
DNA Damage and Its Role in Cancer Therapeutics


Abstract
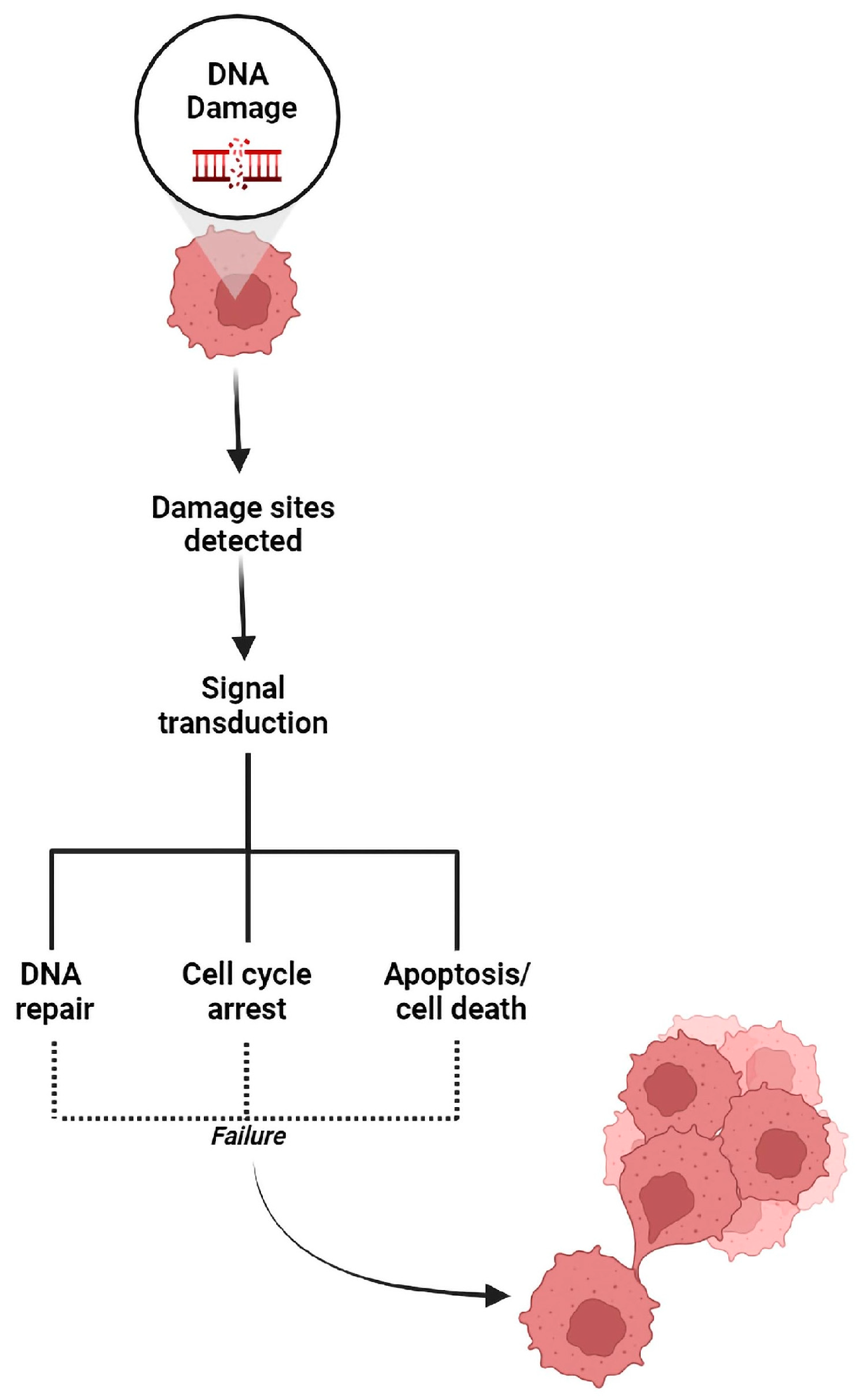
1. Introduction
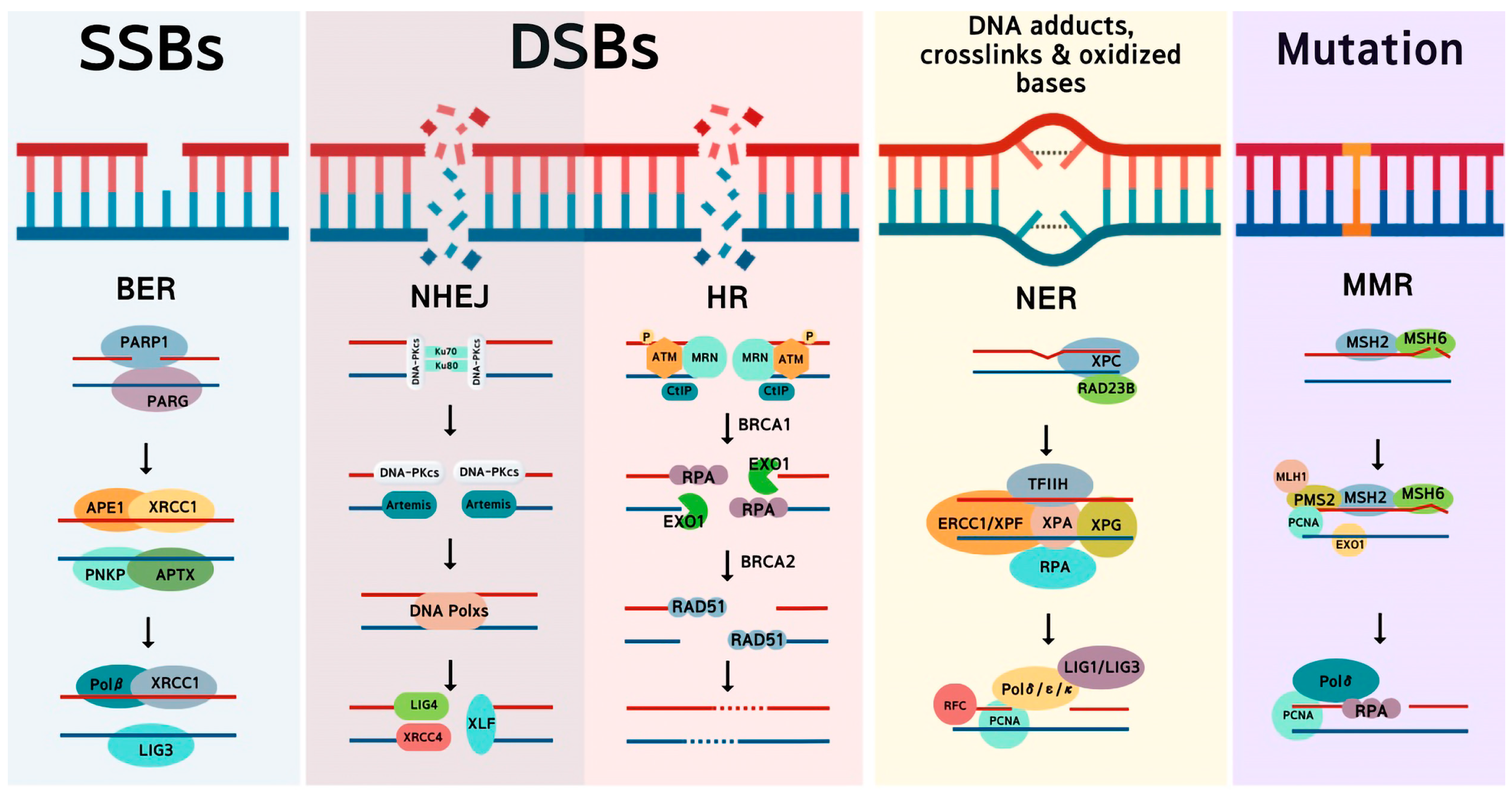
2. Main DNA Repair Pathways in Cancer Cells
2.1. Homologous Recombination
2.2. Non-Homologous End Joining
2.3. Microhomology-Mediated End Joining
2.4. Base Excision Repair
2.5. Nucleotide Excision Repair
2.6. Mismatch Repair
2.7. DNA-Protein Crosslinks
3. DNA Repair Pathways Are the Achilles Heel of Cancer under Treatment

{kind=link}
{kind=link}
| DDR Pathways | DDR Genes | Type of Alterations | Cancer Type | Reference |
|---|---|---|---|---|
| HR | BRCA1 | c.190T>C (cysteine to arginine) | Breast cancer | Wang et al. [117] |
| BRCA2 | c.6408delA (deletion) | |||
| BRCA1 | c.4837A>G (serine to glycine) | Ovarian cancer | ||
| BRCA1 | c.2612C>T (proline to leucine) | |||
| BRCA2 | c.677delC (deletion) | Esophagus cancer | ||
| NHEJ | Ku80 | Ku80 polymorphism G-1401T (SNP rs828907) | Oral, colon, gastric, breast, and bladder cancer | Sischc and Davis [100] |
| Ku70 | Ku70 C-61G polymorphism (SNP rs2267437) | Breast, prostate, and lung cancer | ||
| BER | POLE | Missense mutation in POLE exonuclease domain | Endometrial cancer | Leon-Castillo et al. [110] |
| MUTYH | G: C to T: A transversions mutation | Villy et al. [118] | ||
| NER | ERCC2 | Somatic ERCC2 missense mutation | Muscle-invasive bladder cancer | Mouw [115] |
| MMR | MLH1 | Loss of MLH1, MSH2, and MSH6 due to MLH1 promoter hypermethylation | Ampullary and colon cancer | Ruemmele et al. [119] |
4. Developing Inhibitors Targeting Key Enzymes in DNA Repair Pathways for Cancer Therapeutics
4.1. DNA-PK Inhibitor
4.2. Poly ADP-Ribose Polymerase Inhibitor
4.3. Ataxia Telangiectasia Mutated Inhibitor
4.4. Ataxia Telangiectasia and Rad3-Related Inhibitor
4.5. Checkpoint Kinase 1 Inhibitor
5. Challenges and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perry, M.; Ghosal, G. Mechanisms and Regulation of DNA-Protein Crosslink Repair During DNA Replication by SPRTN Protease. Front Mol. Biosci. 2022, 9, 916697. [Google Scholar] [CrossRef]
- Cervelli, T.; Borghini, A.; Galli, A.; Andreassi, M.G. DNA damage and repair in atherosclerosis: Current insights and future perspectives. Int. J. Mol. Sci. 2012, 13, 16929–16944. [Google Scholar] [CrossRef] [PubMed]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef] [PubMed]
- Ekundayo, B.; Bleichert, F. Origins of DNA replication. PLoS Genet. 2019, 15, e1008320. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.R.; Flynn, T.S.; Freudenthal, B.D. Base excision repair of oxidative DNA damage: From mechanism to disease. Front. Biosci. (Landmark Ed.) 2017, 22, 1493–1522. [Google Scholar] [PubMed]
- Khanna, A. DNA damage in cancer therapeutics: A boon or a curse? Cancer Res. 2015, 75, 2133–2138. [Google Scholar] [CrossRef]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2020, 11, 629266. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; Ao, D. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. MedComm (2020) 2021, 2, 654–691. [Google Scholar] [CrossRef]
- Kieffer, S.R.; Lowndes, N.F. Immediate-Early, Early, and Late Responses to DNA Double Stranded Breaks. Front. Genet. 2022, 13, 793884. [Google Scholar] [CrossRef]
- van den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef]
- Siamof, C.M.; Goel, S.; Cai, W. Moving Beyond the Pillars of Cancer Treatment: Perspectives From Nanotechnology. Front. Chem. 2020, 8, 598100. [Google Scholar] [CrossRef] [PubMed]
- Seebacher, N.A.; Stacy, A.E.; Porter, G.M.; Merlot, A.M. Clinical development of targeted and immune based anti-cancer therapies. J. Exp. Clin. Cancer Res. 2019, 38, 156. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. 2021, 6, 254. [Google Scholar] [CrossRef]
- Thompson, L.H.; Schild, D. Homologous recombinational repair of DNA ensuRes. mammalian chromosome stability. Mutat. Res. 2001, 477, 131–153. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Dev, H.; Chiang, T.W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.M.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Kim, W.; Gao, H.Y.; Liu, C.; Zhang, Y.; Chen, Y.P.; Deng, M.; Zhou, Q.; Huang, J.Z.; Hu, Q.; et al. ASTE1 promotes shieldin-complex-mediated DNA repair by attenuating end resection. Nat. Cell Biol. 2021, 23, 894. [Google Scholar] [CrossRef]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef]
