
Vitiligo, from Pathogenesis to Therapeutic Advances: State of the Art

 , ,
, , {kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Identify Keywords
2.2. Conduct Research
2.3. Review Abstract and Article
2.4. Document Results
3. Results
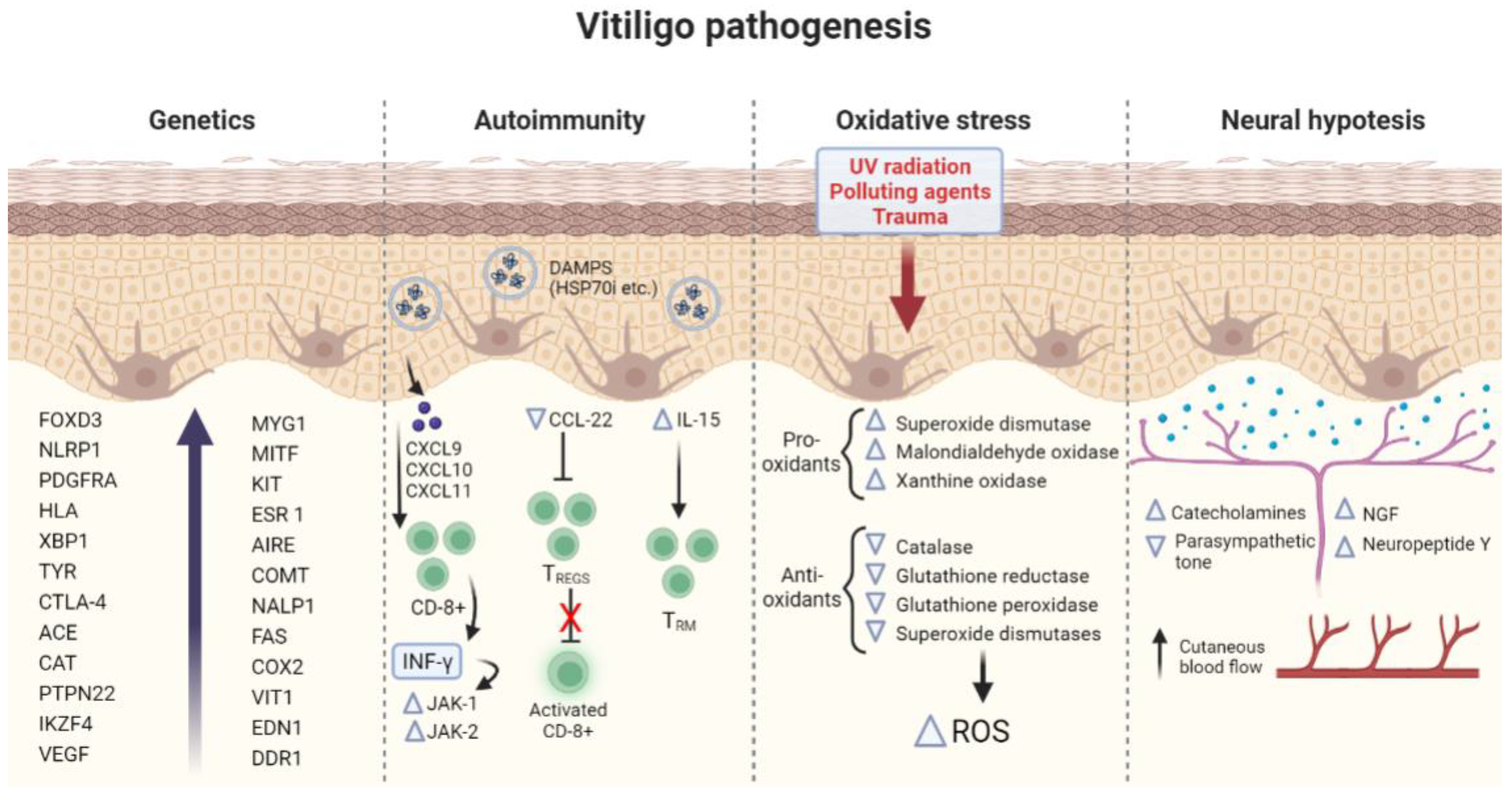
3.1. Pathogenesis of Vitiligo
3.1.1. Genetics
3.1.2. Autoimmunity
3.1.3. Oxidative Stress Hypothesis
3.1.4. Neural Hypothesis
3.2. Treatments of Vitiligo: Past, Present and Future
3.2.1. Afamelanotide
3.2.2. Prostaglandins and Analogues
3.2.3. Janus Kinase Inhibitors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ezzedine, K.; Eleftheriadou, V.; Whitton, M.; van Geel, N. Vitiligo. Lancet 2015, 386, 74. [Google Scholar] [CrossRef]
- Krüger, C.; Schallreuter, K.U. Stigmatisation, Avoidance Behaviour and Difficulties in Coping Are Common among Adult Patients with Vitiligo. Acta Derm. Venereol. 2015, 95, 553. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I.; Silverberg, N.B. Quality of life impairment in children and adolescents with vitiligo. Pediatr. Dermatol. 2014, 31, 309. [Google Scholar] [CrossRef] [PubMed]
- Radi, G.; Simonetti, O.; Diotallevi, F.; Campanati, A.; Brisigotti, V.; Molinelli, E.; Offidani, A. How can I take care of you? The dermatologist meets patients’ needs during the COVID-19 pandemic. Dermatol. Ther. 2020, 33, e13740. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, C.; Ezzedine, K. Vitiligo: A focus on pathogenesis and its therapeutic implications. J. Dermatol. 2021, 48, 252–270. [Google Scholar] [CrossRef]
- Arksey, H.; O’Malley, L. Scoping studies: Towards a methodological framework. Int. J. Soc. Res. Methodol. 2005, 8, 19–32. [Google Scholar] [CrossRef]
- Ezzedine, K.; Lim, H.W.; Suzuki, T.; Katayama, I.; Hamzavi, I.; Lan, C.C.; Goh, B.K.; Anbar, T.; Silva de Castro, C.; Lee, A.Y.; et al. Vitiligo Global Issue Consensus Conference Panelists. Revised classification/nomenclature of vitiligo and related issues: The Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012, 25, E1–E13. [Google Scholar] [CrossRef]
- Bergqvist, C.; Ezzedine, K. Vitiligo: A Review. Dermatology 2020, 236, 571–592. [Google Scholar] [CrossRef]
- Kundu, R.V.; Mhlaba, J.M.; Rangel, S.M.; Le Poole, I.C. The convergence theory for vitiligo: A reappraisal. Exp. Dermatol. 2019, 28, 647–655. [Google Scholar] [CrossRef]
- Ongenae, K.; Van Geel, N.; Naeyaert, J.M. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003, 16, 90–100. [Google Scholar] [CrossRef]
- Nath, S.K.; Majumder, P.P.; Nordlund, J.J. Genetic epidemiology of vitiligo: Multilocus recessivity cross-validated. Am. J. Hum. Genet. 1994, 55, 981–990. [Google Scholar] [PubMed]
- Alkhateeb, A.; Fain, P.R.; Thody, A.; Bennett, D.C.; Spritz, R.A. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. 2003, 16, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.P.; Nordlund, J.J.; Nath, S.K. Pattern of familial aggregation of vitiligo. Arch. Dermatol. 1993, 129, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Liu, J.B.; Gui, J.P.; Li, M.; Xiong, Q.G.; Wu, H.B.; Li, J.-X.; Yang, S.; Wang, H.-Y.; Gao, M.; et al. Characteristics of genetic epidemiology and genetic models for vitiligo. J. Am. Acad. Dermatol. 2004, 51, 383–390. [Google Scholar] [CrossRef]
- Jin, Y.; Andersen, G.; Yorgov, D.; Ferrara, T.M.; Ben, S.; Brownson, K.M.; Holland, P.J.; Birlea, S.A.; Siebert, J.; Hartmann, A.; et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat. Genet. 2016, 48, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Ren, Y.Q.; Xiang, L.H.; Sun, L.D.; Xu, A.E.; Gao, X.-H.; Chen, H.-D.; Pu, X.-M.; Wu, R.-N.; Liang, C.-Z.; et al. Genome-wide association study for vitiligo identifies susceptibility loci at 6q27 and the MHC. Nat. Genet. 2010, 42, 614–618. [Google Scholar] [CrossRef]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Ferrara, T.M.; Bem, S.; Riccardi, S.L.; Cole, J.B.; Gowan, K.; Holland, P.J.; Bennett, D.C.; et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 2012, 44, 676–680. [Google Scholar] [CrossRef]
