
The Pathological Activation of Microglia Is Modulated by Sexually Dimorphic Pathways
Abstract
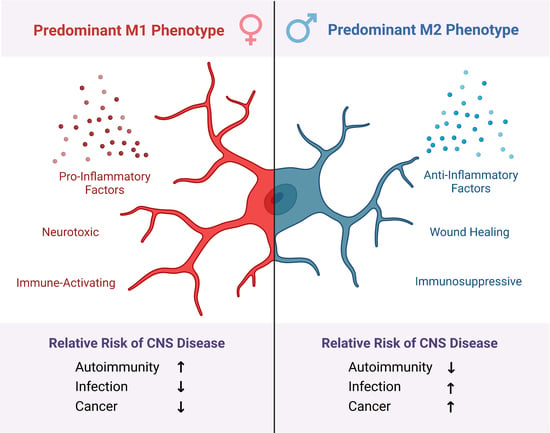
1. Introduction
2. Historical Definition of Microglia
3. Developmental Origin of Microglia
4. Microglia Are Phagocytes of the CNS
5. Microglial Roles in Neuronal Architecture
6. Microglial Activation States
7. Microglia Assume Diverse Morphologies in Disease or Injury
8. Sexually Dimorphic Development of the Neuroimmune System
9. Sex Differentiated Microglial Function through Adulthood
10. Microglial Sexual Dimorphism as a Mediator of Divergent CNS Disorder Outcomes
11. Female Predominance in Autoimmune Disorders Is Mediated by M1 Microglia
11.1. Multiple Sclerosis and Mechanisms of Microglial Involvement
11.2. Neuromyelitis Optica Spectrum Disorders and Mechanisms of Microglial Involvement
12. Worse Outcomes in Male CNS Infections Are Mediated by M2 Microglia
Mechanism of Microglial Involvement in Cryptococcal Meningitis
13. Male Predominance of Glioblastoma Multiforme Is Mediated by M2 Microglia
Mechanisms of Microglial Involvement in GBM
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thompson, K.; Tsirka, S. The Diverse Roles of Microglia in the Neurodegenerative Aspects of Central Nervous System (CNS) Autoimmunity. Int. J. Mol. Sci. 2017, 18, 504. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, I.; Scharler, C.; Zrzavy, T.; Kadowaki, T.; Mödlagl, V.; Rojc, K.; Tröscher, A.R.; Kitic, M.; Ueda, S.; Bradl, M.; et al. Microglia Pre-Activation and Neurodegeneration Precipitate Neuroinflammation without Exacerbating Tissue Injury in Experimental Autoimmune Encephalomyelitis. Acta Neuropathol. Commun. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.N.; Abels, E.R.; van de Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma Hijacks Microglial Gene Expression to Support Tumor Growth. J. Neuroinflamm. 2020, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Béchade, C.; D’Andrea, I.; St-Pierre, M.-K.; Henry, M.S.; Roumier, A.; Tremblay, M.-E. Microglia Gone Rogue: Impacts on Psychiatric Disorders across the Lifespan. Front. Mol. Neurosci. 2018, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Ruytinx, P.; Proost, P.; van Damme, J.; Struyf, S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 2018, 9, 1930. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The Semantics of Microglia Activation: Neuroinflammation, Homeostasis, and Stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef]
- Virchow, R. Gesammelte Abhandlungen Zur Wissenschaftlichen Medicin; Verlag von Meidinger Sohn & Comp.: Frankfurt, Germany, 1856. [Google Scholar]
- Weigert. Beitrage Zur Kenntnis Der Normalen Menschlichen Glia; Verlag Moritz ¨ Diesterweg: Frankfurt, Germany, 1895. [Google Scholar]
- Sierra, A.; de Castro, F.; del Río-Hortega, J.; Rafael Iglesias-Rozas, J.; Garrosa, M.; Kettenmann, H. The “Big-Bang” for Modern Glial Biology: Translation and Comments on Pío Del Río-Hortega 1919 Series of Papers on Microglia. Glia 2016, 64, 1801–1840. [Google Scholar] [CrossRef]
- Kongsui, R.; Beynon, S.B.; Johnson, S.J.; Walker, F.R. Quantitative Assessment of Microglial Morphology and Density Reveals Remarkable Consistency in the Distribution and Morphology of Cells within the Healthy Prefrontal Cortex of the Rat. J. Neuroinflamm. 2014, 11, 182. [Google Scholar] [CrossRef]
- Brennan, F.H.; Li, Y.; Wang, C.; Ma, A.; Guo, Q.; Li, Y.; Pukos, N.; Campbell, W.A.; Witcher, K.G.; Guan, Z.; et al. Microglia Coordinate Cellular Interactions during Spinal Cord Repair in Mice. Nat. Commun. 2022, 13, 4096. [Google Scholar] [CrossRef]
- dos Santos, S.E.; Medeiros, M.; Porfirio, J.; Tavares, W.; Pessôa, L.; Grinberg, L.; Leite, R.E.P.; Ferretti-Rebustini, R.E.L.; Suemoto, C.K.; Filho, W.J.; et al. Similar Microglial Cell Densities across Brain Structures and Mammalian Species: Implications for Brain Tissue Function. J. Neurosci. 2020, 40, 4622–4643. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barres, B.A. Microglia and Macrophages in Brain Homeostasis and Disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Palis, J.; Robertson, S.; Kennedy, M.; Wall, C.; Keller, G. Development of Erythroid and Myeloid Progenitors in the Yolk Sac and Embryo Proper of the Mouse. Development 1999, 126, 5073–5084. [Google Scholar] [CrossRef]
- Ginhoux, F.; Garel, S. The Mysterious Origins of Microglia. Nat. Neurosci. 2018, 21, 897–899. [Google Scholar] [CrossRef] [PubMed]
- Askew, K.; Gomez-Nicola, D. A Story of Birth and Death: Insights into the Formation and Dynamics of the Microglial Population. Brain Behav. Immun. 2018, 69, 9–17. [Google Scholar] [CrossRef]
- Hattori, Y. The Behavior and Functions of Embryonic Microglia. Anat. Sci. Int. 2022, 97, 1–14. [Google Scholar] [CrossRef]
- Schlegelmilch, T.; Henke, K.; Peri, F. Microglia in the Developing Brain: From Immunity to Behaviour. Curr. Opin. Neurobiol. 2011, 21, 5–10. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Zhu, H.; Qiao, X.; Liu, W.; Wang, C.; Zhao, Y. Microglia Play an Essential Role in Synapse Development and Neuron Maturation in Tissue-Engineered Neural Tissues. Front. Neurosci. 2020, 14, 586452. [Google Scholar] [CrossRef] [PubMed]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia Remodel Synapses by Presynaptic Trogocytosis and Spine Head Filopodia Induction. Nat Commun 2018, 9, 1228. [Google Scholar] [CrossRef] [PubMed]
- Cserép, C.; Pósfai, B.; Dénes, Á. Shaping Neuronal Fate: Functional Heterogeneity of Direct Microglia-Neuron Interactions. Neuron 2021, 109, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, P.; Oller, G.; Hoeffel, G.; Pont-Lezica, L.; Rostaing, P.; Low, D.; Bessis, A.; Ginhoux, F.; Garel, S. Microglia Modulate Wiring of the Embryonic Forebrain. Cell Rep. 2014, 8, 1271–1279. [Google Scholar] [CrossRef]
- Diaz-Aparicio, I.; Paris, I.; Sierra-Torre, V.; Plaza-Zabala, A.; Rodríguez-Iglesias, N.; Márquez-Ropero, M.; Beccari, S.; Huguet, P.; Abiega, O.; Alberdi, E.; et al. Microglia Actively Remodel Adult Hippocampal Neurogenesis through the Phagocytosis Secretome. J. Neurosci. 2020, 40, 1453–1482. [Google Scholar] [CrossRef]
- Oyarce, K.; Cepeda, M.Y.; Lagos, R.; Garrido, C.; Vega-Letter, A.M.; Garcia-Robles, M.; Luz-Crawford, P.; Elizondo-Vega, R. Neuroprotective and Neurotoxic Effects of Glial-Derived Exosomes. Front. Cell Neurosci. 2022, 16, 920686. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Kenneth Baillie, J.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial Brain Regionâ ’dependent Diversity and Selective Regional Sensitivities to Aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef]
- Nimmerjahn, A. Optical Window Preparation for Two-Photon Imaging of Microglia in Mice. Cold Spring Harb. Protoc. 2012, 7, 587–593. [Google Scholar] [CrossRef]
- Nimmerjahn, A. Surgical Implantation of a Head Plate in Mice in Preparation for in Vivo Two-Photon Imaging of Microglia. Cold Spring Harb. Protoc. 2012, 7, 583–586. [Google Scholar] [CrossRef]
- Li, T.; Qin, K.; Li, N.; Han, C.; Cao, X. An Endosomal LAPF Is Required for Macrophage Endocytosis and Elimination of Bacteria. Proc. Natl. Acad. Sci. USA 2019, 116, 12958–12963. [Google Scholar] [CrossRef]
- Solé-Domènech, S.; Cruz, D.L.; Capetillo-Zarate, E.; Maxfield, F.R. The Endocytic Pathway in Microglia during Health. Aging Alzheimer’s Dis. 2016, 32, 89–103. [Google Scholar]