- McMahill, M.S.; Sham, C.W.; Bishop, D.K. Synthesis-dependent strand annealing in meiosis. PLoS Biol. 2007, 5, e299. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Callebaut, I.; Malivert, L.; Fischer, A.; Mornon, J.P.; Revy, P.; de Villartay, J.P. Cernunnos interacts with the XRCC4 x DNA-ligase IV complex and is homologous to the yeast nonhomologous end-joining factor Nej1. J. Biol. Chem. 2006, 281, 13857–13860. [Google Scholar] [CrossRef]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. BioChem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar]
- Parrella, P.; Mazzarelli, P.; Signori, E.; Perrone, G.; Marangi, G.F.; Rabitti, C.; Delfino, M.; Prencipe, M.; Gallo, A.P.; Rinaldi, M.; et al. Expression and heterodimer-binding activity of Ku70 and Ku80 in human non-melanoma skin cancer. J. Clin. Pathol. 2006, 59, 1181–1185. [Google Scholar] [CrossRef]
- Xing, J.; Wu, X.; Vaporciyan, A.A.; Spitz, M.R.; Gu, J. Prognostic significance of ataxia-telangiectasia mutated, DNA-dependent protein kinase catalytic subunit, and Ku heterodimeric regulatory complex 86-kD subunit expression in patients with nonsmall cell lung cancer. Cancer 2008, 112, 2756–2764. [Google Scholar] [CrossRef]
- Vaidya, A.; Mao, Z.; Tian, X.; Spencer, B.; Seluanov, A.; Gorbunova, V. Knock-in reporter mice demonstrate that DNA repair by non-homologous end joining declines with age. PLoS Genet. 2014, 10, e1004511. [Google Scholar] [CrossRef]
- Chen, C.; Umezu, K.; Kolodner, R.D. Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol. Cell 1998, 2, 9–22. [Google Scholar] [CrossRef]
- Yu, X.; Gabriel, A. Ku-dependent and Ku-independent end-joining pathways lead to chromosomal rearrangements during double-strand break repair in Saccharomyces cerevisiae. Genetics 2003, 163, 843–856. [Google Scholar] [CrossRef]
- Weinstock, D.M.; Brunet, E.; Jasin, M. Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70. Nat. Cell Biol. 2007, 9, 978–981. [Google Scholar] [CrossRef]
- Welcker, A.J.; de Montigny, J.; Potier, S.; Souciet, J.L. Involvement of very short DNA tandem repeats and the influence of the RAD52 gene on the occurrence of deletions in Saccharomyces cerevisiae. Genetics 2000, 156, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Lu, H.; Gu, J.; Schwarz, K. Flexibility in the order of action and in the enzymology of the nuclease, polymerases, and ligase of vertebrate non-homologous DNA end joining: Relevance to cancer, aging, and the immune system. Cell Res. 2008, 18, 125–133. [Google Scholar] [CrossRef]
- Roth, D.B.; Wilson, J.H. Nonhomologous recombination in mammalian cells: Role for short sequence homologies in the joining reaction. Mol. Cell Biol. 1986, 6, 4295–4304. [Google Scholar]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef]
- Roerink, S.F.; van Schendel, R.; Tijsterman, M. Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res. 2014, 24, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, J.; van Schendel, R.; den Dunnen, J.T.; Tijsterman, M. Templated Insertions: A Smoking Gun for Polymerase Theta-Mediated End Joining. Trends Genet. 2019, 35, 632–644. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A.; Kent, T.; Deng, S.K.; McDevitt, S.; Kashkina, E.; Hoang, T.M.; Pomerantz, R.T.; Sfeir, A. The helicase domain of Poltheta counteracts RPA to promote alt-NHEJ. Nat. Struct Mol. Biol. 2017, 24, 1116–1123. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends BioChem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef]
- Wood, R.D.; Doublie, S. DNA polymerase theta (POLQ), double-strand break repair, and cancer. DNA Repair (Amst.) 2016, 44, 22–32. [Google Scholar] [CrossRef]
- Zhang, Y.; Jasin, M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct Mol. Biol. 2011, 18, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Liu, Y.; Prasad, R.; Beard, W.A.; Kedar, P.S.; Hou, E.W.; Shock, D.D.; Wilson, S.H. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J. Biol. Chem. 2007, 282, 13532–13541. [Google Scholar] [CrossRef]
- Lindahl, T.; Satoh, M.S.; Poirier, G.G.; Klungland, A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends BioChem. Sci. 1995, 20, 405–411. [Google Scholar] [CrossRef]
- Caldecott, K.W. DNA single-strand break repair. Exp. Cell Res. 2014, 329, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Fromme, J.C.; Banerjee, A.; Verdine, G.L. DNA glycosylase recognition and catalysis. Curr. Opin. Struct. Biol. 2004, 14, 43–49. [Google Scholar] [CrossRef]