- Tang, X.F.; Zhang, Z.; Hu, D.Y.; Xu, A.E.; Zhou, H.S.; Sun, L.D.; Gao, M.; Gao, T.-W.; Gao, X.-H.; Chen, H.-D.; et al. Association analyses identify three susceptibility Loci for vitiligo in the Chinese Han population. J. Investig. Dermatol. 2013, 133, 403–410. [Google Scholar] [CrossRef]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Gowan, K.; Riccardi, S.L.; Holland, P.J.; Mailloux, C.M.; Sufit, A.J.D.; Hutton, S.M.; Amadi-Myers, A.; et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N. Engl. J. Med. 2010, 362, 1686–1697. [Google Scholar] [CrossRef]
- Dahir, A.M.; Thomsen, S.F. Comorbidities in vitiligo: Comprehensive review. Int J Dermatol. 2018, 57, 1157–1164. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, R.; Guo, X.; Zou, Y.; Chen, S.; Zhou, K.; Chen, Y.; Li, Y.; Gao, S.; Wu, Y. Identification of TYR, TYRP1, DCT and LARP7 as related biomarkers and immune infiltration characteristics of vitiligo via comprehensive strategies. Bioengineered 2021, 12, 2214–2227. [Google Scholar] [CrossRef] [PubMed]
- Spritz, R.A. The genetics of vitiligo. J. Investig. Dermatol. 2011, 131, E18–E20. [Google Scholar] [CrossRef]
- Alkhateeb, A.; Fain, P.R.; Spritz, R.A. Candidate functional promoter variant in the FOXD3 melanoblast developmental regulator gene in autosomal dominant vitiligo. J. Investig. Dermatol. 2005, 125, 388–391. [Google Scholar] [CrossRef]
- Levandowski, C.B.; Mailloux, C.M.; Ferrara, T.M.; Gowan, K.; Ben, S.; Jin, Y.; McFann, K.K.; Holland, P.J.; Fain, P.R.; Dinarello, C.A.; et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1β processing via the NLRP1 inflammasome. Proc. Natl. Acad. Sci. USA 2013, 110, 2952–2956. [Google Scholar] [CrossRef]
- Singh, R.K.; Lee, K.M.; Vujkovic-Cvijin, I.; Ucmak, D.; Farahnik, B.; Abrouk, M.; Nakamura, M.; Zhu, T.H.; Bhutani, T.; Wei, M.; et al. The role of IL-17 in vitiligo: A review. Autoimmun. Rev. 2016, 15, 397–404. [Google Scholar] [CrossRef]
- Kim, H.J.; Del Duca, E.; Pavel, A.B.; Singer, G.K.; Abittan, B.J.; Chima, M.A.; Kimmel, G.; Bares, J.; Baum, D.; Gagliotti, M.; et al. Apremilast and narrowband ultra-violet B combination therapy suppresses Th17 axis and promotes melanogenesis in vitiligo skin: A randomized, split-body, pilot study in skin types IV–VI. Arch. Dermatol. Res. 2023, 315, 215–221. [Google Scholar] [CrossRef]
- Bernardini, N.; Skroza, N.; Tolino, E.; Mambrin, A.; Anzalone, A.; Balduzzi, V.; Colapietra, D.; Marchesiello, A.; Michelini, S.; Proietti, I.; et al. IL-17 and its role in inflammatory, autoimmune, and oncological skin diseases: State of art. Int. J. Dermatol. 2020, 59, 406–411. [Google Scholar] [CrossRef]
- Le, T.V.T.; Ngoc Phan, H.; Dang, T.N.; Pham, L.D. Increased Circulatory Interleukin-17A Levels in Patients with Progressive and Leukotrichial Vitiligo. Dermatol. Res. Pract. 2021, 2021, 5524566. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhou, Y.; Yang, S.; Ren, Y.; Zhang, C.; Quan, C.; Gao, M.; He, C.; Chen, H.; Hhan, J.; et al. Platelet-derived growth factor receptor alpha gene mutations in vitiligo vulgaris. Acta Derm. Venereol. 2010, 90, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Al-Shobaili, H.A. Update on the genetics characterization of vitiligo. Int. J. Health Sci. 2011, 5, 167–179. [Google Scholar]
- Ren, Y.; Yang, S.; Xu, S.; Gao, M.; Huang, W.; Gao, T.; Fang, Q.; Quan, C.; Zhang, C.; Sun, L.; et al. Genetic variation of promoter sequence modulates XBP1 expression and genetic risk for vitiligo. PLoS Genet. 2009, 5, e1000523. [Google Scholar] [CrossRef] [PubMed]
- Marchioro, H.Z.; Silva de Castro, C.C.; Fava, V.M.; Sakiyama, P.H.; Dellatorre, G.; Miot, H.A. Update on the pathogenesis of vitiligo. An. Bras. Dermatol. 2022, 97, 478–490. [Google Scholar] [CrossRef]