- Greenhalgh, A.D.; David, S. Differences in the Phagocytic Response of Microglia and Peripheral Macrophages after Spinal Cord Injury and Its Effects on Cell Death. J. Neurosci. 2014, 34, 6316–6322. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Saito, T.; Saido, T.C.; Barron, A.M.; Ruedl, C. Microglia and CD206+ Border-Associated Mouse Macrophages Maintain Their Embryonic Origin during Alzheimer’s Disease. eLife 2021, 10, e71879. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M.V. Local Self-Renewal Can Sustain CNS Microglia Maintenance and Function throughout Adult Life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Ivan, D.C.; Walthert, S.; Berve, K.; Steudler, J.; Locatelli, G. Dwellers and Trespassers: Mononuclear Phagocytes at the Borders of the Central Nervous System. Front. Immunol. 2020, 11, 609921. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in Neurodegenerative Diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Garaschuk, O.; Verkhratsky, A. Physiology of Microglia. Methods Mol. Biol. 2019, 2034, 27–40. [Google Scholar] [CrossRef]
- Amici, S.A.; Dong, J.; Guerau-de-Arellano, M. Molecular Mechanisms Modulating the Phenotype of Macrophages and Microglia. Front. Immunol. 2017, 8, 1520. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New Tools for Studying Microglia in the Mouse and Human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a Unique TGF-β–Dependent Molecular and Functional Signature in Microglia. Nat. Neurosci. 2013, 17, 131–143. [Google Scholar] [CrossRef]
- Boada-Romero, E.; Martinez, J.; Heckmann, B.L.; Green, D.R. The Clearance of Dead Cells by Efferocytosis. Nature Reviews. Mol. Cell Biol. 2020, 21, 398–414. [Google Scholar]
- Morioka, S.; Maueröder, C.; Ravichandran, K.S. Living on the Edge: Efferocytosis at the Interface of Homeostasis and Pathology. Immunity 2019, 50, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Eaten Alive! Cell Death by Primary Phagocytosis: “Phagoptosis”. Trends Biochem. Sci. 2012, 37, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Beginnings of a Good Apoptotic Meal: The Find-Me and Eat-Me Signaling Pathways. Immunity 2011, 35, 445–455. [Google Scholar] [CrossRef]
- Lian, H.; Roy, E.; Zheng, H. Microglial Phagocytosis Assay. Bio Protocol 2016, 6, e1988. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of Microglial Phagocytosis Is Sufficient to Prevent Inflammatory Neuronal Death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef]
- Rajbhandari, L.; Tegenge, M.A.; Shrestha, S.; Ganesh Kumar, N.; Malik, A.; Mithal, A.; Hosmane, S.; Venkatesan, A. Toll-like Receptor 4 Deficiency Impairs Microglial Phagocytosis of Degenerating Axons. Glia 2014, 62, 1982–1991. [Google Scholar] [CrossRef]
- Koenigsknecht, J.; Landreth, G. Microglial Phagocytosis of Fibrillar Beta-Amyloid through a Beta1 Integrin-Dependent Mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia Clear Neuron-Released α-Synuclein via Selective Autophagy and Prevent Neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS Beta-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional Impairment of Microglia Coincides with Beta-Amyloid Deposition in Mice with Alzheimer-like Pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.; el Khoury, J. Microglial Scavenger Receptors and Their Roles in the Pathogenesis of Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2012, 2012, 489456. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zheng, Y.; Xu, D.; Sun, Z.; Yang, H.; Yin, Q. Galectin-3: A Key Player in Microglia-Mediated Neuroinflammation and Alzheimer’s Disease. Cell Biosci. 2021, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, H.; Riese, S.; Régnier-Vigouroux, A. Functional Characterization of Mannose Receptor Expressed by Immunocompetent Mouse Microglia. Glia 2003, 42, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Du, H. TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics. Cells 2021, 10, 321. [Google Scholar] [CrossRef]
- Hanke, M.L.; Kielian, T. Toll-like Receptors in Health and Disease in the Brain: Mechanisms and Therapeutic Potential. Clin. Sci. 2011, 121, 367–387. [Google Scholar] [CrossRef]
- Janda, E.; Boi, L.; Carta, A.R. Microglial Phagocytosis and Its Regulation: A Therapeutic Target in Parkinson’s Disease? Front. Mol. Neurosci. 2018, 11, 144. [Google Scholar] [CrossRef]
- Azam, S.; Haque, M.E.; Kim, I.S.; Choi, D.K. Microglial Turnover in Ageing-Related Neurodegeneration: Therapeutic Avenue to Intervene in Disease Progression. Cells 2021, 10, 150. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, D.R.; Blanco-Luquin, I.; Mendioroz, M. The Participation of Microglia in Neurogenesis: A Review. Brain Sci 2021, 11, 658. [Google Scholar] [CrossRef]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Folick, A.; Koliwad, S.K.; Valdearcos, M. Microglial Lipid Biology in the Hypothalamic Regulation of Metabolic Homeostasis. Front. Endocrinol. 2021, 12, 591. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia Actively Regulate the Number of Functional Synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef]
- Lim, T.K.Y.; Ruthazer, E.S. Microglial Trogocytosis and the Complement System Regulate Axonal Pruning in Vivo. eLife 2021, 10, e62167. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.; Danver, J.; Ji, K.; Miyauchi, J.T.; Chen, D.; Anderson, M.E.; West, B.L.; Robinson, J.K.; Tsirka, S.E. Dynamic Microglial Modulation of Spatial Learning and Social Behavior. Brain Behav. Immun. 2016, 55, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and Microglia Mediate Early Synapse Loss in Alzheimer Mouse Models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia Contact Induces Synapse Formation in Developing Somatosensory Cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C.; et al. Astrocyte-Derived Interleukin-33 Promotes Microglial Synapse Engulfment and Neural Circuit Development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef]
- Blinzinger, K.; Kreutzberg, G. Displacement of Synaptic Terminals from Regenerating Motoneurons by Microglial Cells. Z. Für Zellforsch. Und Mikrosk. Anat. 1968, 85, 145–157. [Google Scholar] [CrossRef]
- Chen, Z.; Jalabi, W.; Hu, W.; Park, H.J.; Gale, J.T.; Kidd, G.J.; Bernatowicz, R.; Gossman, Z.C.; Chen, J.T.; Dutta, R.; et al. Microglial Displacement of Inhibitory Synapses Provides Neuroprotection in the Adult Brain. Nat. Commun. 2014, 5, 5486. [Google Scholar] [CrossRef]
- Avignone, E.; Lepleux, M.; Angibaud, J.; Nägerl, U.V. Altered Morphological Dynamics of Activated Microglia after Induction of Status Epilepticus. J. Neuroinflamm. 2015, 12, 202. [Google Scholar] [CrossRef] [PubMed]
- Abdolhoseini, M.; Kluge, M.G.; Walker, F.R.; Johnson, S.J. Segmentation, Tracing, and Quantification of Microglial Cells from 3D Image Stacks. Sci. Rep. 2019, 9, 8557. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.; Nilsson, M.; Jones, K. Acute and Chronic Stress-Induced Disturbances of Microglial Plasticity, Phenotype and Function. Curr. Drug Targets 2013, 14, 1262–1276. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.; Benjaminsen, J.; Moritz, C.; Henke, K.; Hartmann, J.; Norlin, N.; Richter, K.; Schieber, N.L.; Franke, T.; Schwab, Y.; et al. Clearance by Microglia Depends on Packaging of Phagosomes into a Unique Cellular Compartment. Dev. Cell 2019, 49, 77–88.e7. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- von Leden, R.E.; Yauger, Y.J.; Khayrullina, G.; Byrnes, K.R. Central Nervous System Injury and Nicotinamide Adenine Dinucleotide Phosphate Oxidase: Oxidative Stress and Therapeutic Targets. J. Neurotrauma 2017, 34, 755–764. [Google Scholar] [CrossRef]
- Zhuang, Z.; Yoshizawa-Smith, S.; Glowacki, A.; Maltos, K.; Pacheco, C.; Shehabeldin, M.; Mulkeen, M.; Myers, N.; Chong, R.; Verdelis, K.; et al. Induction of M2 Macrophages Prevents Bone Loss in Murine Periodontitis Models. J. Dent. Res. 2019, 98, 200–208. [Google Scholar] [CrossRef]