- Demple, B.; Herman, T.; Chen, D.S. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: Definition of a family of DNA repair enzymes. Proc. Natl. Acad. Sci. USA 1991, 88, 11450–11454. [Google Scholar] [CrossRef]
- Cappelli, E.; Taylor, R.; Cevasco, M.; Abbondandolo, A.; Caldecott, K.; Frosina, G. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J. Biol. Chem. 1997, 272, 23970–23975. [Google Scholar] [CrossRef]
- Pascucci, B.; Stucki, M.; Jonsson, Z.O.; Dogliotti, E.; Hubscher, U. Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J. Biol. Chem. 1999, 274, 33696–33702. [Google Scholar] [CrossRef]
- Dreher, D.; Junod, A.F. Role of oxygen free radicals in cancer development. Eur. J. Cancer 1996, 32A, 30–38. [Google Scholar] [CrossRef]
- Karihtala, P.; Soini, Y. Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies. APMIS 2007, 115, 81–103. [Google Scholar] [CrossRef]
- Mohammed, M.Z.; Vyjayanti, V.N.; Laughton, C.A.; Dekker, L.V.; Fischer, P.M.; Wilson, D.M., 3rd; Abbotts, R.; Shah, S.; Patel, P.M.; Hickson, I.D.; et al. Development and evaluation of human AP endonuclease inhibitors in melanoma and glioma cell lines. Br J. Cancer 2011, 104, 653–663. [Google Scholar] [CrossRef]
- Zhong, S.; Chen, X.; Zhu, X.; Dziegielewska, B.; Bachman, K.E.; Ellenberger, T.; Ballin, J.D.; Wilson, G.M.; Tomkinson, A.E.; MacKerell, A.D., Jr. Identification and validation of human DNA ligase inhibitors using computer-aided drug design. J. Med. Chem. 2008, 51, 4553–4562. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, S.; Zhu, X.; Dziegielewska, B.; Ellenberger, T.; Wilson, G.M.; MacKerell, A.D., Jr.; Tomkinson, A.E. Rational design of human DNA ligase inhibitors that Target. cellular DNA replication and repair. Cancer Res. 2008, 68, 3169–3177. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Gillet, L.C.; Scharer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Degitz, K.; Besch, R.; Berking, C. Differential expression of melanoma-associated growth factors in keratinocytes and fibroblasts by ultraviolet A and ultraviolet B radiation. Br. J. Dermatol. 2005, 153, 733–739. [Google Scholar] [CrossRef]
- Marrot, L.; Meunier, J.R. Skin DNA photodamage and its biological consequences. J. Am. Acad. Dermatol. 2008, 58 (Suppl. 2), S139–S148. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. SignatuRes. of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Sakoda, L.C.; Loomis, M.M.; Doherty, J.A.; Julianto, L.; Barnett, M.J.; Neuhouser, M.L.; Thornquist, M.D.; Weiss, N.S.; Goodman, G.E.; Chen, C. Germ line variation in nucleotide excision repair genes and lung cancer risk in smokers. Int. J. Mol. Epidemiol. Genet. 2012, 3, 1–17. [Google Scholar] [PubMed]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA mismatch repair: Functions and mechanisms. Chem. Rev. 2006, 106, 302–323. [Google Scholar] [CrossRef]
- Larrea, A.A.; Lujan, S.A.; Kunkel, T.A. SnapShot: DNA mismatch repair. Cell 2010, 141, 730.e1. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Furgason, J.M.; Bahassiel, M. Targeting DNA repair mechanisms in cancer. Pharmacol. Ther. 2013, 137, 298–308. [Google Scholar] [CrossRef]
- Guillotin, D.; Martin, S.A. Exploiting DNA mismatch repair deficiency as a therapeutic strategy. Exp. Cell Res. 2014, 329, 110–115. [Google Scholar] [CrossRef]
- Swenberg, J.A.; Lu, K.; Moeller, B.C.; Gao, L.; Upton, P.B.; Nakamura, J.; Starr, T.B. Endogenous versus exogenous DNA adducts: Their role in carcinogenesis, epidemiology, and risk assessment. Toxicol. Sci. 2011, 120 (Suppl. 1), S130–S145. [Google Scholar] [CrossRef]
- Nakano, T.; Miyamoto-Matsubara, M.; Shoulkamy, M.I.; Salem, A.M.; Pack, S.P.; Ishimi, Y.; Ide, H. Translocation and stability of replicative DNA helicases upon encountering DNA-protein cross-links. J. Biol. Chem. 2013, 288, 4649–4658. [Google Scholar] [CrossRef]
- Noguchi, C.; Grothusen, G.; Anandarajan, V.; Martinez-Lage Garcia, M.; Terlecky, D.; Corzo, K.; Tanaka, K.; Nakagawa, H.; Noguchi, E. Genetic controls of DNA damage avoidance in response to acetaldehyde in fission yeast. Cell Cycle 2017, 16, 45–58. [Google Scholar] [CrossRef]
- Dellarco, V.L. A mutagenicity assessment of acetaldehyde. Mutat. Res. 1988, 195, 1–20. [Google Scholar] [CrossRef]
- Lorenti Garcia, C.; Mechilli, M.; Proietti De Santis, L.; Schinoppi, A.; Kobos, K.; Palitti, F. Relationship between DNA lesions, DNA repair and chromosomal damage induced by acetaldehyde. Mutat. Res. 2009, 662, 3–9. [Google Scholar] [CrossRef]