- Okamoto, M.; Watanabe, M.; Inoue, N.; Ogawa, K.; Hidaka, Y.; Iwatani, Y. Gene polymorphisms of VEGF and VEGFR2 are associated with the severity of Hashimoto’s disease and the intractability of Graves’ disease, respectively. Endocr. J. 2020, 67, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zhou, T.; Zhong, Z.; Zhong, H. Meta-analysis of associations of vascular endothelial growth factor protein levels and −634G/C polymorphism with systemic lupus erythematosus susceptibility. BMC Med. Genet. 2019, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Hong, Y.J.; Kim, M. Angiogenesis in Chronic Inflammatory Skin Disorders. Int. J. Mol. Sci. 2021, 22, 12035. [Google Scholar] [CrossRef]
- Mabeta, P.; Steenkamp, V. The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15585. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, O.; Lucarini, G.; Campanati, A.; Goteri, G.; Zizzi, A.; Marconi, B.; Ganzetti, G.; Minardi, D.; Di Primio, R.; Offidani, A. VEGF, survivin and NOS overexpression in psoriatic skin: Critical role of nitric oxide synthases. J. Dermatol. Sci. 2009, 54, 205–208. [Google Scholar] [CrossRef]
- Simonetti, O.; Lucarini, G.; Rubini, C.; Goteri, G.; Zizzi, A.; Staibano, S.; Campanati, A.; Ganzetti, G.; Di Primio, R.; Offidani, A. Microvessel density and VEGF, HIF-1α expression in primary oral melanoma: Correlation with prognosis. Oral Dis. 2013, 19, 620–627. [Google Scholar] [CrossRef]
- Almasi-Nasrabadi, M.; Amoli, M.M.; Robati, R.M.; Rajabi, F.; Parichehreh Dizaji, S. Is the +405 G/C single nucleotide polymorphism of the vascular endothelial growth factor (VEGF) gene associated with late-onset vitiligo? Int. J. Immunogenet. 2019, 46, 241–246. [Google Scholar] [CrossRef]
- Roberts, G.H.L.; Santorico, S.A.; Spritz, R.A. Deep genotype imputation captures virtually all heritability of autoimmune vitiligo. Hum. Mol. Genet. 2020, 29, 859–863. [Google Scholar] [CrossRef]
- Sandru, F.; Carsote, M.; Albu, S.E.; Dumitrascu, M.C.; Valea, A. Vitiligo and chronic autoimmune thyroiditis. J. Med. Life 2021, 14, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Lommerts, J.E.; Bekkenk, M.W.; Luiten, R.M. Vitiligo induced by immune checkpoint in-hibitors in melanoma patients: An expert opinion. Expert Opin. Drug Saf. 2021, 20, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Xu, R.; Fan, B.; Chen, J.; Li, X.; Mao, W.; Hua, S.; Li, B. PD-L1 reverses depigmentation in pmel-1 vitiligo mice by increasing the abundance of tregs in the skin. Sci. Rep. 2018, 8, 1605. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chapman, N.M.; Zhang, B.; Li, M.; Fan, M.; Laribee, R.N.; Zaidi, M.R.; Pfeffer, L.M.; Chi, H.; Wu, Z.-H. Upregulation of PD-L1 via HMGB1-activated IRF3 and NF-κB contributes to UV radiation-induced immune suppression. Cancer Res. 2019, 79, 2909–2922. [Google Scholar] [CrossRef]
- Iqbal, S.; Premalatha, S.; Zahra, A. Dermatoglyphics in vitiligo. Int. J. Dermatol. 1985, 24, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Schallreuter, K.U.; Lemke, R.; Brandt, O.; Schwartz, R.; Westhofen, M.; Montz, R.; Berger, J. Vitiligo and other diseases: Coexistence or true association? Hamburg study on 321 patients. Dermatology 1994, 188, 269–275. [Google Scholar] [CrossRef]
- Birlea, S.A.; Fain, P.R.; Spritz, R.A. A Romanian population isolate with high frequency of vitiligo and associated autoimmune diseases. Arch. Dermatol. 2008, 144, 310–316. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Bahadoran, P.; Picardo, M.; Slominski, A.; Elassiuty, Y.E.; Kemp, E.H.; Giachino, C.; Liu, J.B.; Luiten, R.M.; Lambe, T.; et al. Vitiligo pathogenesis: Autoimmune disease, genetic defect, excessive reactive oxygen species, calcium imbalance, or what else? Exp. Dermatol. 2008, 17, 139–140, discussion 141–160. [Google Scholar] [CrossRef]
- Jin, Y.; Mailloux, C.M.; Gowan, K.; Riccardi, S.L.; LaBerge, G.; Bennett, D.C.; Fain, P.R.; Spritz, R.A. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 2007, 356, 1216–1225. [Google Scholar] [CrossRef]
- Wong, P.M.; Yang, L.; Yang, L.; Wu, H.; Li, W.; Ma, X.; Katayama, I.; Zhang, H. New insight into the role of exosomes in vitiligo. Autoimmun. Rev. 2020, 19, 102664. [Google Scholar] [CrossRef]
- Liu, L.S.P.; Yi, X.; Li, C.; Gao, T. 067 Serum-derived exosomes contribute to abnormal melanocyte function in patients with active vitiligo. J. Investig. Dermatol. 2016, 136, S12. [Google Scholar] [CrossRef]