- Czeh, M.; Gressens, P.; Kaindl, A.M. The Yin and Yang of Microglia. Dev. Neurosci. 2011, 33, 199–209. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Li, J.; Shui, X.; Sun, R.; Wan, L.; Zhang, B.; Xiao, B.; Luo, Z. Microglial Phenotypic Transition: Signaling Pathways and Influencing Modulators Involved in Regulation in Central Nervous System Diseases. Front Cell Neurosci 2021, 15, 359. [Google Scholar] [CrossRef] [PubMed]
- Verkhrasky, A.; Krishtal, O.A.; Burnstock, G. Purinoceptors on Neuroglia. Mol. Neurobiol. 2009, 39, 190–208. [Google Scholar] [CrossRef] [PubMed]
- Franke, H.; Verkhratsky, A.; Burnstock, G.; Illes, P. Pathophysiology of Astroglial Purinergic Signalling. Purinergic Signal. 2012, 8, 629. [Google Scholar] [CrossRef] [PubMed]
- Mead, E.L.; Mosley, A.; Eaton, S.; Dobson, L.; Heales, S.J.; Pocock, J.M. Microglial Neurotransmitter Receptors Trigger Superoxide Production in Microglia; Consequences for Microglial-Neuronal Interactions. J. Neurochem. 2012, 121, 287–301. [Google Scholar] [CrossRef]
- Davis, B.M.; Salinas-Navarro, M.; Cordeiro, M.F.; Moons, L.; Groef, L. de Characterizing Microglia Activation: A Spatial Statistics Approach to Maximize Information Extraction. Sci. Rep. 2017, 7, 1576. [Google Scholar] [CrossRef]
- Woollacott, I.O.C.; Toomey, C.E.; Strand, C.; Courtney, R.; Benson, B.C.; Rohrer, J.D.; Lashley, T. Microglial Burden, Activation and Dystrophy Patterns in Frontotemporal Lobar Degeneration. J. Neuroinflamm. 2020, 17, 234. [Google Scholar] [CrossRef]
- Shahidehpour, R.K.; Higdon, R.E.; Crawford, N.G.; Neltner, J.H.; Ighodaro, E.T.; Patel, E.; Price, D.; Nelson, P.T.; Bachstetter, A.D. Dystrophic Microglia Are Associated with Neurodegenerative Disease and Not Healthy Aging in the Human Brain. Neurobiol. Aging 2021, 99, 19–27. [Google Scholar] [CrossRef]
- Ohm, D.T.; Fought, A.J.; Martersteck, A.; Coventry, C.; Sridhar, J.; Gefen, T.; Weintraub, S.; Bigio, E.; Mesulam, M.M.; Rogalski, E.; et al. Accumulation of Neurofibrillary Tangles and Activated Microglia Is Associated with Lower Neuron Densities in the Aphasic Variant of Alzheimer’s Disease. Brain Pathol. 2021, 31, 189–204. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of Senescent Glial Cells Prevents Tau-Dependent Pathology and Cognitive Decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Safwan-Zaiter, H.; Wagner, N.; Wagner, K.D. P16INK4A-More Than a Senescence Marker. Life 2022, 12, 1332. [Google Scholar] [CrossRef]
- Au, N.P.B.; Ma, C.H.E. Recent Advances in the Study of Bipolar/Rod-Shaped Microglia and Their Roles in Neurodegeneration. Front. Aging Neurosci. 2017, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.E.; Morganti-Kossmann, C.; Lifshitz, J.; Ziebell, J.M. Rod Microglia: A Morphological Definition. PLoS ONE 2014, 9, e97096. [Google Scholar] [CrossRef] [PubMed]
- Giordano, K.R.; Denman, C.R.; Dubisch, P.S.; Akhter, M.; Lifshitz, J. An Update on the Rod Microglia Variant in Experimental and Clinical Brain Injury and Disease. Brain Commun. 2021, 3, fcaa227. [Google Scholar] [CrossRef] [PubMed]
- Lier, J.; Streit, W.J.; Bechmann, I. Beyond Activation: Characterizing Microglial Functional Phenotypes. Cells 2021, 10, 2236. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; el Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial Signatures and Their Role in Health and Disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Fitz, N.F.; Nam, K.N.; Wolfe, C.M.; Letronne, F.; Playso, B.E.; Iordanova, B.E.; Kozai, T.D.Y.; Biedrzycki, R.J.; Kagan, V.E.; Tyurina, Y.Y.; et al. Phospholipids of APOE Lipoproteins Activate Microglia in an Isoform-Specific Manner in Preclinical Models of Alzheimer’s Disease. Nat. Commun. 2021, 12, 3416. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- McCarthy, M.M. A New View of Sexual Differentiation of Mammalian Brain. J. Comp. Physiol. Neuroethol. Sens. Neural Behav. Physiol. 2020, 206, 369–378. [Google Scholar] [CrossRef]
- Brooks, W.H.; Renaudineau, Y. Epigenetics and Autoimmune Diseases: The X Chromosome-Nucleolus Nexus. Front. Genet. 2015, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Forest, M.G.; Cathiard, A.M.; Bertrand, J.A. Total and Unbound Testosterone Levels in the Newborn and in Normal and Hypogonadal Children: Use of a Sensitive Radioimmunoassay for Testosterone. J. Clin. Endocrinol. Metab. 1973, 36, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Reyes, F.I.; Winter, S.D.; Faiman, C. Studies on Human Sexual Development. I. Fetal Gonadal and Adrenal Sex Steroids. J. Clin Endocrinol Metab 1973, 37, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Vegeto, E.; Poletti, A.; Maggi, A. Estrogens, Neuroinflammation, and Neurodegeneration. Endocr. Rev. 2016, 37, 372–402. [Google Scholar] [CrossRef] [PubMed]
- Nugent, B.M.; Wright, C.L.; Shetty, A.C.; Hodes, G.E.; Lenz, K.M.; Mahurkar, A.; Russo, S.J.; Devine, S.E.; McCarthy, M.M. Brain Feminization Requires Active Repression of Masculinization via DNA Methylation. Nat. Neurosci. 2015, 18, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Carmignoto, G.; Pasti, L.; Vesce, S.; Rossi, D.; Rizzini, B.L.; Pozzant, T.; Volterra, A. Prostaglandins Stimulate Calcium-Dependent Glutamate Release in Astrocytes. Nature 1998, 391, 281–285. [Google Scholar] [CrossRef]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2016, 42, 193–215. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Cantaut-Belarif, Y.; Antri, M.; Pizzarelli, R.; Colasse, S.; Vaccari, I.; Soares, S.; Renner, M.; Dallel, R.; Triller, A.; Bessis, A. Microglia Control the Glycinergic but Not the GABAergic Synapses via Prostaglandin E2 in the Spinal Cord. J. Cell Biol. 2017, 216, 2979–2989. [Google Scholar] [CrossRef]
- Lenz, K.M.; McCarthy, M.M. A Starring Role for Microglia in Brain Sex Differences. Neuroscientist 2015, 21, 306. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “Quad-Partite” Synapse: Microglia-Synapse Interactions in the Developing and Mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Ceasrine, A.M.; Bilbo, S.D. Microglia and Sexual Differentiation of the Developing Brain: A Focus on Ontogeny and Intrinsic Factors. Glia 2020, 68, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Sholar, P.W.; Bilbo, S.D. Sex Differences in Microglial Colonization of the Developing Rat Brain. J. Neurochem 2012, 120, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.S. Sexual Steroids and Their Receptors Affect Microglia-Mediated Neuroinflammation in Neurodegenerative Diseases. Biomed. J. Sci. Tech. Res. 2020, 25. [Google Scholar] [CrossRef]
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P. Maternal Immune Activation and Abnormal Brain Development across CNS Disorders. Nat Rev Neurol 2014, 10, 643–660. [Google Scholar] [CrossRef]
- Jiang, N.M.; Cowan, M.; Moonah, S.N.; Petri, W.A. The Impact of Systemic Inflammation on Neurodevelopment. Trends Mol. Med. 2018, 24, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Hanamsagar, R.; Alter, M.D.; Block, C.S.; Sullivan, H.; Bolton, J.L.; Bilbo, S.D. Generation of a Microglial Developmental Index in Mice and in Humans Reveals a Sex Difference in Maturation and Immune Reactivity. Glia 2017, 65, 1504–1520. [Google Scholar] [CrossRef]
- Mangold, C.A.; Wronowski, B.; Du, M.; Masser, D.R.; Hadad, N.; Bixler, G.V.; Brucklacher, R.M.; Ford, M.M.; Sonntag, W.E.; Freeman, W.M. Sexually Divergent Induction of Microglial-Associated Neuroinflammation with Hippocampal Aging. J. Neuroinflammation 2017, 14, 141. [Google Scholar] [CrossRef]
- Murtaj, V.; Belloli, S.; di Grigoli, G.; Pannese, M.; Ballarini, E.; Rodriguez-Menendez, V.; Marmiroli, P.; Cappelli, A.; Masiello, V.; Monterisi, C.; et al. Age and Sex Influence the Neuro-Inflammatory Response to a Peripheral Acute LPS Challenge. Front. Aging Neurosci. 2019, 11, 299. [Google Scholar] [CrossRef]