- Joenje, H.; Patel, K.J. The emerging genetic and molecular basis of Fanconi anaemia. Nat. Rev. Genet. 2001, 2, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.D.; D’Andrea, A.D. The Fanconi Anemia/BRCA pathway: New faces in the crowd. Genes Dev. 2005, 19, 2925–2940. [Google Scholar] [CrossRef] [PubMed]
- Kee, Y.; D’Andrea, A.D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010, 24, 1680–1694. [Google Scholar] [CrossRef]
- Stingele, J.; Bellelli, R.; Boulton, S.J. Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Ridpath, J.R.; Nakamura, A.; Tano, K.; Luke, A.M.; Sonoda, E.; Arakawa, H.; Buerstedde, J.M.; Gillespie, D.A.; Sale, J.E.; Yamazoe, M.; et al. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007, 67, 11117–11122. [Google Scholar] [CrossRef]
- Martin, L.J. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.D. Mechanisms of mutagenesis induced by DNA lesions: Multiple factors affect mutations in translesion DNA synthesis. Crit. Rev. BioChem. Mol. 2020, 55, 219–251. [Google Scholar] [CrossRef] [PubMed]
- Pilger, D.; Seymour, L.W.; Jackson, S.P. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Gene Dev. 2021, 35, 602–618. [Google Scholar] [CrossRef] [PubMed]
- Nakad, R.; Schumacher, B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal. Transduct. Tar. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Hélène Gaillard, T.G.-M.A.A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Czajkowski, D.; Szmyd, R.; Gee, H.E. Impact of DNA damage response defects in cancer cells on response to immunotherapy and radiotherapy. J. Med. Imag. Radiat. Oncol. 2022, 66, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, N.; Ryder, S.; Forbes, C.; Ross, J.; Quek, R.G.W. A systematic review of the international prevalence of BRCA mutation in breast cancer. Clin. Epidemiol. 2019, 11, 543–561. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.W.; Zhang, Y.; Deng, Q.; Liang, H. Expression and mutations of BRCA in breast cancer and ovarian cancer: Evidence from bioinformatics analyses. Int. J. Mol. Med. 2018, 42, 3542–3550. [Google Scholar] [CrossRef]
- Turner, N.C.; Tutt, A.N.J. Platinum chemotherapy for BRCA1-related breast cancer: Do we need more evidence? Breast Cancer Res. 2012, 14, 115. [Google Scholar] [CrossRef]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A.; Piccart, M. An update on PARP inhibitors-moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41. [Google Scholar] [CrossRef]
- Binder, K.A.R.; Mick, R.; O’Hara, M.; Teitelbaum, U.; Karasic, T.; Schneider, C.; O’Dwyer, P.J.; Carpenter, E.; Pantel, A.; Makvandi, M.; et al. A Phase II, single arm study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic mutation in BRCA1, BRCA2, PALB2. Cancer Res. 2019, 79, 2497–2505. [Google Scholar] [CrossRef]
- Chan, D.; Clarke, S.; Gill, A.J.; Chantrill, L.; Samra, J.; Li, B.T.; Barnes, T.; Nahar, K.; Pavlakis, N. Pathogenic PALB2 mutation in metastatic pancreatic adenocarcinoma and neuroendocrine tumour: A case report. Mol. Clin. Oncol. 2015, 3, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Matsunaga, Y.; Tsurutani, J.; Akashi-Tanaka, S.; Masuda, H.; Ide, Y.; Hashimoto, R.; Inuzuka, M.; Watanabe, C.; Taruno, K.; et al. BRCAness as a prognostic indicator in patients with early breast cancer. Sci. Rep.-UK 2020, 10, 21173. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.Y.; Zhao, F.; Cui, G.F.; Zhang, Y.; Deshpande, R.A.; Chen, Y.P.; Deng, M.; Kloeber, J.A.; Shi, Y.; Zhou, Q.; et al. METTL16 antagonizes MRE11-mediated DNA end resection and confers synthetic lethality to PARP inhibition in pancreatic ductal adenocarcinoma. Nat. Cancer 2022, 3, 1088. [Google Scholar]
- Stok, C.; Kok, Y.P.; van den Tempel, N.; van Vugt, M.A.T.M. Shaping the BRCAness mutational landscape by alternative double-strand break repair, replication stress and mitotic aberrancies. Nucleic Acids Res. 2021, 49, 4239–4257. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Singh, N.; Behera, D.; Sharma, S. Role of polymorphic XRCC6 (Ku70)/XRCC7 (DNA-PKcs) genes towards susceptibility and prognosis of lung cancer patients undergoing platinum based doublet chemotherapy. Mol. Biol. Rep. 2018, 45, 253–261. [Google Scholar]
- Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef]
