

Characterization of Genes That Exhibit Genotype-Dependent Allele-Specific Expression and Its Implications for the Development of Maize Kernel

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
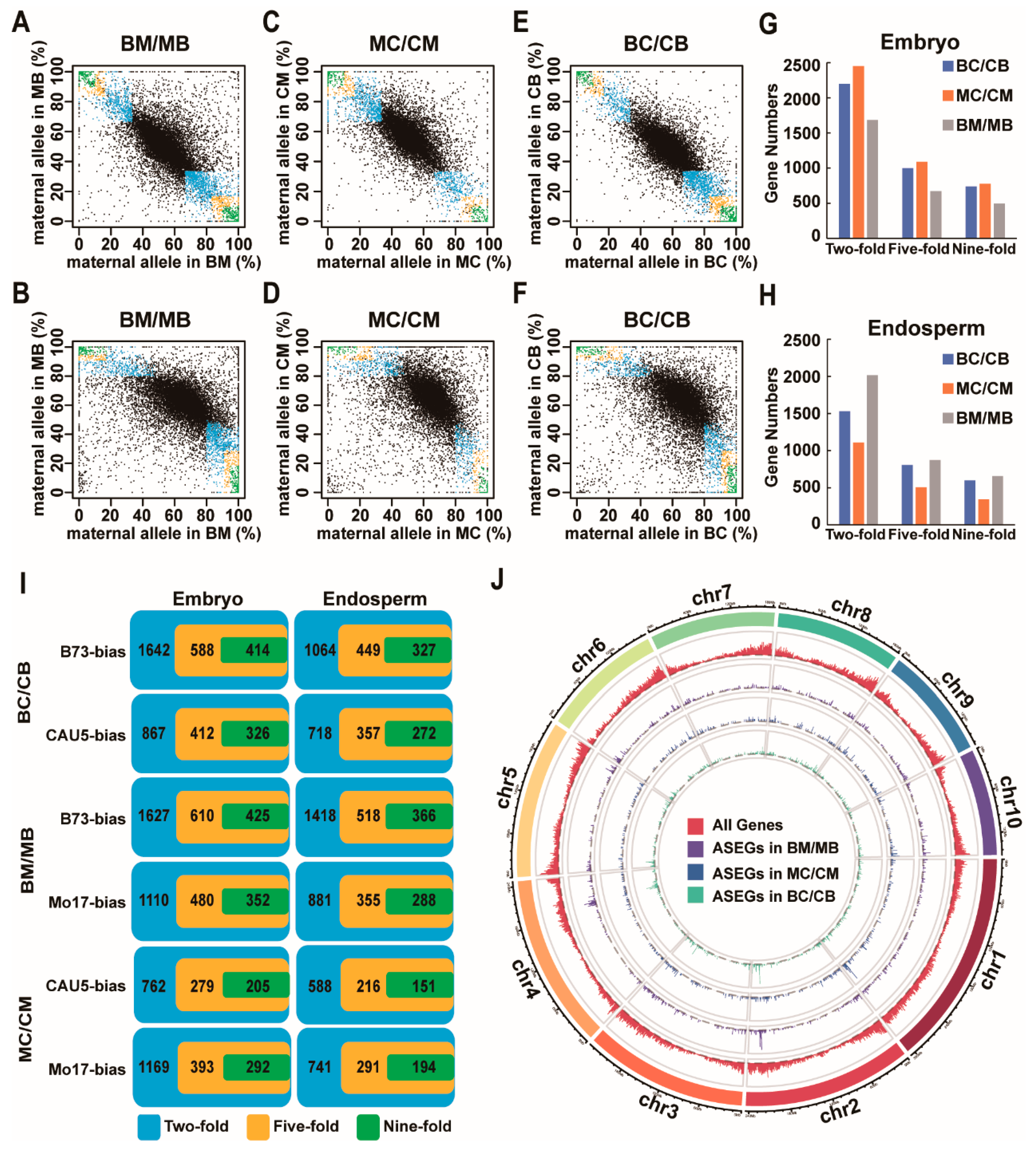
2.1. Identification of ASEGs in the Embryo and Endosperm of Three Reciprocal Crosses
2.2. Most of the ASEGs Were Consistent ASE across Different Tissues
2.3. Half of the ASEGs Were Consistent ASE across Different Crosses
2.4. Mutant of One ASEG Zm00001d046765 Affects Kernel Development
2.5. The Allele Pattern of Methylation Level around ASEG
3. Discussion
3.1. ASEGs in the Different Tissues and Hybrid Crosses
3.2. The Potential Function of Genotype-Dependent ASEGs
3.3. Methylation Plays an Important Role in the Regulation of Allelic Expression of Genotype-Dependent ASEGs
4. Materials and Methods
4.1. Plant Materials
4.2. Library Construction for RNA-Seq and MethylC-Seq
4.3. Read Mapping, Gene Expression Analysis, and SNP Calling
4.4. Identification of Genotype-Dependent ASEGs
4.5. GO Term Enrichment and Functional Category Analysis
4.6. Pipeline for MethylC-Seq Analysis
4.7. Genetic Transformation of Maize
4.8. The Method of Measuring Kernel Area
4.9. Primers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BM and MB | represent the crosses of B73 × Mo17 and Mo17 × B73, respectively. |
| BC and CB | represent the crosses of B73 × CAU5 and CAU5 × B73, respectively. |
| MC and CM | represent the crosses of Mo17 × CAU5 and CAU5 × Mo17, respectively. |
| DEGs | represent differentially expressed genes. |
| ASE | represents allele-specific expression. |
| ASEGs | represent allele-specific expression genes. |
| GO | represents gene ontology. |
| DAP | represents days after pollination. |
| WT | represents a wild type. |
References
- Schnable, P.S.; Springer, N.M. Progress toward understanding heterosis in crop plants. Plant Biol. 2013, 64, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, S.; Gong, J.; Zhao, Q.; Feng, Q.; Zhan, Q.; Zhao, Y.; Li, W.; Cheng, B.; Xia, J.; et al. Genomic architecture of heterosis for yield traits in rice. Nature 2016, 537, 629–633. [Google Scholar] [CrossRef]
- Goff, S.A.; Zhang, Q. Heterosis in elite hybrid rice: Speculation on the genetic and biochemical mechanisms. Plant Biol. 2013, 16, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.P.; Xing, Y.Z.; Xu, C.G.; Sun, X.L.; Yu, S.B.; Zhang, Q. Genetic Dissection of an Elite Rice Hybrid Revealed That Heterozygotes Are Not Always Advantageous for Performance. Genetics 2002, 162, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, X.; Ren, D.; Huang, H.; Xu, M.; He, G.; Deng, X.W. Genomic architecture of biomass heterosis in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, 8101–8106. [Google Scholar] [CrossRef]