- Yanguas-Casás, N.; Crespo-Castrillo, A.; de Ceballos, M.L.; Chowen, J.A.; Azcoitia, I.; Arevalo, M.A.; Garcia-Segura, L.M. Sex Differences in the Phagocytic and Migratory Activity of Microglia and Their Impairment by Palmitic Acid. Glia 2018, 66, 522–537. [Google Scholar] [CrossRef]
- Hind, L.E.; Lurier, E.B.; Dembo, M.; Spiller, K.L.; Hammer, D.A. Effect of M1-M2 Polarization on the Motility and Traction Stresses of Primary Human Macrophages. Cell Mol. Bioeng. 2016, 9, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.S.; Ase, A.R.; Kinsara, A.; Rao, V.T.S.; Robinson, M.M.; Leong, S.Y.; Butovsky, O.; Ludwin, S.K.; Seguela, P.; Bar-Or, A.; et al. P2Y12 Expression and Function in Alternatively Activated Human Microglia. Neurol. (R) Neuroimmunol. Neuroinflamm. 2015, 2, e80. [Google Scholar] [CrossRef] [PubMed]
- Guneykaya, D.; Ivanov, A.; Hernandez, D.P.; Haage, V.; Wojtas, B.; Meyer, N.; Maricos, M.; Jordan, P.; Buonfiglioli, A.; Gielniewski, B.; et al. Transcriptional and Translational Differences of Microglia from Male and Female Brains. Cell Rep. 2018, 24, 2773–2783.e6. [Google Scholar] [CrossRef] [PubMed]
- Barreto, G.; Veiga, S.; Azcoitia, I.; Garcia-Segura, L.M.; Garcia-Ovejero, D. Testosterone Decreases Reactive Astroglia and Reactive Microglia after Brain Injury in Male Rats: Role of Its Metabolites, Oestradiol and Dihydrotestosterone. Eur. J. Neurosci. 2007, 25, 3039–3046. [Google Scholar] [CrossRef]
- Yilmaz, C.; Karali, K.; Fodelianaki, G.; Gravanis, A.; Chavakis, T.; Charalampopoulos, I.; Alexaki, V.I. Neurosteroids as Regulators of Neuroinflammation. Front. Neuroendocrinol. 2019, 55, 788. [Google Scholar] [CrossRef]
- Patil, C.N.; Wallace, K.; la Marca, B.D.; Moulana, M.; Lopez-Ruiz, A.; Soljancic, A.; Juncos, L.A.; Grande, J.P.; Reckelhoff, J.F. Low-Dose Testosterone Protects against Renal Ischemia-Reperfusion Injury by Increasing Renal IL-10-to-TNF-α Ratio and Attenuating T-Cell Infiltration. Am. J. Physiol. Renal Physiol. 2016, 311, F395–F403. [Google Scholar] [CrossRef]
- Mohamad, N.V.; Wong, S.K.; Wan Hasan, W.N.; Jolly, J.J.; Nur-Farhana, M.F.; Ima-Nirwana, S.; Chin, K.Y. The Relationship between Circulating Testosterone and Inflammatory Cytokines in Men. Aging Male 2018, 22, 129–140. [Google Scholar] [CrossRef]
- Cutolo, M.; Capellino, S.; Sulli, A.; Serioli, B.; Secchi, M.E.; Villaggio, B.; Straub, R.H. Estrogens and Autoimmune Diseases. Ann. N. Y. Acad. Sci. 2006, 1089, 538–547. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, J.; Huang, M.; Kosonen, R.; Lee, J.E. CCR4 and CCR5 Involvement in Monocyte-Derived Macrophage Migration in Neuroinflammation. Front. Immunol. 2022, 13, 1885. [Google Scholar] [CrossRef]
- Grimaldi, C.M.; Jeganathan, V.; Diamond, B. Hormonal Regulation of B Cell Development: 17 Beta-Estradiol Impairs Negative Selection of High-Affinity DNA-Reactive B Cells at More than One Developmental Checkpoint. J. Immunol. 2006, 176, 2703–2710. [Google Scholar] [CrossRef]
- Mo, R.; Chen, J.; Grolleau-Julius, A.; Murphy, H.S.; Richardson, B.C.; Yung, R.L. Estrogen Regulates CCR Gene Expression and Function in T Lymphocytes. J. Immunol. 2005, 174, 6023–6029. [Google Scholar] [CrossRef] [PubMed]
- Loram, L.C.; Sholar, P.W.; Taylor, F.R.; Wiesler, J.L.; Babb, J.A.; Strand, K.A.; Berkelhammer, D.; Day, H.E.W.; Maier, S.F.; Watkins, L.R. Sex and Estradiol Influence Glial Pro-Inflammatory Responses to Lipopolysaccharide in Rats. Psychoneuroendocrinology 2012, 37, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nugent, B.M.; Haliyur, R.; McCarthy, M.M. Microglia Are Essential to Masculinization of Brain and Behavior. J. Neurosci. 2013, 33, 2761. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Gelosa, P.; Castiglioni, L.; Cimino, M.; Rizzi, N.; Pepe, G.; Lolli, F.; Marcello, E.; Sironi, L.; Vegeto, E.; et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018, 23, 3501–3511. [Google Scholar] [CrossRef]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Microglia Express Distinct M1 and M2 Phenotypic Markers in the Postnatal and Adult Central Nervous System in Male and Female Mice. J. Neurosci. Res. 2013, 91, 1143–1151. [Google Scholar] [CrossRef]
- Oyola, M.G.; Handa, R.J. Hypothalamic-Pituitary-Adrenal and Hypothalamic-Pituitary-Gonadal Axes: Sex Differences in Regulation of Stress Responsivity. Stress 2017, 20, 476–494. [Google Scholar] [CrossRef]
- Trumble, B.C.; Blackwell, A.D.; Stieglitz, J.; Thompson, M.E.; Suarez, I.M.; Kaplan, H.; Gurven, M. Associations between Male Testosterone and Immune Function in a Pathogenically Stressed Forager-Horticultural Population. Am. J. Phys. Anthropol. 2016, 161, 494–505. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, E67–E492. [Google Scholar] [CrossRef]
- Peacock, J.L.; Marston, L.; Marlow, N.; Calvert, S.A.; Greenough, A. Neonatal and Infant Outcome in Boys and Girls Born Very Prematurely. Pediatr. Res. 2012, 71, 305–310. [Google Scholar] [CrossRef]
- Salminen, S.; Vuoksimaa, E.; Rose, R.J.; Kaprio, J. Age, Sex, and Genetic and Environmental Effects on Unintentional Injuries in Young and Adult Twins. Twin Res. Hum. Genet. 2018, 21, 502–506. [Google Scholar] [CrossRef]
- Santos, S.; Ferreira, H.; Martins, J.; Gonçalves, J.; Castelo-Branco, M. Male Sex Bias in Early and Late Onset Neurodevelopmental Disorders: Shared Aspects and Differences in Autism Spectrum Disorder, Attention Deficit/Hyperactivity Disorder, and Schizophrenia. Neurosci. Biobehav. Rev. 2022, 135, 104577. [Google Scholar] [CrossRef] [PubMed]
- Baizabal-Carvallo, J.F.; Jankovic, J. Sex Differences in Patients with Tourette Syndrome. CNS Spectr. 2022, 16, 1–7. [Google Scholar] [CrossRef]
- Arnett, A.B.; Pennington, B.F.; Peterson, R.L.; Willcutt, E.G.; DeFries, J.C.; Olson, R.K. Explaining the Sex Difference in Dyslexia. J. Child Psychol. Psychiatry 2017, 58, 719. [Google Scholar] [CrossRef] [PubMed]
- Drayna, D.; Kilshaw, J.; Kelly, J. The Sex Ratio in Familial Persistent Stuttering. Am. J. Hum. Genet. 1999, 65, 1473–1475. [Google Scholar] [CrossRef] [PubMed]
- Sommer, I.E.; Tiihonen, J.; van Mourik, A.; Tanskanen, A.; Taipale, H. The Clinical Course of Schizophrenia in Women and Men-a Nation-Wide Cohort Study. NPJ Schizophr. 2020, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Striegel-Moore, R.H.; Rosselli, F.; Perrin, N.; DeBar, L.; Wilson, G.T.; May, A.; Kraemer, H.C. Gender Difference in the Prevalence of Eating Disorder Symptoms. Int. J. Eat. Disord. 2009, 42, 471–474. [Google Scholar] [CrossRef]
- Bove, R.M.; Healy, B.; Augustine, A.; Musallam, A.; Gholipour, T.; Chitnis, T. Effect of Gender on Late-Onset Multiple Sclerosis. Mult. Scler. 2012, 18, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender Differences in Autoimmune Disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef]
- Ahlgren, C.; Odén, A.; Lycke, J. High Nationwide Prevalence of Multiple Sclerosis in Sweden. Mult. Scler. 2011, 17, 901–908. [Google Scholar] [CrossRef]
- Wallin, M.T.; Culpepper, W.J.; Coffman, P.; Pulaski, S.; Maloni, H.; Mahan, C.M.; Haselkorn, J.K.; Kurtzke, J.F. The Gulf War Era Multiple Sclerosis Cohort: Age and Incidence Rates by Race, Sex and Service. Brain 2012, 135, 1778–1785. [Google Scholar] [CrossRef]
- Gold, S.M.; Willing, A.; Leypoldt, F.; Paul, F.; Friese, M.A. Sex Differences in Autoimmune Disorders of the Central Nervous System. Semin. Immunopathol. 2019, 41, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Sati, P.; Masuzzo, F.; Nair, G.; Sethi, V.; Kolb, H.; Ohayon, J.; Wu, T.; Cortese, I.C.M.; Reich, D.S. Association of Chronic Active Multiple Sclerosis Lesions With Disability In Vivo. JAMA Neurol 2019, 76, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Metz, I.; Weigand, S.D.; Popescu, B.F.G.; Frischer, J.M.; Parisi, J.E.; Guo, Y.; Lassmann, H.; Brück, W.; Lucchinetti, C.F. Pathologic Heterogeneity Persists in Early Active Multiple Sclerosis Lesions. Ann. Neurol 2014, 75, 728–738. [Google Scholar] [CrossRef]