- Medova, M.; Medo, M.; Hovhannisyan, L.; Munoz-Maldonado, C.; Aebersold, D.M.; Zimmer, Y. DNA-PK in human malignant disorders: Mechanisms and implications for pharmacological interventions. Pharmacol. Ther. 2020, 215, 107617. [Google Scholar] [CrossRef]
- Koike, M. Dimerization, translocation and localization of Ku70 and Ku80 proteins. J. Radiat. Res. 2002, 43, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef]
- Zighelboim, I.; Schmidt, A.P.; Gao, F.; Thaker, P.H.; Powell, M.A.; Rader, J.S.; Gibb, R.K.; Mutch, D.G.; Goodfellow, P.J. ATR Mutation in Endometrioid Endometrial Cancer Is Associated with Poor Clinical Outcomes. J. Clin. Oncol. 2009, 27, 3091–3096. [Google Scholar] [CrossRef]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef]
- Gerlinger, M. Immunotherapy Sensitivity of Mismatch Repair-Deficient Cancer: Mutation Load Is Not Enough. Cancer Cell 2021, 39, 16–18. [Google Scholar] [CrossRef]
- Henninger, E.E.; Pursell, Z.F. DNA Polymerase ε and Its Roles in Genome Stability. Lubmb Life 2014, 66, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.S.; Thomas, L.E.; Abascal, F.; Jung, H.; Harvey, L.M.R.; West, H.D.; Olafsson, S.; Lee, B.C.H.; Coorens, T.H.H.; Lee-Six, H.; et al. Inherited MUTYH mutations cause elevated somatic mutation rates and distinctive mutational signatuRes. in normal human cells. Nat. Commun. 2022, 13, 3949. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Li, S.H.; Tang, X.M.; Wang, Y.; Guo, W.W.; Cao, G.J.; Chen, K.; Zhang, M.; Guan, M.; Yang, D. Copy Number Amplification of DNA Damage Repair Pathways Potentiates Therapeutic Resistance in Cancer. Theranostics 2020, 10, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- Leon-Castillo, A.; Britton, H.; McConechy, M.K.; McAlpine, J.N.; Nout, R.; Kommoss, S.; Brucker, S.Y.; Carlson, J.W.; Epstein, E.; Rau, T.; et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J. Pathol. 2020, 250, 323–335. [Google Scholar] [CrossRef]
- Dong, S.; Zakaria, H.; Hsiehchen, D. Non-Exonuclease Domain POLE Mutations Associated with Immunotherapy Benefit. Oncologist 2022, 27, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Maria Cristina Curia, T.C.; Aceto, G.M. MUTYH: Not just polyposis. World J. Clin. Oncol. 2020, 11, 428–449. [Google Scholar] [CrossRef] [PubMed]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. Ebiomedicine 2017, 20, 39–49. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, Q.; Wang, R.K.; Li, Y.; Sun, Y.; Yang, L. Targeting DNA Damage Repair for Immune CheckpoInt. Inhibition: Mechanisms and Potential Clinical Applications. Front. Oncol. 2021, 11, 648687. [Google Scholar] [CrossRef]
- Mouw, K.W. DNA Repair Pathway Alterations in Bladder Cancer. Cancers 2017, 9, 28. [Google Scholar] [CrossRef]
- Miyagi, H.; Kwenda, E.; Ramnaraign, B.H.; Chatzkel, J.A.; Brisbane, W.G.; O’Malley, P.; Crispen, P.L. Predicting Complete Response to Neoadjuvant Chemotherapy in Muscle-Invasive Bladder Cancer. Cancers 2023, 15, 168. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, J.L.; Xiao, J.J.; Huang, M.L.; Li, N.L.; Ling, R. Mutational analysis of BRCA1 and BRCA2 in northwest Chinese breast cancer patients. Transl. Cancer Res. 2019, 8, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Villy, M.C.; Masliah-Planchon, J.; Buecher, B.; Beaulaton, C.; Vincent-Salomon, A.; Stoppa-Lyonnet, D.; Colas, C. Endometrial cancer may be part of the MUTYH-associated polyposis cancer spectrum. Eur. J. Med. Genet. 2022, 65, 104385. [Google Scholar] [CrossRef]
- Ruemmele, P.; Dietmaier, W.; Terracciano, L.; Tornillo, L.; Bataille, F.; Kaiser, A.; Wuensch, P.H.; Heinmoeller, E.; Homayounfar, K.; Luettges, J.; et al. Histopathologic FeatuRes. and Microsatellite Instability of Cancers of the Papilla of Vater and Their Precursor Lesions. Am. J. Surg Pathol. 2009, 33, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Traganos, F.; Wlodkowic, D. Impaired DNA damage response—An Achilles’ heel sensitizing cancer to chemotherapy and radiotherapy. Eur. J. Pharmacol. 2009, 625, 143–150. [Google Scholar] [CrossRef]
- Guo, G.S.; Zhang, F.M.; Gao, R.J.; Delsite, R.; Feng, Z.H.; Powell, S.N. DNA repair and synthetic lethality. Int. J. Oral Sci. 2011, 3, 176–179. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. Lubmb Life 2017, 69, 929–937. [Google Scholar] [CrossRef]