- Mosenson, J.A.; Eby, J.M.; Hernandez, C.; Le Poole, I.C. A central role for inducible heat-shock protein 70 in autoimmune vitiligo. Exp. Dermatol. 2013, 22, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Henning, S.W.; Fernandez, M.F.; Mahon, J.P.; Duff, R.; Azarafrooz, F.; Guevara-Patiño, J.A.; Rademaker, A.W.; Salzman, A.L.; Le Poole, I.C. HSP70iQ435Aencoding DNA repigments vitiligo lesions in sinclair swine. J. Investig. Dermatol. 2018, 138, 2531–2539. [Google Scholar] [CrossRef]
- Van den Boorn, J.G.; Konijnenberg, D.; Dellemijn, T.A.M.; Van Der Veen, J.P.W.; Bos, J.D.; Melief, C.J.M.; Vyth-Dreese, F.A.; Luiten, R.M. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J. Investig. Dermatol. 2009, 129, 2220–2232. [Google Scholar] [CrossRef] [PubMed]
- Le Poole, I.C.; van den Wijngaard, R.M.; Westerhof, W.; Das, P.K. Presence of T cells and macrophages in inflammatory vitiligo skin parallels melanocyte disappearance. Am. J. Pathol. 1996, 148, 1219–1228. [Google Scholar]
- Strassner, J.P.; Rashighi, M.; Refat, M.A.; Harris, J.E. Suction blistering the lesional skin of vitiligo patients reveal useful biomarkers of disease activity. J. Am. Acad. Dermatol. 2017, 76, 847–855. [Google Scholar] [CrossRef]
- Xie, H.; Zhou, F.; Liu, L.; Zhu, G.; Li, Q.; Li, C.; Gao, T. Vitiligo: How do oxidative stress-induced autoantigens trigger autoimmunity? J. Dermatol. Sci. 2016, 81, 3–9. [Google Scholar] [CrossRef]
- Harris, J.E.; Harris, T.H.; Weninger, W.; Wherry, E.J.; Hunter, C.A.; Turka, L.A. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin. J. Investig. Dermatol. 2012, 132, 1869–1876. [Google Scholar] [CrossRef]
- Rashighi, M.; Agarwal, P.; Richmond, J.M.; Harris, T.H.; Dresser, K.; Su, M.-W.; Zhou, Y.; Deng, A.; Hunter, C.A.; Luster, A.D.; et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci. Transl. Med. 2014, 6, 223ra23. [Google Scholar]
- Abdallah, M.; El-Mofty, M.; Anbar, T.; Rasheed, H.; Esmat, S.; Al-Tawdy, A.; Fawzy, M.M.; Abdel-Halim, D.; Hegazy, R.; Gawdat, H.; et al. CXCL-10 and Interleukin-6 are reliable serum markers for vitiligo activity: A multicenter cross-sectional study. Pigment Cell Melanoma Res. 2018, 31, 330–336. [Google Scholar] [CrossRef]
- Abdou, A.G.; Maraee, A.; Yassien, H.; Sarhan, M. Immunohistochemistry of Janus Kinase 1 (JAK1) expression in vitiligo. J. Pathol. Transl. Med. 2018, 52, 363–368. [Google Scholar] [CrossRef]
- Howell, M.D.; Kuo, F.I.; Smith, P.A. Targeting the Janus Kinase family in autoimmune skin diseases. Front. Immunol. 2019, 10, 2342. [Google Scholar] [CrossRef]
- Dwivedi, M.; Kemp, E.H.; Laddha, N.C.; Mansuri, M.S.; Weetman, A.P.; Begum, R. Regulatory T cells in vitiligo: Implications for pathogenesis and therapeutics. Autoimmun. Rev. 2015, 14, 49–56. [Google Scholar] [CrossRef]
- Klarquist, J.; Denman, C.J.; Hernandez, C.; Wainwright, D.; Strickland, F.M.; Overbeck, A.; Mehrotra, S.; Nishimura, M.I.; Le Poole, I.C. Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res. 2010, 23, 276–286. [Google Scholar] [CrossRef]
- Eby, J.M.; Kang, H.K.; Tully, S.T.; Bindeman, W.E.; Peiffer, D.S.; Chatterjee, S.; Mehrotra, S.; Le Poole, I.C. CCL22 to activate Treg migration and suppress depigmentation in vitiligo. J. Investig. Dermatol. 2015, 135, 1574–1580. [Google Scholar] [CrossRef]
- Nicolaidou, E.; Antoniou, C.; Stratigos, A.J.; Stefanaki, C.; Katsambas, A.D. Efficacy, predictors of response, and long-term follow-up in patients with vitiligo treated with narrowband UVB phototherapy. J. Am. Acad. Dermatol. 2007, 56, 274–278. [Google Scholar] [CrossRef]
- Boniface, K.; Jacquemin, C.; Darrigade, A.-S.; Dessarthe, B.; Martins, C.; Boukhedouni, N.; Vernisse, C.; Grasseau, A.; Thiolat, D.; Rambert, J.; et al. Vitiligo skin is imprinted with resident memory CD8 T cells expressing CXCR3. J. Investig. Dermatol. 2018, 138, 355–364. [Google Scholar] [CrossRef]