- Hua, J.; Xing, Y.; Wu, W.; Xu, C.; Sun, X.; Yu, S.; Zhang, Q. Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 2003, 100, 2574–2579. [Google Scholar] [CrossRef]
- Yu, D.; Gu, X.; Zhang, S.; Dong, S.; Miao, H.; Gebretsadik, K.; Bo, K. Molecular basis of heterosis and related breeding strategies reveal its importance in vegetable breeding. Hortic. Res. 2021, 8, 120. [Google Scholar] [CrossRef]
- Niayale, R.; Cui, Y.; Adzitey, F. Male hybrid sterility in the cattle-yak and other bovines: A review. Biol. Reprod. 2021, 104, 495–507. [Google Scholar] [CrossRef]
- Guo, M.; Rupe, M.A.; Zinselmeier, C.; Habben, J.; Bowen, B.A.; Smith, O.S. Allelic variation of gene expression in maize hybrids. Plant Cell 2004, 16, 1707–1716. [Google Scholar] [CrossRef]
- Guo, M.; Rupe, M.A.; Yang, X.; Crasta, O.; Zinselmeier, C.; Smith, O.S.; Bowen, B. Genome-wide transcript analysis of maize hybrids: Allelic additive gene expression and yield heterosis. Theor. Appl. Genet. 2006, 113, 831–845. [Google Scholar] [CrossRef]
- Springer, N.M.; Stupar, R.M. Allelic variation and heterosis in maize: How do two halves make more than a whole? Genome Res. 2007, 17, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Paschold, A.; Jia, Y.; Marcon, C.; Lund, S.; Larson, N.B.; Yeh, C.T.; Ossowski, S.; Lanz, C.; Nettleton, D.; Schnable, P.S.; et al. Complementation contributes to transcriptome complexity in maize (Zea mays L.) hybrids relative to their inbred parents. Genome Res. 2012, 22, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Paschold, A.; Larson, N.B.; Marcon, C.; Schnable, J.C.; Yeh, C.T.; Lanz, C.; Nettleton, D.; Piepho, H.P.; Schnable, P.S.; Hochholdinger, F. Nonsyntenic genes drive highly dynamic complementation of gene expression in Maize hybrids. Plant Cell 2014, 26, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.J.; Makarevitch, I.; Noshay, J.; Burghardt, L.T.; Hirsch, C.N.; Hirsch, C.D.; Springer, N.M. Natural variation for gene expression responses to abiotic stress in maize. Plant J. 2017, 89, 706–717. [Google Scholar] [CrossRef]
- Zhang, X.; Borevitz, J.O. Global analysis of allele-specific expression in Arabidopsis thaliana. Genetics 2009, 182, 943–954. [Google Scholar] [CrossRef]
- Song, G.; Guo, Z.; Liu, Z.; Cheng, Q.; Qu, X.; Chen, R.; Jiang, D.; Liu, C.; Wang, W.; Sun, Y.; et al. Global RNA sequencing reveals that genotype-dependent allele-specific expression contributes to differential expression in rice F1 hybrids. BMC Plant Biol. 2013, 13, 221. [Google Scholar] [CrossRef]
- He, G.; Chen, B.; Wang, X.; Li, X.; Li, J.; He, H.; Yang, M.; Lu, L.; Qi, Y.; Wang, X.; et al. Conservation and divergence of transcriptomic and epigenomic variation in maize hybrids. Genome Biol. 2013, 14, R57. [Google Scholar] [CrossRef]
- Von Korff, M.; Radovic, S.; Choumane, W.; Stamati, K.; Udupa, S.M.; Grando, S.; Ceccarelli, S.; MacKay, I.; Powell, W.; Baum, M.; et al. Asymmetric allele-specific expression in relation to developmental variation and drought stress in barley hybrids. Plant J. 2009, 59, 14–26. [Google Scholar] [CrossRef]