- Shaheen, M.; Allen, C.; Nickoloff, J.A.; Hromas, R. Synthetic lethality: Exploiting the addiction of cancer to DNA repair. Blood 2011, 117, 6074–6082. [Google Scholar] [CrossRef]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA damage response: Next-generation anti-cancer therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1758835918786658. [Google Scholar] [CrossRef] [PubMed]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Durocher, D.; Jackson, S.P. DNA-PK, ATM and ATR as sensors of DNA damage: Variations on a theme? Curr. Opin. Cell Biol. 2001, 13, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar]
- Allen, C.; Halbrook, J.; Nickoloff, J.A. Interactive competition between homologous recombination and non-homologous end joining. Mol. Cancer Res. 2003, 1, 913–920. [Google Scholar] [PubMed]
- Carlsen, L.; El-Deiry, W.S. Anti-cancer immune responses to DNA damage response inhibitors: Molecular mechanisms and progress toward clinical translation. Front. Oncol. 2022, 12, 998388. [Google Scholar] [CrossRef] [PubMed]
- Collis, S.J.; DeWeese, T.L.; Jeggo, P.A.; Parker, A.R. The life and death of DNA-PK. Oncogene 2005, 24, 949–961. [Google Scholar] [CrossRef]
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Herceg, Z.; Wang, Z.Q. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. 2001, 477, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Gao, Z.; Li, H.Z.; Zhang, B.F.; Wang, G.; Zhang, Q.; Pei, D.S.; Zheng, J.N. DNA damage response—A double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015, 358, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Barnieh, F.; Loadman, P.; Falconer, R. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef]
- Wengner, A.M.; Scholz, A.; Haendler, B. Targeting DNA Damage Response in Prostate and Breast Cancer. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef]
- Rogers, R.F.; Walton, M.I.; Cherry, D.L.; Collins, I.; Clarke, P.A.; Garrett, M.D.; Workman, P. CHK1 Inhibition Is Synthetically Lethal with Loss of B-Family DNA Polymerase Function in Human Lung and Colorectal Cancer Cells. Cancer Res. 2020, 80, 1735–1761. [Google Scholar] [CrossRef]
- Pescatori, S.; Leone, S.; Cipolletti, M.; Bartoloni, S.; di Masi, A.; Acconcia, F. Clinically relevant CHK1 inhibitors abrogate wild-type and Y537S mutant ERalpha expression and proliferation in luminal primary and metastatic breast cancer cells. J. Exp. Clin. Cancer Res. 2022, 41, 141. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. RadioTher. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- He, M.; Cao, C.G.; Ni, Z.H.; Liu, Y.B.; Song, P.L.; Hao, S.; He, Y.N.; Sun, X.Y.; Rao, Y. PROTACs: Great opportunities for academia and industry (an update from 2020 to 2021). Signal. Transduct. Tar. 2022, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, Y.J.; Yang, S.B.; Chen, W.J.; Xing, D.M. PROTACs for BRDs proteins in cancer therapy: A review. J. Enzym. Inhib. Med. Chem. 2022, 37, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef] [PubMed]


| Inhibitor Target | Clinical Trials. National Library of Medicine (NLM) | Clinical Phase | Disease | Intervention Title |
|---|---|---|---|---|
| DNA-PK | NCT02516813 | Phase 1 | Advanced Solid Tumors | Phase 1 Trial of MSC2490484A, an Inhibitor of a DNA-dependent Protein Kinase, in Combination with Radiotherapy |
| NCT02316197 | Phase 1 | Advanced Solid Tumors Chronic Lymphocytic Leukemia | Clinical Phase I Study Investigating MSC2490484A, an Inhibitor of a DNA-dependent Protein Kinase, in Advanced Solid Tumors or Chronic Lymphocytic Leukemia | |
| NCT03770689 | Phase 1 Phase 2 | Locally Advanced Rectal Cancer | Study of Peposertib in Combination with Capecitabine and RT in Rectal Cancer | |
| NCT03907969 | Phase 1 Phase 2 | Advanced Malignancies | Clinical Trial to Evaluate AZD7648 Alone and in Combination with Other Anti-cancer Agents in Patients with Advanced Cancers | |
| NCT03724890 | Phase 1 | Oncology Solid Tumors | Study of Avelumab-M3814 Combinations | |
| NCT05002140 | Phase 1 | Metastasis Locally Advanced Solid Tumor Recurrent Cancer | Trial of XRD-0394, a Kinase Inhibitor, in Combination with Palliative Radiotherapy in Advanced Cancer Patients | |
| PARP | NCT01844986 | Phase 3 | Newly Diagnosed Advanced Ovarian Cancer FIGO Stage III-IV BRCA Mutation Complete Response Partial Response First Line Platinum Chemotherapy | Olaparib Maintenance Monotherapy in Patients with BRCA Mutated Ovarian Cancer Following First Line Platinum Based Chemotherapy (SOLO-1) |
