obese mice. J. Surg. Res. 2009, 153, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Meeh, K.v. Ober flahenmessungen des mensch-lichen kopers. Z. Fur Biol. 1879, 15, S. 425–S. 458. [Google Scholar]
- Zacchigna, S.; Paldino, A.; Falcao-Pires, I.; Daskalopoulos, E.P.; Dal Ferro, M.; Vodret, S.; Lesizza, P.; Cannata, A.; Miranda-Silva, D.; Lourenco, A.P.; et al. Toward standardization of echocardiography for the evaluation of left ventricular function in adult rodents: A position paper of the ESC Working Group on Myocardial Function. Cardiovasc. Res. 2020, 117, 43–59. [Google Scholar] [CrossRef]
- Settelmeier, S.; Rassaf, T.; Hendgen-Cotta, U.B. Revealing Subtle Changes in Cardiac Function using Transthoracic Dobutamine Stress Echocardiography in Mice. J. Vis. Exp. 2021, 168, e62019. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korste, S.; Settelmeier, S.; Michel, L.; Odersky, A.; Stock, P.; Reyes, F.; Haj-Yehia, E.; Anker, M.S.; Grüneboom, A.; Hendgen-Cotta, U.B.; et al. Anthracycline Therapy Modifies Immune Checkpoint Signaling in the Heart. Int. J. Mol. Sci. 2023, 24, 6052. https://doi.org/10.3390/ijms24076052
Korste S, Settelmeier S, Michel L, Odersky A, Stock P, Reyes F, Haj-Yehia E, Anker MS, Grüneboom A, Hendgen-Cotta UB, et al. Anthracycline Therapy Modifies Immune Checkpoint Signaling in the Heart. International Journal of Molecular Sciences. 2023; 24(7):6052. https://doi.org/10.3390/ijms24076052
Chicago/Turabian StyleKorste, Sebastian, Stephan Settelmeier, Lars Michel, Andrea Odersky, Pia Stock, Fabrice Reyes, Elias Haj-Yehia, Markus S. Anker, Anika Grüneboom, Ulrike B. Hendgen-Cotta, and et al. 2023. "Anthracycline Therapy Modifies Immune Checkpoint Signaling in the Heart" International Journal of Molecular Sciences 24, no. 7: 6052. https://doi.org/10.3390/ijms24076052
APA StyleKorste, S., Settelmeier, S., Michel, L., Odersky, A., Stock, P., Reyes, F., Haj-Yehia, E., Anker, M. S., Grüneboom, A., Hendgen-Cotta, U. B., Rassaf, T., & Totzeck, M. (2023). Anthracycline Therapy Modifies Immune Checkpoint Signaling in the Heart. International Journal of Molecular Sciences, 24(7), 6052. https://doi.org/10.3390/ijms24076052