- He, G.; Zhu, X.; Elling, A.A.; Chen, L.; Wang, X.; Guo, L.; Liang, M.; He, H.; Zhang, H.; Chen, F.; et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 2010, 22, 17–33. [Google Scholar] [CrossRef]
- Chodavarapu, R.K.; Feng, S.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.; Wang, G.L.; Meyers, B.C.; Jacobsen, S.E.; Pellegrini, P.M. Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045. [Google Scholar] [CrossRef]
- Todesco, M.; Balasubramanian, S.; Hu, T.T.; Traw, M.B.; Horton, M.; Epple, P.; Kuhns, C.; Sureshkumar, S.; Schwartz, C.; Lanz, C.; et al. Natural allelic variation underlying a major fitness trade-off in Arabidopsis thaliana. Nature 2010, 465, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Xing, F.; Xu, C.; Zhang, Q.; Che, J.; Wang, X.; Song, J.; Li, X.; Xiao, J.; Chen, L.L.; et al. Patterns of genome-wide allele-specific expression in hybrid rice and the implications on the genetic basis of heterosis. Proc. Natl. Acad. Sci. USA 2019, 116, 5653–5658. [Google Scholar] [CrossRef] [PubMed]
- Pignatta, D.; Novitzky, K.; Satyaki, P.R.V.; Gehring, M. A variably imprinted epiallele impacts seed development. PLOS Genetics 2018, 14, 1007469. [Google Scholar] [CrossRef]
- Chen, Z.J. Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet. 2013, 14, 471–482. [Google Scholar] [CrossRef]
- Lv, Z.; Zhang, W.; Wu, Y.; Huang, S.; Zhou, Y.; Zhang, A.; Deng, X.; Xu, C.; Xu, Z.; Gong, L.; et al. Extensive allele-level remodeling of histone methylation modification in reciprocal F 1 hybrids of rice subspecies. Plant J. 2019, 97, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, A.M.B.; Fuchs, J.; Czauderna, T.; Houben, A.; Mette, M.F. Intraspecific hybrids of Arabidopsis thaliana revealed no gross alterations in endopolyploidy, DNA methylation, histone modifications and transcript levels. Theor. Appl. Genet. 2010, 120, 215–226. [Google Scholar] [CrossRef]
- Jackson, S.A. Epigenomics: Dissecting hybridization and polyploidization. Genome Biol. 2017, 18, 117. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Saze, H.; Kakutani, T. Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. EMBO J. 2007, 26, 3641–3652. [Google Scholar] [CrossRef]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef]
- Tsukahara, S.; Kobayashi, A.; Kawabe, A.; Mathieu, O.; Miura, A.; Kakutani, T. Bursts of retrotransposition reproduced in Arabidopsis. Nature 2009, 461, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Lauss, K.; Wardenaar, R.; Oka, R.; van Hulten, M.H.A.; Guryev, V.; Keurentjes, J.J.B.; Stam, M.; Johannes, F. Parental DNA methylation states are associated with heterosis in epigenetic hybrids. Plant Physiol. 2018, 176, 1627–1645. [Google Scholar] [CrossRef]