- Richmond, J.M.; Strassner, J.P.; Rashighi, M.; Agarwal, P.; Garg, M.; Essien, K.I.; Pell, L.S.; Harris, J.E. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J. Investig. Dermatol. 2019, 139, 769–778. [Google Scholar] [CrossRef]
- Richmond, J.M.; Strassner, J.P.; Zapata, L., Jr.; Garg, M.; Riding, R.L.; Refat, M.A.; Fan, X.; Azzolino, V.; Tovar-Garza, A.; Tsurushita, N.; et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci. Transl. Med. 2018, 10, eaam7710. [Google Scholar] [CrossRef]
- Kroon, M.W.; Kemp, E.H.; Wind, B.S.; Krebbers, G.; Bos, J.D.; Gawkrodger, D.J.; Wolkerstorfer, A.; van der Veen, J.P.W.; Luiten, R.M. Melanocyte antigen-specific antibodies cannot be used as markers for recent disease activity in patients with vitiligo. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1172–1175. [Google Scholar] [CrossRef]
- Migayron, L.; Boniface, K.; Seneschal, J. Vitiligo, From Physiopathology to Emerging Treatments: A Review. Dermatol. Ther. 2020, 10, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Denat, L.; Kadekaro, A.L.; Marrot, L.; Leachman, S.A.; Abdel-Malek, Z.A. Melanocytes as instigators and victims of oxidative stress. J. Investig. Dermatol. 2014, 134, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, I.C.; Batcioglu, K.; Karatas, F.; Hazneci, E.; Genc, M. Comparison of plasma malondialdehyde, glutathione, glutathione peroxidase, hydroxyproline and selenium levels in patients with vitiligo and healthy controls. Indian J. Dermatol. 2008, 53, 106–110. [Google Scholar] [PubMed]
- Sravani, P.V.; Babu, N.K.; Gopal, K.V.T.; Rao, G.R.R.; Rao, A.R.; Moorthy, B.; Rao, T.R. Determination of oxidative stress in vitiligo by measuring superoxide dismutase and catalase levels in vitiliginous and non-vitiliginous skin. Indian J. Dermatol. Venereol. Leprol. 2009, 75, 268–271. [Google Scholar] [PubMed]
- Xuan, Y.; Yang, Y.; Xiang, L.; Zhang, C. The Role of Oxidative Stress in the Pathogenesis of Vitiligo: A Culprit for Melanocyte Death. Oxid. Med. Cell. Longev. 2022, 2022, 8498472. [Google Scholar] [CrossRef] [PubMed]
- Al-Shobaili, H.A.; Rasheed, Z. Oxidized tyrosinase: A possible antigenic stimulus for non-segmental vitiligo autoantibodies. J. Dermatol. Sci. 2015, 79, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Passeron, T.; Ortonne, J.P. Activation of the unfolded protein response in vitiligo: The missing link? J. Investig. Dermatol. 2012, 132, 2502–2504. [Google Scholar] [CrossRef]
- Toosi, S.; Orlow, S.J.; Manga, P. Vitiligo-inducing phenols activate the unfolded protein response in melanocytes resulting in upregulation of IL6 and IL8. J. Investig. Dermatol. 2012, 132, 2601–2609. [Google Scholar] [CrossRef]
- Kang, P.; Zhang, W.; Chen, X.; Yi, X.; Song, P.; Chang, Y.; Zhang, S.; Gao, T.; Li, C.; Li, S. TRPM2 mediates mitochondria-dependent apoptosis of melanocytes under oxidative stress. Free Radic. Biol. Med. 2018, 126, 259–268. [Google Scholar] [CrossRef]
- Maresca, V.; Roccella, M.; Roccella, F.; Camera, E.; Del Porto, G.; Passi, S.; Grammatico, P.; Picardo, M. Increased sensitivity to peroxidative agents as a possible pathogenic factor of melanocyte damage in vitiligo. J. Investig. Dermatol. 1997, 109, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Zedan, H.; Abdel-Motaleb, A.A.; Kassem, N.M.; Hafeez, H.A.; Hussein, M.R. Low glutathione peroxidase activity levels in patients with vitiligo. J. Cutan. Med. Surg. 2015, 19, 144–148. [Google Scholar] [CrossRef]
- Jian, Z.; Li, K.; Song, P.; Zhu, G.; Zhu, L.; Cui, T.; Liu, B.; Tang, L.; Wang, X.; Wang, G.; et al. Impaired activation of the Nrf2-ARE signaling pathway undermines H2O2-induced oxidative stress response: A possible mechanism for melanocyte degeneration in vitiligo. J. Investig. Dermatol. 2014, 134, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Meng, X.; Song, Z.; Lin, J. Nuclear factor erythroid 2-related factor 2 (Nrf2) as a potential therapeutic target for vitiligo. Arch. Biochem. Biophys. 2020, 696, 108670. [Google Scholar] [CrossRef]