- Pozzilli, C.; Tomassini, V.; Marinelli, F.; Paolillo, A.; Gasperini, C.; Bastianello, S. “Gender Gap” in Multiple Sclerosis: Magnetic Resonance Imaging Evidence. Eur. J. Neurol 2003, 10, 95–97. [Google Scholar] [CrossRef]
- Rodriguez-mogeda, C.; Lorenzo, S.R.; Attia, J.; van Horssen, J.; Witte, M.E.; de Vries, H.E. Breaching Brain Barriers: B Cell Migration in Multiple Sclerosis. Biomolecules 2022, 12, 800. [Google Scholar] [CrossRef]
- Confavreux, C.; Vukusic, S.; Adeleine, P. Early Clinical Predictors and Progression of Irreversible Disability in Multiple Sclerosis: An Amnesic Process. Brain 2003, 126, 770–782. [Google Scholar] [CrossRef]
- Chitnis, T. The Role of Testosterone in MS Risk and Course. Mult. Scler. 2018, 24, 36–41. [Google Scholar] [CrossRef]
- Bove, R.; Malik, M.T.; Diaz-Cruz, C.; Chua, A.; Saraceno, T.J.; Bargiela, D.; Greeke, E.; Glanz, B.I.; Healy, B.C.; Chitnis, T. The 2D:4D Ratio, a Proxy for Prenatal Androgen Levels, Differs in Men with and without MS. Neurology 2015, 85, 1209. [Google Scholar] [CrossRef]
- Jarius, S.; Wildemann, B.; Paul, F. Neuromyelitis Optica: Clinical Features, Immunopathogenesis and Treatment. Clin. Exp. Immunol. 2014, 176, 149–164. [Google Scholar] [CrossRef]
- Chan, K.H.; Tse, C.T.; Chung, C.P.; Lee, R.L.C.; Kwan, J.S.C.; Ho, P.W.L.; Ho, J.W.M. Brain Involvement in Neuromyelitis Optica Spectrum Disorders. Arch. Neurol. 2011, 68, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Höftberger, R.; Fujihara, K.; Wimmer, I.; Takai, Y.; Nishiyama, S.; Nakashima, I.; Konno, H.; Bradl, M.; Garzuly, F.; et al. Presence of Six Different Lesion Types Suggests Diverse Mechanisms of Tissue Injury in Neuromyelitis Optica. Acta Neuropathol. 2013, 125, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Asgari, N.; Lillevang, S.T.; Skejoe, H.P.B.; Falah, M.; Stenager, E.; Kyvik, K.O. A Population-Based Study of Neuromyelitis Optica in Caucasians. Neurology 2011, 76, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, D.I.; Sveinsson, O.; Hakim, R.; Brundin, L. Epidemiology of NMOSD in Sweden from 1987 to 2013: A Nationwide Population-Based Study. Neurology 2019, 93, e181. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, M.; Aldea, M.; Escudero, D.; Llufriu, S.; Arrambide, G.; Otero-Romero, S.; Sastre-Garriga, J.; Romero-Pinel, L.; Martínez-Yélamos, S.; Sola-Valls, N.; et al. Epidemiology of NMOSD in Catalonia: Influence of the New 2015 Criteria in Incidence and Prevalence Estimates. Mult. Scler. 2018, 24, 1843–1851. [Google Scholar] [CrossRef]
- Borisow, N.; Kleiter, I.; Gahlen, A.; Fischer, K.; Wernecke, K.D.; Pache, F.; Ruprecht, K.; Havla, J.; Krumbholz, M.; Kümpfel, T.; et al. Influence of Female Sex and Fertile Age on Neuromyelitis Optica Spectrum Disorders. Mult. Scler. J. 2016, 23, 1092–1103. [Google Scholar] [CrossRef]
- Murtonen, A.; Sumelahti, M.L. Multiple Sclerosis Prevalence in 2000 and 2010 in Western Finland. Acta Neurol. Scand. 2020, 141, 311–318. [Google Scholar] [CrossRef]
- Papp, V.; Illes, Z.; Magyari, M.; Koch-Henriksen, N.; Kant, M.; Pfleger, C.C.; Roemer, S.F.; Jensen, M.B.; Petersen, A.E.; Nielsen, H.H.; et al. Nationwide Prevalence and Incidence Study of Neuromyelitis Optica Spectrum Disorder in Denmark. Neurology 2018, 91, e2265. [Google Scholar] [CrossRef]
- Flanagan, E.P.; Cabre, P.; Weinshenker, B.G.; Sauver, J.S.; Jacobson, D.J.; Majed, M.; Lennon, V.A.; Lucchinetti, C.F.; McKeon, A.; Matiello, M.; et al. Epidemiology of Aquaporin-4 Autoimmunity and Neuromyelitis Optica Spectrum. Ann. Neurol. 2016, 79, 775. [Google Scholar] [CrossRef]
- Quek, A.M.; McKeon, A.; Lennon, V.A.; Mandrekar, J.N.; Iorio, R.; Jiao, Y.; Costanzi, C.; Weinshenker, B.G.; Wingerchuk, D.M.; Lucchinetti, C.F.; et al. Effects of Age and Sex on Aquaporin-4 Autoimmunity. Archives of Neurology. 2012, 69, 1039–1043. [Google Scholar] [CrossRef]
- Kim, S.M.; Waters, P.; Woodhall, M.; Kim, Y.J.; Kim, J.A.; Cheon, S.Y.; Lee, S.; Jo, S.R.; Kim, D.G.; Jung, K.C.; et al. Gender Effect on Neuromyelitis Optica Spectrum Disorder with Aquaporin4-Immunoglobulin G. Mult. Scler. 2017, 23, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Moinfar, Z.; Zamvil, S.S. Microglia Complement Astrocytes in Neuromyelitis Optica. J. Clin. Investig. 2020, 130, 3961. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, S.; Gordon, P.M.K.; Lee, B.H.; Read, J.; Workentine, M.L.; Sharkey, K.A.; Pittman, Q.J. Characterization of Microglial Transcriptomes in the Brain and Spinal Cord of Mice in Early and Late Experimental Autoimmune Encephalomyelitis Using a RiboTag Strategy. Sci. Rep. 2021, 11, 14319. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH Oxidase Expression in Active Multiple Sclerosis Lesions in Relation to Oxidative Tissue Damage and Mitochondrial Injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef]
- Fischer, M.T.; Wimmer, I.; Höftberger, R.; Gerlach, S.; Haider, L.; Zrzavy, T.; Hametner, S.; Mahad, D.; Binder, C.J.; Krumbholz, M.; et al. Disease-Specific Molecular Events in Cortical Multiple Sclerosis Lesions. Brain 2013, 136, 1799–1815. [Google Scholar] [CrossRef]
- Nally, F.K.; de Santi, C.; McCoy, C.E. Nanomodulation of Macrophages in Multiple Sclerosis. Cells 2019, 8, 543. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 Microglia and Macrophages Drive Oligodendrocyte Differentiation during CNS Remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]
- Kaskow, B.J.; Baecher-Allan, C. Effector T Cells in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Nissen, J.C.; Tsirka, S.E. Tuftsin-Driven Experimental Autoimmune Encephalomyelitis Recovery Requires Neuropilin-1. Glia 2016, 64, 923–936. [Google Scholar] [CrossRef]
- Wu, M.; Nissen, J.C.; Chen, E.I.; Tsirka, S.E. Tuftsin Promotes an Anti-Inflammatory Switch and Attenuates Symptoms in Experimental Autoimmune Encephalomyelitis. PLoS ONE 2012, 7, e34933. [Google Scholar] [CrossRef]
- Thompson, K.K.; Nissen, J.C.; Pretory, A.; Tsirka, S.E. Tuftsin Combines With Remyelinating Therapy and Improves Outcomes in Models of CNS Demyelinating Disease. Front. Immunol. 2018, 9, 2784. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.C.; Thompson, K.K.; West, B.L.; Tsirka, S.E. Csf1R Inhibition Attenuates Experimental Autoimmune Encephalomyelitis and Promotes Recovery. Exp. Neurol. 2018, 307, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhu, K.; Zhou, K.; Hakim, R.; Sankavaram, S.R.; Blomgren, K.; Lund, H.; Zhang, X.M.; Harris, R.A. Sex-Specific Effects of Microglia-Like Cell Engraftment during Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci 2020, 21, 6824. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pasamar, S.; Abad, E.; Moreno, B.; Velez de Mendizabal, N.; Martinez-Forero, I.; Garcia-Ojalvo, J.; Villoslada, P. Dynamic Cross-Regulation of Antigen-Specific Effector and Regulatory T Cell Subpopulations and Microglia in Brain Autoimmunity. BMC Syst. Biol. 2013, 7, 34. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-Cell Follicles in Secondary Progressive Multiple Sclerosis Associate with Early Onset of Disease and Severe Cortical Pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.W.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Serafini, B.; Aloisi, F.; Reynolds, R. A Gradient of Neuronal Loss and Meningeal Inflammation in Multiple Sclerosis. Ann. Neurol. 2010, 68, 477–493. [Google Scholar] [CrossRef]
- Jie, Z.; Ko, C.J.; Wang, H.; Xie, X.; Li, Y.; Gu, M.; Zhu, L.; Yang, J.Y.; Gao, T.; Ru, W.; et al. Microglia Promote Autoimmune Inflammation via the Noncanonical NF-ΚB Pathway. Sci. Adv. 2021, 7, eabh0609. [Google Scholar] [CrossRef]