- Zhang, M.; Xie, S.; Dong, X.; Zhao, X.; Zeng, B.; Chen, J.; Li, H.; Yang, W.; Zhao, H.; Wang, G.; et al. Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize. Genome Res. 2017, 24, 167–176. [Google Scholar] [CrossRef]
- Ibarra, C.A.; Feng, X.; Schoft, V.K.; Hsieh, T.F.; Uzawa, R.; Rodrigues, J.A.; Zemach, A.; Chumak, N.; Machlicova, A.; Nishimura, T.; et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 2012, 337, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.A.; Ruan, R.; Nishimura, T.; Sharma, M.K.; Sharma, R.; Ronald, P.C.; Fischer, R.L.; Zilberman, D. Imprinted expression of genes and small RNA is associated with localized hypomethylation of the maternal genome in rice endosperm. Proc. Natl. Acad. Sci. USA 2013, 110, 7934–7939. [Google Scholar] [CrossRef] [PubMed]
- Wolff, P.; Weinhofer, I.; Seguin, J.; Roszak, P.; Beisel, C.; Donoghue, M.T.A.; Spillane, C.; Nordborg, M.; Rehmsmeier, M.; Köhler, C. High-Resolution analysis of parent-of-origin allelic expression in the Arabidopsis endosperm. PLOS Genet. 2011, 7, 1002126. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.F.; Shin, J.; Uzawa, R.; Silva, P.; Cohen, S.; Bauer, M.J.; Hashimoto, M.; Kirkbride, R.C.; Harada, J.J.; Zilberman, D.; et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc. Natl. Acad. Sci. USA 2011, 108, 1755–1762. [Google Scholar] [CrossRef]
- Ma, X.; Xing, F.; Jia, Q.; Zhang, Q.; Hu, T.; Wu, B.; Shao, L.; Zhao, Y.; Zhang, Q.; Zhou, D.X. Parental variation in CHG methylation is associated with allelic-specific expression in elite hybrid rice. Plant Physiol. 2021, 186, 1025–1041. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Sun, S.; Zhou, Y.; Chen, J.; Shi, J.; Zhao, H.; Zhao, H.; Song, W.; Zhang, M.; Cui, Y.; Dong, X.; et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 2018, 50, 1289–1295. [Google Scholar] [CrossRef]
- Springer, N.M.; Stupar, R.M. Allele-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize. Plant Cell 2007, 19, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, L.; Xing, F.; Kudrna, D.A.; Yao, W.; Copetti, D.; Mu, T.; Li, W.; Song, J.; Xie, W.; et al. Extensive sequence divergence between the reference genomes of two elite Indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. USA 2016, 113, E5163–E5171. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Hirsch, C.; Briggs, S.; Springer, N. Dynamic Patterns of gene expression additivity and regulatory variation throughout maize development. Mol. Plant 2019, 12, 410–425. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Crow, T.; Nojoomi, S.; Schulz, A.; Estévez-Palmas, J.; Hufford, M.; Flint-Garcia, S.; Sawers, R.; Rellán-Álvarez, R.; Ross-Ibarra, J.; et al. Allele-specific expression reveals multiple paths to highland adaptation in maize. Mol. Biol. Evol. 2022, 39, msac239. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Hedden, P.; Zhang, Q. Heterosis in rice seedlings: Its relationship to gibberellin content and expression of gibberellin metabolism and signaling genes. Plant Physiol. 2011, 156, 1905–1920.47. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell. 2016, 61, 199–209. [Google Scholar] [CrossRef]