- Zhang, C.F.; Gruber, F.; Ni, C.; Mildner, M.; Koening, U.; Karner, S.; Barresi, C.; Rossiter, H.; Narzt, M.-S.; Nagelreiter, I.M.; et al. Suppression of autophagy dysregulates the antioxidant response and causes premature senescence of melanocytes. J. Investig. Dermatol. 2015, 135, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Phiske, M.M.; Jerajani, H.R. Evaluation of safety and efficacy of topical prostaglandin E2 in treatment of vitiligo. Br. J. Dermatol. 2009, 160, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, Y.; Cario-Andre, M.; Lepreux, S.; Pain, C.; Taieb, A. Melanocyte detachment after skin friction in non lesional skin of patients with generalized vitiligo. Br. J. Dermatol. 2003, 148, 95–101. [Google Scholar]
- Bertolotti, A.; Boniface, K.; Vergier, B.; Mossalayi, D.; Taieb, A.; Ezzedine, K.; Seneschal, J. Type I interferon signature in the initiation of the immune response in vitiligo. Pigment Cell Melanoma Res. 2014, 27, 398–407. [Google Scholar] [CrossRef]
- Karsli, N.; Akcali, C.; Ozgoztasi, O.; Kirtak, N.; Inaloz, S. Role of oxidative stress in the pathogenesis of vitiligo with special emphasis on the antioxidant action of narrowband ultraviolet B phototherapy. J. Int. Med. Res. 2014, 42, 799–805. [Google Scholar] [CrossRef]
- Speeckaert, R.; Dugardin, J.; Lambert, J.; Lapeere, H.; Verghaehe, E.; Speeckaert, M.M.; van Geel, N. Critical appraisal of the oxidative stress pathway in vitiligo: A systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1089–1098. [Google Scholar] [CrossRef]
- Wan, J.; Lin, F.; Zhang, W.; Xu, A.; DeGiorgis, J.; Lu, H.; Wan, Y. Novel approaches to vitiligo treatment via modulation of mTOR and NF-κB pathways in human skin melanocytes. Int. J. Biol. Sci. 2017, 13, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.W.; Kelly, R.; Winans, T.; Marchena, I.; Shadakshari, A.; Yu, J.; Dawood, M.; Garcia, R.; Tily, H.; Francis, L.; et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: A single-arm, open-label, phase 1/2 trial. Lancet 2018, 391, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Eby, J.M.; Al-Khami, A.A.; Soloshchenko, M.; Kang, H.K.; Kaur, N.; Naga, O.S.; Murali, A.; Nishimura, M.I.; Le Poole, I.C.; et al. A quantitative increase in regulatory T cells controls development of vitiligo. J. Investig. Dermatol. 2014, 134, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, D.; He, M.; Lin, M.; Tu, C.; Zhang, B. Polymeric nanoparticles containing rapamycin and autoantigen induce antigen-specific immunological tolerance for preventing vitiligo in mice. Hum. Vaccines Immunother. 2021, 17, 1923–1929. [Google Scholar] [CrossRef]
- Njoo, M.D.; Westerhof, W. Vitiligo. Pathogenesis and treatment. Am. J. Clin. Dermatol. 2001, 2, 167–181. [Google Scholar] [CrossRef]
- Nordlund, J.J. The significance of depigmentation. Pigment Cell Res. 1992, 2, 237–241. [Google Scholar] [CrossRef]
- Al’Abadie, M.S.; Senior, H.J.; Bleehen, S.S.; Gawkrodger, D.J. Neuropeptide and neuronal marker studies in vitiligo. Br. J. Dermatol. 1994, 131, 160–165. [Google Scholar] [CrossRef]
- Bose, S.K. Probable mechanisms of loss of Merkel cells in completely depigmented skin of stable vitiligo. J. Dermatol. 1994, 21, 725–728. [Google Scholar] [CrossRef]
- Simons, R.E.; Zevy, D.L.; Jafferany, M. Psychodermatology of vitiligo: Psychological impact and consequences. Dermatol. Ther. 2020, 33, e13418. [Google Scholar] [CrossRef]
- Wu, C.S.; Yu, H.S.; Chang, H.R.; Yu, C.L.; Yu, C.L.; Wu, B.N. Cutaneous blood flow and adrenoceptor response increase in segmental-type vitiligo lesions. J. Dermatol. Sci. 2000, 23, 53–62. [Google Scholar] [CrossRef]
- Morrone, A.; Picardo, M.; de Luca, C.; Terminali, O.; Passi, S.; Ippolito, F. Catecholamines and vitiligo. Pigment Cell Res. 1992, 5, 58–62. [Google Scholar] [CrossRef]
- Lazarova, R.; Hristakieva, E.; Lazarov, N.; Shani, J. Vitiligo-related neuropeptides in nerve fibers of the skin. Arch. Physiol. Biochem. 2000, 108, 262–267. [Google Scholar] [PubMed]
- Rateb, A.A.H.; Azzam, O.A.; Rashed, L.A.; El-Guindy, N.M.; El-Din, M.S. The role of nerve growth factor in the pathogenesis of vitligo. JEWDS 2005, 1, 18–24. [Google Scholar]