- Zepp, J.; Wu, L.; Li, X. IL-17 Receptor Signaling and T Helper 17-Mediated Autoimmune Demyelinating Disease. Trends Immunol. 2011, 32, 232–239. [Google Scholar] [CrossRef]
- Hamilton, J.A. GM-CSF in Inflammation. J. Exp. Med. 2020, 217, e20190945. [Google Scholar] [CrossRef]
- Qie, S.; Ran, Y.; Lu, X.; Su, W.; Li, W.; Xi, J.; Gong, W.; Liu, Z. Candesartan Modulates Microglia Activation and Polarization via NF-ΚB Signaling Pathway. Int. J. Immunopathol. Pharmacol. 2020, 34. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C. NF-ΚB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.M.; Richman, A.R.; Boyd, J.R.; Sabikunnahar, B.; Lahue, K.G.; Montgomery, T.L.; Caldwell, S.; Varnum, S.; Frietze, S.; Krementsov, D.N. P38 MAP Kinase Signaling in Microglia Plays a Sex-Specific Protective Role in CNS Autoimmunity and Regulates Microglial Transcriptional States. Front. Immunol. 2021, 12, 4122. [Google Scholar] [CrossRef] [PubMed]
- Murphy, Á.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H.G. Infiltration of Th1 and Th17 Cells and Activation of Microglia in the CNS during the Course of Experimental Autoimmune Encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Vogel, D.Y.S.; Vereyken, E.J.F.; Glim, J.E.; Heijnen, P.D.A.M.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in Inflammatory Multiple Sclerosis Lesions Have an Intermediate Activation Status. J. Neuroinflamm. 2013, 10, 809. [Google Scholar] [CrossRef] [PubMed]
- Hiremath, M.M.; Chen, V.S.; Suzuki, K.; Ting, J.P.Y.; Matsushima, G.K. MHC Class II Exacerbates Demyelination in Vivo Independently of T Cells. J. Neuroimmunol. 2008, 203, 23–32. [Google Scholar] [CrossRef]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic Lymphoid-like Structures in Infection, Cancer and Autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef]
- Chen, T.; Lennon, V.A.; Liu, Y.U.; Bosco, D.B.; Li, Y.; Yi, M.H.; Zhu, J.; Wei, S.; Wu, L.J. Astrocyte-Microglia Interaction Drives Evolving Neuromyelitis Optica Lesion. J. Clin. Investig. 2020, 130, 4025–4038. [Google Scholar] [CrossRef]
- Traka, M.; Podojil, J.R.; Mccarthy, D.P.; Miller, S.D.; Popko, B. Oligodendrocyte Death Results in Immune-Mediated CNS Demyelination. Nat. Neurosci. 2015, 19, 65–74. [Google Scholar] [CrossRef]
- Ding, M.; Lang, Y.; Cui, L. AQP4-IgG Positive Paraneoplastic NMOSD: A Case Report and Review. Brain Behav. 2021, 11, e2282. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Guo, Y.; Popescu, B.F.G.; Fujihara, K.; Itoyama, Y.; Misu, T. The Pathology of an Autoimmune Astrocytopathy: Lessons Learned from Neuromyelitis Optica. Brain Pathol. 2014, 24, 83. [Google Scholar] [CrossRef]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal Inflammation Is Widespread and Linked to Cortical Pathology in Multiple Sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef] [PubMed]
- Lennon, P.V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A Serum Autoantibody Marker of Neuromyelitis Optica: Distinction from Multiple Sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Içöz, S.; Tüzün, E.; Kürtüncü, M.; Durmuş, H.; Mutlu, M.; Eraksoy, M.; Akman-Demir, G. Enhanced IL-6 Production in Aquaporin-4 Antibody Positive Neuromyelitis Optica Patients. Int. J. Neurosci. 2010, 120, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Varrin-Doyer, M.; Spencer, C.M.; Schulze-Topphoff, U.; Nelson, P.A.; Stroud, R.M.; Bruce, B.A.; Zamvil, S.S. Aquaporin 4-Specific T Cells in Neuromyelitis Optica Exhibit a Th17 Bias and Recognize Clostridium ABC Transporter. Ann. Neurol. 2012, 72, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Kitic, M.; Hochmeister, S.; Wimmer, I.; Bauer, J.; Misu, T.; Mader, S.; Reindl, M.; Fujihara, K.; Lassmann, H.; Bradl, M. Intrastriatal Injection of Interleukin-1 Beta Triggers the Formation of Neuromyelitis Optica-like Lesions in NMO-IgG Seropositive Rats. Acta Neuropathol. Commun. 2013, 1, 5. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-Mediated Neuroinflammation in Neurodegenerative Diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a Keystone Cytokine in Health and Disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Hsu, M.P.; Frausto, R.; Rose-John, S.; Campbell, I.L. Analysis of IL-6/Gp130 Family Receptor Expression Reveals That in Contrast to Astroglia, Microglia Lack the Oncostatin M Receptor and Functional Responses to Oncostatin M. Glia 2015, 63, 132–141. [Google Scholar] [CrossRef]
- West, P.K.; Viengkhou, B.; Campbell, I.L.; Hofer, M.J. Microglia Responses to Interleukin-6 and Type I Interferons in Neuroinflammatory Disease. Glia 2019, 67, 1821–1841. [Google Scholar] [CrossRef]
- Howe, C.L.; Kaptzan, T.; Magaña, S.M.; Ayers-Ringler, J.R.; Lafrance-Corey, R.G.; Lucchinetti, C.F. Neuromyelitis Optica IgG Stimulates an Immunological Response in Rat Astrocyte Cultures. Glia 2014, 62, 692–708. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Nakashima, I.; Kanbayashi, T.; Konno, M.; Takahashi, T.; Fujihara, K.; Misu, T.; Takeda, A.; Shiga, Y.; Ogawa, H.; et al. Narcolepsy as an Initial Manifestation of Neuromyelitis Optica with Anti-Aquaporin-4 Antibody. J. Neurol. 2009, 256, 287–288. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Weinshenker, B.G.; Lucchinetti, C.F.; Wingerchuk, D.M.; Corboy, J.R.; Lennon, V.A. Neuromyelitis Optica Brain Lesions Localized at Sites of High Aquaporin 4 Expression. Arch. Neurol. 2006, 63, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Roemer, S.F.; Parisi, J.E.; Lennon, V.A.; Benarroch, E.E.; Lassmann, H.; Bruck, W.; Mandler, R.N.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; et al. Pattern-Specific Loss of Aquaporin-4 Immunoreactivity Distinguishes Neuromyelitis Optica from Multiple Sclerosis. Brain 2007, 130, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Weigand, S.D.; Popescu, B.F.; Lennon, V.A.; Parisi, J.E.; Pittock, S.J.; Parks, N.E.; Clardy, S.L.; Howe, C.L.; Lucchinetti, C.F. Pathogenic Implications of Cerebrospinal Fluid Barrier Pathology in Neuromyelitis Optica. Acta Neuropathol. 2017, 133, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Bosco, D.B.; Ying, Y.; Tian, D.S.; Wu, L.J. The Emerging Role of Microglia in Neuromyelitis Optica. Front. Immunol. 2021, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Verkman, A.S. Longitudinally Extensive NMO Spinal Cord Pathology Produced by Passive Transfer of NMO-IgG in Mice Lacking Complement Inhibitor CD59. J. Autoimmun. 2014, 53, 67–77. [Google Scholar] [CrossRef]
- Zhang, Y.; Bao, Y.; Qiu, W.; Peng, L.; Fang, L.; Xu, Y.; Yang, H. Structural and Visual Functional Deficits in a Rat Model of Neuromyelitis Optica Spectrum Disorders Related Optic Neuritis. Exp. Eye Res. 2018, 175, 124–132. [Google Scholar] [CrossRef]
- Duan, T.; Smith, A.J.; Verkman, A.S. Complement-Independent Bystander Injury in AQP4-IgG Seropositive Neuromyelitis Optica Produced by Antibody-Dependent Cellular Cytotoxicity. Acta Neuropathol. Commun. 2019, 7, 112. [Google Scholar] [CrossRef]
- Yick, L.W.; Ma, O.K.F.; Ng, R.C.L.; Kwan, J.S.C.; Chan, K.H. Aquaporin-4 Autoantibodies From Neuromyelitis Optica Spectrum Disorder Patients Induce Complement-Independent Immunopathologies in Mice. Front. Immunol. 2018, 9, 1438. [Google Scholar] [CrossRef]
- Hardy, T.A.; Reddel, S.W.; Barnett, M.H.; Palace, J.; Lucchinetti, C.F.; Weinshenker, B.G. Atypical Inflammatory Demyelinating Syndromes of the CNS. Lancet Neurol 2016, 15, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.; Hecker, C.; Martin, E.; Langui, D.; Gliem, M.; Stankoff, B.; Lubetzki, C.; Gruchot, J.; Göttle, P.; Issberner, A.; et al. Increased Remyelination and Proregenerative Microglia Under Siponimod Therapy in Mechanistic Models. Neurol. (R) Neuroimmunol. Neuroinflamm. 2022, 9, e1161. [Google Scholar] [CrossRef]