- Liu, Y.; Qu, J.; Zhang, L.; Xu, X.; Wei, G.; Zhao, Z.; Ren, M.; Cao, M. Identification and characterization of the TCA cycle genes in maize. BMC Plant Biol. 2019, 19, 592. [Google Scholar] [CrossRef]
- Klosinska, M.; Picard, C.L.; Gehring, M. Conserved imprinting associated with unique epigenetic signatures in the Arabidopsis genus. Nat. Plants 2016, 2, 16145. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, M.; Chen, J.; Peng, L.; Zhang, N.; Wang, X.; Lai, J. Dynamic and antagonistic allele-specific epigenetic modifications controlling the expression of imprinted genes in maize endosperm. Mol. Plant 2017, 10, 442–455. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotech. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential Gene and Transcript Expression Analysis of RNA-Seq Experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Ishida, Y.; Hiei, Y.; Komari, T. Agrobacterium-mediated transformation of maize. Nat. Protoc. 2007, 2, 1614–1621. [Google Scholar] [CrossRef]
- Yang, L.; Xing, F.; He, Q.; Qamar, M.T.U.; Chen, L.L.; Xing, Y. Conserved imprinted genes between intra-subspecies and inter-subspecies are involved in energy metabolism and seed development in rice. Int. J. Mol. Sci. 2020, 21, 9618. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, X.; Luo, H.; Yao, J.; Guo, Q.; Yu, S.; Zhang, X.; Cheng, X.; Meng, D. Characterization of Genes That Exhibit Genotype-Dependent Allele-Specific Expression and Its Implications for the Development of Maize Kernel. Int. J. Mol. Sci. 2023, 24, 4766. https://doi.org/10.3390/ijms24054766
Dong X, Luo H, Yao J, Guo Q, Yu S, Zhang X, Cheng X, Meng D. Characterization of Genes That Exhibit Genotype-Dependent Allele-Specific Expression and Its Implications for the Development of Maize Kernel. International Journal of Molecular Sciences. 2023; 24(5):4766. https://doi.org/10.3390/ijms24054766
Chicago/Turabian StyleDong, Xiaomei, Haishan Luo, Jiabin Yao, Qingfeng Guo, Shuai Yu, Xiaoyu Zhang, Xipeng Cheng, and Dexuan Meng. 2023. "Characterization of Genes That Exhibit Genotype-Dependent Allele-Specific Expression and Its Implications for the Development of Maize Kernel" International Journal of Molecular Sciences 24, no. 5: 4766. https://doi.org/10.3390/ijms24054766
APA StyleDong, X., Luo, H., Yao, J., Guo, Q., Yu, S., Zhang, X., Cheng, X., & Meng, D. (2023). Characterization of Genes That Exhibit Genotype-Dependent Allele-Specific Expression and Its Implications for the Development of Maize Kernel. International Journal of Molecular Sciences, 24(5), 4766. https://doi.org/10.3390/ijms24054766