- Al’Abadie, M.S.; Warren, M.A.; Bleehen, S.S.; Gawkrodger, D.J. Morphologic observations on the dermal nerves in vitiligo: An ultrastructural study. Int. J. Dermatol. 1995, 34, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Ezzedine, K.; Hamzavi, I.; Pandya, A.G.; Harris, J.E. Vitiligo Working Group. Current and emerging treatments for vitiligo. J. Am. Acad. Dermatol. 2017, 77, 17–29. [Google Scholar] [CrossRef]
- Minder, E.I.; Barman-Aksoezen, J.; Schneider-Yin, X. Pharmacokinetics and Pharmacodynamics of Afamelanotide and Its Clinical Use in Treating Dermatologic Disorders. Clin. Pharmacokinet. 2017, 56, 815–823. [Google Scholar] [CrossRef]
- Lim, H.W.; Grimes, P.E.; Agbai, O.; Hamzavi, I.; Henderson, M.; Haddican, M.; Linkner, R.V.; Lebwohl, M. Afamelanotide and narrowband UV-B phototherapy for the treatment of vitiligo: A randomized multicenter trial. JAMA Dermatol. 2015, 151, 42–50. [Google Scholar] [CrossRef]
- Karagaiah, P.; Valle, Y.; Sigova, J.; Zerbinati, N.; Vojvodic, P.; Parsad, D.; Schwartz, R.A.; Grabbe, S.; Goldust, M.; Lotti, T. Emerging drugs for the treatment of vitiligo. Expert Opin. Emerg. Drugs 2020, 25, 7–24. [Google Scholar] [CrossRef]
- Grimes, P.E.; Hamzavi, I.; Lebwohl, M.; Ortonne, J.P.; Lim, H.W. The efficacy of afamelanotide and narrowband UV-B phototherapy for repigmentation of vitiligo. JAMA Dermatol. 2013, 149, 68–73. [Google Scholar] [CrossRef]
- Toh, J.J.H.; Chuah, S.Y.; Jhingan, A.; Chong, W.S.; Thng, S.T.G. Afamelanotide implants and narrow-band ultraviolet B phototherapy for the treatment of nonsegmental vitiligo in Asians. J. Am. Acad. Dermatol. 2020, 82, 1517–1519. [Google Scholar] [CrossRef]
- Passeron, T. Indications and limitations of afamelanotide for treating vitiligo. JAMA Dermatol. 2015, 151, 349–350. [Google Scholar] [CrossRef]
- Lim, H.W.; Grimes, P.E.; Lebwohl, M. Indications and limitations of afamelanotide for treating vitiligo-reply. JAMA Dermatol. 2015, 151, 350. [Google Scholar] [CrossRef]
- Zubair, R.; Hamzavi, I.H. Phototherapy for Vitiligo. Dermatol. Clin. 2020, 38, 55–62. [Google Scholar] [CrossRef]
- Parsad, D.; Pandhi, R.; Dogra, S.; Kumar, B. Topical prostaglandin analog (PGE2) in vitiligo--a preliminary study. Int. J. Dermatol. 2002, 41, 942–945. [Google Scholar] [CrossRef]
- Hempel, L.; Wessels, D.A. Prostaglandin E2 synthesis after oxidant stress is dependent on cell glutathione content. Am. J. Physiol. Cell Physiol. 1994, 266, 1392–1399. [Google Scholar] [CrossRef]
- Kapur, R.; Osmanovic, S.; Toyran, S.; Edward, D.P. Bimatoprost-induced periocular skin hyperpigmentation: Histopathological study. Arch. Ophthalmol. 2005, 123, 1541. [Google Scholar] [CrossRef]
- Narang, G. Efficacy and Safety of Topical Bimatoprost Solution 0.03% in Stable Vitiligo: A Prelliminary Study. In Proceedings of the World Congress of Dermatology, Seoul, Republic of Korea, 22–29 May 2011. [Google Scholar]
- Kanokrungsee, S.; Pruettivorawongse, D.; Rajatanavin, N. Clinical outcomes of topical bimatoprost for nonsegmental facial vitiligo: A preliminary study. J. Cosmet. Dermatol. 2021, 20, 812–818. [Google Scholar] [CrossRef]
- Grimes, P.E. Bimatoprost 0.03% Solution for the Treatment of Nonfacial Vitiligo. J. Drugs Dermatol. 2016, 15, 703. [Google Scholar]
- Anbar, T.S.; El-Ammawi, T.S.; Abdel-Rahman, A.T.; Hanna, M.R. The effect of latanoprost on vitiligo: A preliminary comparative study. Int. J. Dermatol. 2015, 54, 587–593. [Google Scholar] [CrossRef]
- Korobko, I.V.; Lomonosov, K.M. A pilot comparative study of topical latanoprost and tacrolimus in combination with narrow-band ultraviolet B phototherapy and microneedling for the treatment of nonsegmental vitiligo. Dermatol. Ther. 2016, 29, 437–441. [Google Scholar] [CrossRef]
- Neinaa, Y.M.E.; Lotfy, S.S.; Ghaly, N.R.; Doghaim, N.N. A comparative study of combined microneedling and narrowband ultraviolet B phototherapy versus their combination with topical latanoprost in the treatment of vitiligo. Dermatol. Ther. 2021, 34, e14813. [Google Scholar] [CrossRef] [PubMed]