- Liddelow, S.A. Development of the Choroid Plexus and Blood-CSF Barrier. Front. Neurosci. 2015, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.; Dziegielewska, K.M.; Fame, R.M.; Lehtinen, M.K.; Liddelow, S.A. The Choroid Plexus: A Missing Link in Our Understanding of Brain Development and Function. Physiol. Rev. 2023, 103, 919–956. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.Y.; Li, J.; Wang, K.F.; Xia, W.W.; Zhu, Z.Q.; Wang, C.R.; Li, X.F.; Liu, H.Y. Blood-Spinal Cord Barrier in Spinal Cord Injury: A Review. J. Neurotrauma 2021, 38, 1203–1224. [Google Scholar] [CrossRef]
- Díaz-Coránguez, M.; Ramos, C.; Antonetti, D.A. The Inner Blood-Retinal Barrier: Cellular Basis and Development. Vis. Res. 2017, 139, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S. Mechanisms of Microbial Traversal of the Blood-Brain Barrier. Nat. Rev. Microbiol. 2008, 6, 625–634. [Google Scholar] [CrossRef]
- Woodham, A.W.; Skeate, J.G.; Sanna, A.M.; Taylor, J.R.; da Silva, D.M.; Cannon, P.M.; Martin Kast, W. Human Immunodeficiency Virus Immune Cell Receptors, Coreceptors, and Cofactors: Implications for Prevention and Treatment. AIDS Patient Care STDS 2016, 30, 291–306. [Google Scholar] [CrossRef]
- Krishnan, S.; Fernandez, G.E.; Sacks, D.B.; Prasadarao, N.V. IQGAP1 Mediates the Disruption of Adherens Junctions to Promote Escherichia Coli K1 Invasion of Brain Endothelial Cells. Cell Microbiol. 2012, 14, 1415–1433. [Google Scholar] [CrossRef]
- Diesselberg, C.; Ribes, S.; Seele, J.; Kaufmann, A.; Redlich, S.; Bunkowski, S.; Hanisch, U.K.; Michel, U.; Nau, R.; Schütze, S. Activin A Increases Phagocytosis of Escherichia Coli K1 by Primary Murine Microglial Cells Activated by Toll-like Receptor Agonists. J. Neuroinflamm. 2018, 15, 175. [Google Scholar] [CrossRef]
- Huang, S.H.; Wass, C.; Fu, Q.; Prasadarao, N.V.; Stins, M.; Kim, K.S. Escherichia Coli Invasion of Brain Microvascular Endothelial Cells in Vitro and in Vivo: Molecular Cloning and Characterization of Invasion Gene Ibe10. Infect. Immun. 1995, 63, 4470–4475. [Google Scholar] [CrossRef]
- Nizet, V.; Kim, K.S.; Stins, M.; Jonas, M.; Chi, E.Y.; Nguyen, D.; Rubens, C.E. Invasion of Brain Microvascular Endothelial Cells by Group B Streptococci. Infect. Immun. 1997, 65, 5074–5081. [Google Scholar] [CrossRef] [PubMed]
- Stoner, T.D.; Weston, T.A.; Trejo, J.; Doran, K.S. Group B Streptococcal Infection and Activation of Human Astrocytes. PLoS ONE 2015, 10, e0128431. [Google Scholar] [CrossRef] [PubMed]
- Edwards, V.E.; Sutherland, J.M.; Tyrer, J.H. Cryptococcosis of the Central Nervous System: Epidemiological, Clinical, and Therapeutic Features. J. Neurol. Neurosurg. Psychiatry 1970, 33, 415. [Google Scholar] [CrossRef]
- Campbell, G.D. Primary Pulmonary Cryptococcosis. Am. Rev. Respir. Dis. 1966, 94, 236–243. [Google Scholar] [CrossRef]
- Guess, T.E.; Rosen, J.A.; McClelland, E.E. An Overview of Sex Bias in C. Neoformans Infections. J. Fungi 2018, 4, 49. [Google Scholar] [CrossRef]
- Lee, Y.C.; Wang, J.T.; Sun, H.Y.; Chen, Y.C. Comparisons of Clinical Features and Mortality of Cryptococcal Meningitis between Patients with and without Human Immunodeficiency Virus Infection. J. Microbiol. Immunol. Infect. 2011, 44, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Guess, T.E.; Rosen, J.; Castro-Lopez, N.; Wormley, F.L.; McClelland, E.E. An Inherent T Cell Deficit in Healthy Males to C. Neoformans Infection May Begin to Explain the Sex Susceptibility in Incidence of Cryptococcosis. Biol. Sex Differ. 2019, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Springer, D.J.; Saini, D.; Byrnes, E.J.; Heitman, J.; Frothingham, R. Development of an Aerosol Model of Cryptococcus Reveals Humidity as an Important Factor Affecting the Viability of Cryptococcus during Aerosolization. PLoS ONE 2013, 8, e69804. [Google Scholar] [CrossRef]
- Mirza, S.A.; Phelan, M.; Rimland, D.; Graviss, E.; Hamill, R.; Brandt, M.E.; Gardner, T.; Sattah, M.; de Leon, G.P.; Baughman, W.; et al. The Changing Epidemiology of Cryptococcosis: An Update from Population-Based Active Surveillance in 2 Large Metropolitan Areas, 1992-2000. Clin. Infect. Dis. 2003, 36, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D.L.; Khine, H.; Abadi, J.; Lindenberg, D.J.; Pirofski, L.A.; Niang, R.; Casadevall, A. Serologic Evidence for Cryptococcus Neoformans Infection in Early Childhood. Pediatrics 2001, 107, e66. [Google Scholar] [CrossRef] [PubMed]
- Al-Odaini, N.; Li, X.Y.; Li, B.K.; Chen, X.C.; Huang, C.Y.; Lv, C.Y.; Pan, K.S.; Zheng, D.Y.; Zheng, Y.Q.; Liao, W.Q.; et al. In Vitro Antifungal Susceptibility Profiles of Cryptococcus Neoformans Var. Grubii and Cryptococcus Gattii Clinical Isolates in Guangxi, Southern China. Front. Microbiol. 2021, 12, 708280. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Lien, C.Y.; Lee, J.J.; Hsiao, W.C.; Huang, C.R.; Tsai, N.W.; Chang, C.C.; Lu, C.H.; Chang, W.N. The Clinical Characteristics and Therapeutic Outcomes of Cryptococcal Meningitis in Elderly Patients: A Hospital-Based Study. BMC Geriatr. 2019, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.H.; Yip, C.W.; Jiang, Y.K.; Zhou, L.H.; Que, C.X.; Luo, Y.; Wang, X.; Zhao, H.Z.; Zhu, L.P. Clinical Predictors Impacting Cryptococcal Dissemination and Poor Outcome in Patients With Cirrhosis. Open Forum Infect. Dis. 2021, 8, ofab296. [Google Scholar] [CrossRef] [PubMed]
- de Pauw, B.; Walsh, T.J.; Donnelly, J.P.; Stevens, D.A.; Edwards, J.E.; Calandra, T.; Pappas, P.G.; Maertens, J.; Lortholary, O.; Kauffman, C.A.; et al. Revised Definitions of Invasive Fungal Disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin. Infect. Dis. 2008, 46, 1813–1821. [Google Scholar] [CrossRef]
- Nsenga, L.; Kajjimu, J.; Olum, R.; Ninsiima, S.; Kyazze, A.P.; Ssekamatte, P.; Kibirige, D.; Baluku, J.B.; Andia-Biraro, I.; Bongomin, F. Cryptococcosis Complicating Diabetes Mellitus: A Scoping Review. Ther. Adv. Infect. Dis. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.M.P.; Sereti, I. Immune Restoration after Antiretroviral Therapy: The Pitfalls of Hasty or Incomplete Repairs. Immunol. Rev. 2013, 254, 343. [Google Scholar] [CrossRef]
- Sloan, D.J.; Parris, V. Cryptococcal Meningitis: Epidemiology and Therapeutic Options. Clin. Epidemiol. 2014, 6, 169–182. [Google Scholar] [CrossRef]
- Yoon, H.A.; Felsen, U.; Wang, T.; Pirofski, L.A. Cryptococcus Neoformans Infection in Human Immunodeficiency Virus (HIV)-Infected and HIV-Uninfected Patients at an Inner-City Tertiary Care Hospital in the Bronx. Med. Mycol. 2020, 58, 434–443. [Google Scholar] [CrossRef]
- Tseng, H.K.; Huang, T.Y.; Wu, A.Y.J.; Chen, H.H.; Liu, C.P.; Jong, A. How Cryptococcus Interacts with the Blood-Brain Barrier. Future Microbiol. 2015, 10, 1669–1682. [Google Scholar] [CrossRef]
- Perfect, J.R.; Dismukes, W.E.; Dromer, F.; Goldman, D.L.; Graybill, J.R.; Hamill, R.J.; Harrison, T.S.; Larsen, R.A.; Lortholary, O.; Nguyen, M.H.; et al. Clinical Practice Guidelines for the Management of Cryptococcal Disease: 2010 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2010, 50, 291–322. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Casadevall, A.; Dickson, D.W. Immunohistochemical Localization of Capsular Polysaccharide Antigen in the Central Nervous System Cells in Cryptococcal Meningoencephalitis. Am. J. Pathol. 1996, 148, 1267. [Google Scholar] [PubMed]
- Tucker, J.S.; Guess, T.E.; McClelland, E.E. The Role of Testosterone and Gibberellic Acid in the Melanization of Cryptococcus Neoformans. Front. Microbiol. 2020, 11, 1921. [Google Scholar] [CrossRef] [PubMed]
- Rifkind, D.; Frey, J.A. Sex Difference in Antibody Response of CFW Mice to Candida Albicans. Infect. Immun. 1972, 5, 695–698. [Google Scholar] [CrossRef]