| NCT02282020 | Phase 3 | Relapsed Ovarian Cancer, BRCA Mutation, Platinum Sensitivity | Olaparib Treatment in Relapsed Germline Breast Cancer Susceptibility Gene (BRCA) Mutated Ovarian Cancer Patients Who Have Progressed at Least 6 Months After Last Platinum Treatment and Have Received at Least 2 Prior Platinum Treatments (SOLO3) | |
| NCT02446704 | Phase 1 Phase 2 | Small Cell Lung Cancer | Study of Olaparib and Temozolomide in Patients with Recurrent Small Cell Lung Cancer Following Failure of Prior Chemotherapy | |
| NCT02789332 | Phase 2 | Breast Cancer Triple Negative Breast Neoplasms HRpos Breast Neoplasms BRCA 1/2 and/or HRD | Assessing the Efficacy of Paclitaxel and Olaparib in Comparison to Paclitaxel/Carboplatin, Followed by Epirubicin/Cyclophosphamide as Neoadjuvant Chemotherapy in Patients with HER2-negative Early Breast Cancer and Homologous Recombination Deficiency (GeparOla) | |
| NCT01989546 | Phase 1 Phase 2 | Advanced Ovarian Cancer Primary Peritoneal Cancer Advanced Breast Cancer Advanced Solid Tumors | Pilot Trial of BMN 673, an Oral PARP Inhibitor, in Patients with Advanced Solid Tumors and Deleterious BRCA Mutations | |
| NCT00494442 | Phase 2 | Ovarian Neoplasm | Study to Assess the Efficacy and Safety of a PARP Inhibitor for the Treatment of BRCA-positive Advanced Ovarian Cancer (ICEBERG 2) | |
| NCT01945775 | Phase 3 | Breast Neoplasms BRCA 1 Gene Mutation BRCA 2 Gene Mutation | A Study Evaluating Talazoparib (BMN 673), a PARP Inhibitor, in Advanced and/or Metastatic Breast Cancer Patients with BRCA Mutation (EMBRACA Study) (EMBRACA) | |
| NCT04174716 | Phase 1 Phase 2 | Solid Tumors Homologous Recombination Repair Gene Mutation Homologous Recombination Deficiency | Basket Trial of IDX-1197, a PARP Inhibitor, in Patients with HRR Mutated Solid Tumors (VASTUS) (VASTUS) | |
| NCT04586335 | Phase 1 | Ovarian Cancer Breast Cancer Solid Tumor Prostate Cancer Endometrial Cancer | Study of CYH33 in Combination with Olaparib, an Oral PARP Inhibitor, in Patients with Advanced Solid Tumors | |
| NCT04972110 | Phase 1 Phase 2 | Advanced Solid Tumor, Adult | Study of RP-3500 with Niraparib or Olaparib in Advanced Solid Tumors (ATTACC) | |
| ATM | NCT04882917 | Phase 1 | Advanced Solid Tumors | First-in-human Study of M4076 in Advanced Solid Tumors (DDRiver Solid Tumors 410) |
| NCT03423628 | Phase 1 | Recurrent Glioblastoma Multiforme Primary Glioblastoma Multiforme Brain Neoplasms, Malignant Leptomeningeal Disease (LMD) | A Study to Assess the Safety and Tolerability of AZD1390 Given with Radiation Therapy in Patients with Brain Cancer | |
| NCT02588105 | Phase 1 | Advanced Solid Tumors | Study to Assess the Safety and Preliminary Efficacy of AZD0156 at Increasing Doses Alone or in Combination with Other Anti-cancer Treatment in Patients with Advanced Cancer (AToM) | |
| NCT03225105 | Phase 1 | Solid Tumors | M3541 in Combination with Radiotherapy in Subjects with Solid Tumors | |
| ATR | NCT03188965 | Phase 1 | Advanced Solid Tumor Non-Hodgkin’s Lymphoma Mantle Cell Lymphoma | First-in-human Study of ATR Inhibitor BAY1895344 in Patients with Advanced Solid Tumors and Lymphomas |
| NCT04267939 | Phase 1 | Advanced Solid Tumors (Excluding Prostate Cancer) Ovarian Cancer | ATR Inhibitor Elimusertib (BAY1895344) Plus Niraparib First phase b Study in Advanced Solid Tumors and Ovarian Cancer | |
| NCT05338346 | Phase 1 | Advanced Solid Tumors Hematological Malignancies | A Study of ATG-018 (ATR Inhibitor) Treatment in Patients with Advanced Solid Tumors and Hematological Malignancies (ATRIUM) | |
| NCT04065269 | Phase 2 | Gynecological Cancers | ATR Inhibitor in Combination with Olaparib in Gynecological Cancers with ARId1A Loss or no Loss (ATARI) | |
| NCT05071209 | Phase 1 Phase 2 | Recurrent Alveolar Rhabdomyosarcoma Recurrent Ewing Sarcoma Recurrent Lymphoma Recurrent Malignant Solid Neoplasm Refractory Alveolar Rhabdomyosarcoma Refractory Ewing Sarcoma Refractory Lymphoma Refractory Malignant Solid Neoplasm | Elimusertib for the Treatment of Relapsed or Refractory Solid Tumors | |
| NCT04972110 | Phase 1 Phase 2 | Advanced Solid Tumor, Adult | Study of RP-3500 with Niraparib or Olaparib in Advanced Solid Tumors (ATTACC) | |
| NCT04497116 | Phase 1 Phase 2 | Advanced Solid Tumor | Study of RP-3500 in Advanced Solid Tumors | |
| NCT05269316 | Phase 1 | Solid Tumor Advanced Solid Tumor | Study to Evaluate IMP9064 as a Monotherapy or in Combination in Patients with Advanced Solid Tumors | |