- Radi, G.; Simonetti, O.; Rizzetto, G.; Diotallevi, F.; Molinelli, E.; Offidani, A. Baricitinib: The First Jak Inhibitor Approved in Europe for the Treatment of Moderate to Severe Atopic Dermatitis in Adult Patients. Healthcare 2021, 9, 1575. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Lu, Y. Advances in vitiligo: Update on therapeutic targets. Front. Immunol. 2022, 13, 986918. [Google Scholar] [CrossRef] [PubMed]
- Birlea, S.A.; Costin, G.E.; Roop, D.R.; Norris, D.A. Trends in Regenerative Medicine: Re-pigmentation in Vitiligo through Melanocyte Stem Cell Mobilization. Med. Res. Rev. 2017, 37, 907–935. [Google Scholar] [CrossRef]
- Harris, J.E.; Rashighi, M.; Nguyen, N.; Jabbari, A.; Ulerio, G.; Clynes, R.; Christiano, A.M.; Mackay-Wiggan, J. Rapid skin repigmentation on oral ruxolitinib in a patient with coexistent vitiligo and alopecia areata (AA). J. Am. Acad. Dermatol. 2016, 74, 370. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Strassner, J.P.; Refat, M.A.; Harris, J.E.; King, B.A. Repigmentation in vitiligo using the Janus kinase inhibitor tofacitinib may require concomitant light exposure. J. Am. Acad. Dermatol. 2017, 77, 675. [Google Scholar] [CrossRef]
- Phan, K.; Phan, S.; Shumack, S.; Gupta, M. Repigmentation in vitiligo using janus kinase (JAK) inhibitors with phototherapy: Systematic review and Meta-analysis. J. Dermatol. Treat. 2022, 33, 173. [Google Scholar] [CrossRef] [PubMed]
- Hesham, H.; Rady, M.; Hathout, R.M.; Abdel-Halim, M.; Mansour, S. The skin delivery of tofacitinib citrate using transethosomes and hybridized ethosomes/nanostructured lipid carriers for vitiligo therapy: Dermatopharmacokinetics and in vivo assays. Int. J. Pharm. 2022, 629, 122387. [Google Scholar] [CrossRef]
- Rothstein, B.; Joshipura, D.; Saraiya, A.; Abdat, R.; Ashkar, H.; Turkowski, Y.; Sheth, V.; Huang, V.; Au, S.C.; Kachuk, C.; et al. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib. J. Am. Acad. Dermatol. 2017, 76, 1054. [Google Scholar] [CrossRef]
- Rosmarin, D.; Pandya, A.G.; Lebwohl, M.; Grimes, P.; Hamzavi, I.; Gottlieb, A.B.; Butler, K.; Kuo, F.; Sun, K.; Ji, T.; et al. Ruxolitinib cream for treatment of vitiligo: A randomised, controlled, phase 2 trial. Lancet 2020, 396, 110. [Google Scholar] [CrossRef]
- Dong, J.; Huang, X.; Ma, L.P.; Qi, F.; Wang, S.N.; Zhang, Z.Q.; Wei, S.N.; Gao, L.; Liu, F. Baricitinib is Effective in Treating Progressing Vitiligo in vivo and in vitro. Dose Response 2022, 20, 15593258221105370. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K.; Ishida, Y.; Otsuka, A.; Kabashima, K. Two cases of vitiligo vulgaris treated with topical Janus kinase inhibitor delgocitinib. Australas. J. Dermatol. 2021, 62, 433–434. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diotallevi, F.; Gioacchini, H.; De Simoni, E.; Marani, A.; Candelora, M.; Paolinelli, M.; Molinelli, E.; Offidani, A.; Simonetti, O. Vitiligo, from Pathogenesis to Therapeutic Advances: State of the Art. Int. J. Mol. Sci. 2023, 24, 4910. https://doi.org/10.3390/ijms24054910
Diotallevi F, Gioacchini H, De Simoni E, Marani A, Candelora M, Paolinelli M, Molinelli E, Offidani A, Simonetti O. Vitiligo, from Pathogenesis to Therapeutic Advances: State of the Art. International Journal of Molecular Sciences. 2023; 24(5):4910. https://doi.org/10.3390/ijms24054910
Chicago/Turabian StyleDiotallevi, Federico, Helena Gioacchini, Edoardo De Simoni, Andrea Marani, Matteo Candelora, Matteo Paolinelli, Elisa Molinelli, Annamaria Offidani, and Oriana Simonetti. 2023. "Vitiligo, from Pathogenesis to Therapeutic Advances: State of the Art" International Journal of Molecular Sciences 24, no. 5: 4910. https://doi.org/10.3390/ijms24054910
APA StyleDiotallevi, F., Gioacchini, H., De Simoni, E., Marani, A., Candelora, M., Paolinelli, M., Molinelli, E., Offidani, A., & Simonetti, O. (2023). Vitiligo, from Pathogenesis to Therapeutic Advances: State of the Art. International Journal of Molecular Sciences, 24(5), 4910. https://doi.org/10.3390/ijms24054910