| NCT03787680 | Phase 2 | Prostate Cancer | Targeting Resistant Prostate Cancer with ATR and PARP Inhibition (TRAP Trial) | |
| CHK1 | NCT02797964 | Phase 1 Phase 2 | Advanced Solid Tumors or Non-Hodgkin’s Lymphoma (NHL) | A First phase/2 Trial of SRA737 in Subjects with Advanced Cancer |
| NCT02797977 | Phase 1 Phase 2 | Advanced Solid Tumors | A First phase/2 Trial of SRA737 in Combination with Gemcitabine Plus Cisplatin or Gemcitabine Alone in Subjects with Advanced Cancer | |
| NCT03057145 | Phase 1 | Solid Tumor | Combination Study of Prexasertib and Olaparib in Patients with Advanced Solid Tumors | |
| NCT00045513 | Phase 1 Phase 2 | Leukemia Lymphoma | Combination Chemotherapy in Treating Patients with Chronic Lymphocytic Leukemia or Lymphocytic Lymphoma | |
| NCT00700336 | Phase 1 Phase 2 | Malignant Pleural Mesothelioma Solid Tumors | Study of CBP501 + Pemetrexed + Cisplatin on MPM (Phase I/II) | |
| NCT00415636 | Phase 1 | Cancer | Safety and Tolerability of IC83/LY2603618 Administered After Pemetrexed 500 mg/m2 Every 21 Days in Patients with Cancer | |
| NCT00839332 | Phase 1 Phase 2 | Pancreatic Neoplasms | A Study for Participants with Pancreatic Cancer | |
| NCT00988858 | Phase 2 | Non-Small Cell Lung Cancer | A Study of Advanced or Metastatic Non-Small Cell Lung Cancer | |
| NCT01296568 | Phase 1 | Advanced Cancer | C14 Study in Oncology Patients with Advanced and/or Metastatic Solid Tumors | |
| NCT01139775 | Phase 1 Phase 2 | Non-Small Cell Lung Cancer | A Study in Non-Small Cell Lung Cancer | |
| NCT01341457 | Phase 1 | Solid Tumors | A Study of LY2603618 in Combination with Gemcitabine in Participants with Solid Tumors | |
| NCT01358968 | Phase 1 | Cancer | A Drug Interaction Study to Assess the Effect of LY2603618 on the Metabolic Pathway of Desipramine | |
| NCT00779584 | Phase 1 | Hodgkin Disease Lymphoma, Non-Hodgkin Neoplasms | A Dose-Escalation Study of MK-8776 (SCH 900776) with and without Gemcitabine in Participants with Solid Tumors or Lymphoma (MK-8776-002/P05248) | |
| NCT01115790 | Phase 1 | Advanced Cancer Squamous Cell Carcinoma Carcinoma, Squamous Cell of Head and Neck Lung Squamous Cell Carcinoma Stage IV Anal Squamous Cell Carcinoma Carcinoma, Non-Small-Cell Lung | A First Phase Study in Participants with Advanced Cancer | |
| NCT04095221 | Phase 1 Phase 2 | Desmoplastic Small Round Cell Tumor Rhabdomyosarcoma | A Study of the Drugs Prexasertib, Irinotecan, and Temozolomide in People with Desmoplastic Small Round Cell Tumor and Rhabdomyosarcoma | |
| NCT03495323 | Phase 1 | Cancer | A Study of Prexasertib (LY2606368), CHK1 Inhibitor, and LY3300054, PD-L1 Inhibitor, in Patients with Advanced Solid Tumors | |
| NCT03057145 | Phase 1 | Solid Tumor | Combination Study of Prexasertib and Olaparib in Patients with Advanced Solid Tumors | |
| NCT02873975 | Phase 2 | Advanced Cancers | A Study of LY2606368 (Prexasertib) in Patients with Solid Tumors with Replicative Stress or Homologous Repair Deficiency | |
| NCT02808650 | Phase 1 | Childhood Solid Neoplasm Recurrent Malignant Solid Neoplasm Recurrent Primary Central Nervous System Neoplasm Refractory Malignant Solid Neoplasm Refractory Primary Central Nervous System Neoplasm | Prexasertib in Treating Pediatric Patients with Recurrent or Refractory Solid Tumors | |
| NCT02514603 | Phase 1 | Neoplasm | A Study of Prexasertib (LY2606368) in Japanese Participants with Advanced Cancers |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, J.; Kitty, I.; Renata, K.; Qin, S.; Zhao, F.; Kim, W. DNA Damage and Its Role in Cancer Therapeutics. Int. J. Mol. Sci. 2023, 24, 4741. https://doi.org/10.3390/ijms24054741
Moon J, Kitty I, Renata K, Qin S, Zhao F, Kim W. DNA Damage and Its Role in Cancer Therapeutics. International Journal of Molecular Sciences. 2023; 24(5):4741. https://doi.org/10.3390/ijms24054741
Chicago/Turabian StyleMoon, Jaeyoung, Ichiwa Kitty, Kusuma Renata, Sisi Qin, Fei Zhao, and Wootae Kim. 2023. "DNA Damage and Its Role in Cancer Therapeutics" International Journal of Molecular Sciences 24, no. 5: 4741. https://doi.org/10.3390/ijms24054741
APA StyleMoon, J., Kitty, I., Renata, K., Qin, S., Zhao, F., & Kim, W. (2023). DNA Damage and Its Role in Cancer Therapeutics. International Journal of Molecular Sciences, 24(5), 4741. https://doi.org/10.3390/ijms24054741