- Arroyo-Mendoza, M.; Peraza, K.; Olson, J.; Adler-Moore, J.P.; Buckley, N.E. Effect of Testosterone and Estrogen Supplementation on the Resistance to Systemic Candida Albicans Infection in Mice. Heliyon 2020, 6, E04437. [Google Scholar] [CrossRef] [PubMed]
- Tiuria, R.; Horii, Y.; Tateyama, S.; Tsuchiya, K.; Nawa, Y. The Indian Soft-Furred Rat, Millardia Meltada, a New Host for Nippostrongylus Brasiliensis, Showing Androgen-Dependent Sex Difference in Intestinal Mucosal Defence. Int. J. Parasitol. 1994, 24, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Rettew, J.A.; Huet-Hudson, Y.M.; Marriott, I. Testosterone Reduces Macrophage Expression in the Mouse of Toll-like Receptor 4, a Trigger for Inflammation and Innate Immunity. Biol. Reprod. 2008, 78, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Butts, A.; Martin, J.A.; DiDone, L.; Bradley, E.K.; Mutz, M.; Krysan, D.J. Structure-Activity Relationships for the Antifungal Activity of Selective Estrogen Receptor Antagonists Related to Tamoxifen. PLoS ONE 2015, 10, e0125927. [Google Scholar] [CrossRef]
- McClelland, E.E.; Hobbs, L.M.; Rivera, J.; Casadevall, A.; Potts, W.K.; Smith, J.M.; Ory, J.J. The Role of Host Gender in the Pathogenesis of Cryptococcus Neoformans Infections. PLoS ONE 2013, 8, 63632. [Google Scholar] [CrossRef]
- Zaragoza, O.; Alvarez, M.; Telzak, A.; Rivera, J.; Casadevall, A. The Relative Susceptibility of Mouse Strains to Pulmonary Cryptococcus Neoformans Infection Is Associated with Pleiotropic Differences in the Immune Response. Infect. Immun. 2007, 75, 2729. [Google Scholar] [CrossRef]
- Merkel, S.M.; Alexander, S.; Zufall, E.; Oliver, J.D.; Huet-Hudson, Y.M. Essential Role for Estrogen in Protection against Vibrio Vulnificus-Induced Endotoxic Shock. Infect. Immun. 2001, 69, 6119–6122. [Google Scholar] [CrossRef] [PubMed]
- Saia, R.S.; Garcia, F.M.; Cárnio, E.C. Estradiol Protects Female Rats against Sepsis Induced by Enterococcus Faecalis Improving Leukocyte Bactericidal Activity. Steroids 2015, 102, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, M.; Ge, Z.; García, A.; Rogers, A.B.; Muthupalani, S.; Taylor, N.S.; Xu, S.; Watanabe, K.; Feng, Y.; Marini, R.P.; et al. 17 β-Estradiol Suppresses Helicobacter Pylori-Induced Gastric Pathology in Male Hypergastrinemic INS-GAS Mice. Carcinogenesis 2011, 32, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Park, M.; Cho, J.Y.; Ahn, S.J.; Yoon, C.; Kim, S.G.; Cho, S.J. Tumorigenic Mechanisms of Estrogen and Helicobacter Pylori Cytotoxin-Associated Gene A in Estrogen Receptor α-Positive Diffuse-Type Gastric Adenocarcinoma. Gastric Cancer 2022, 25, 678–696. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Honstettre, A.; Lepidi, H.; Capo, C.; Bayard, F.; Raoult, D.; Mege, J.L. Effect of Sex on Coxiella Burnetii Infection: Protective Role of 17beta-Estradiol. J. Infect. Dis. 2004, 189, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.; Melenotte, C.; Lopez, A.; Desnues, B.; Raoult, D.; Leone, M.; Mezouar, S.; Mege, J.L. Impact of Sex Hormones on Macrophage Responses to Coxiella Burnetii. Front. Immunol. 2021, 12, 5510. [Google Scholar] [CrossRef]
- Lee, S.C.; Kress, Y.; Zhao, M.L.; Dickson, D.W.; Casadevall, A. Cryptococcus Neoformans Survive and Replicate in Human Microglia. Laboratory Investigation. J. Tech. Methods Pathol. 1995, 73, 871–879. [Google Scholar]
- Biondo, C.; Midiri, A.; Messina, L.; Tomasello, F.; Garufi, G.; Catania, M.R.; Bombaci, M.; Beninati, C.; Teti, G.; Mancuso, G. MyD88 and TLR2, but Not TLR4, Are Required for Host Defense against Cryptococcus Neoformans. Eur. J. Immunol. 2005, 35, 870–878. [Google Scholar] [CrossRef]
- Yauch, L.E.; Mansour, M.K.; Shoham, S.; Rottman, J.B.; Levitz, S.M. Involvement of CD14, Toll-like Receptors 2 and 4, and MyD88 in the Host Response to the Fungal Pathogen Cryptococcus Neoformans in Vivo. Infect. Immun. 2004, 72, 5373–5382. [Google Scholar] [CrossRef]
- da Silva-Junior, E.B.; Firmino-Cruz, L.; Guimarães-de-Oliveira, J.C.; De-Medeiros, J.V.R.; de Oliveira Nascimento, D.; Freire-de-Lima, M.; de Brito-Gitirana, L.; Morrot, A.; Previato, J.O.; Mendonça-Previato, L.; et al. The Role of Toll-like Receptor 9 in a Murine Model of Cryptococcus Gattii Infection. Sci. Rep. 2021, 11, 1407. [Google Scholar] [CrossRef]
- Giles, S.S.; Dagenais, T.R.T.; Botts, M.R.; Keller, N.P.; Hull, C.M. Elucidating the Pathogenesis of Spores from the Human Fungal Pathogen Cryptococcus Neoformans. Infect. Immun. 2009, 77, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Kozel, T.R.; Highison, B.; Stratton, C.J. Localization on Encapsulated Cryptococcus Neoformans of Serum Components Opsonic for Phagocytosis by Macrophages and Neutrophils. Infect. Immun. 1984, 43, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.C.; Ley, S.C. Mitogen-Activated Protein Kinases in Innate Immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Neal, L.M.; Xing, E.; Xu, J.; Kolbe, J.L.; Osterholzer, J.J.; Segal, B.M.; Williamson, P.R.; Olszewski, M.A. CD4+ T Cells Orchestrate Lethal Immune Pathology despite Fungal Clearance during Cryptococcus Neoformans Meningoencephalitis. mBio 2017, 8. [Google Scholar] [CrossRef]
- Weinstein, J.R.; Quan, Y.; Hanson, J.F.; Colonna, L.; Iorga, M.; Honda, S.; Shibuya, K.; Shibuya, A.; Elkon, K.B.; Möller, T. IgM-Dependent Phagocytosis in Microglia Is Mediated by Complement Receptor 3, Not Fcα/μ Receptor. J. Immunol. 2015, 195, 5309. [Google Scholar] [CrossRef]
- Zhou, Q.; Gault, R.A.; Kozel, T.R.; Murphy, W.J. Protection from Direct Cerebral Cryptococcus Infection by Interferon-Gamma-Dependent Activation of Microglial Cells. J. Immunol. 2007, 178, 5753–5761. [Google Scholar] [CrossRef]
- Kleinschek, M.A.; Muller, U.; Brodie, S.J.; Stenzel, W.; Kohler, G.; Blumenschein, W.M.; Straubinger, R.K.; McClanahan, T.; Kastelein, R.A.; Alber, G. IL-23 Enhances the Inflammatory Cell Response in Cryptococcus Neoformans Infection and Induces a Cytokine Pattern Distinct from IL-12. J. Immunol. 2006, 176, 1098–1106. [Google Scholar] [CrossRef]
- Chen, H.; Jin, Y.; Chen, H.; Liao, I.; Yan, W.; Chen, J. MicroRNA-Mediated Inflammatory Responses Induced by Cryptococcus Neoformans Are Dependent on the NF-ΚB Pathway in Human Monocytes. Int. J. Mol. Med. 2017, 39, 1525–1532. [Google Scholar] [CrossRef]
- Song, X.; Tanaka, S.; Cox, D.; Lee, S.C. Fcgamma Receptor Signaling in Primary Human Microglia: Differential Roles of PI-3K and Ras/ERK MAPK Pathways in Phagocytosis and Chemokine Induction. J. Leukoc. Biol. 2004, 75, 1147–1155. [Google Scholar] [CrossRef]
- Preissler, J.; Grosche, A.; Lede, V.; le Duc, D.; Krügel, K.; Matyash, V.; Szulzewsky, F.; Kallendrusch, S.; Immig, K.; Kettenmann, H.; et al. Altered Microglial Phagocytosis in GPR34-Deficient Mice. Glia 2015, 63, 206–215. [Google Scholar] [CrossRef]
- Buchanan, K.L.; Doyle, H.A. Requirement for CD4(+) T Lymphocytes in Host Resistance against Cryptococcus Neoformans in the Central Nervous System of Immunized Mice. Infect Immun 2000, 68, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D.; Song, X.; Kitai, R.; Casadevall, A.; Zhao, M.L.; Lee, S.C. Cryptococcus Neoformans Induces Macrophage Inflammatory Protein 1alpha (MIP-1alpha) and MIP-1beta in Human Microglia: Role of Specific Antibody and Soluble Capsular Polysaccharide. Infect. Immun. 2001, 69, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, K.; Crowe, J.; Haas, A.; Smith, J. Resistance to Cryptococcus Neoformans Infection in the Absence of CD4 + T Cells. Med. Mycol. 2004, 42, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef]
- Shimoda, M.; Jones, V.C.; Kobayashi, M.; Suzuki, F. Microglial Cells from Psychologically Stressed Mice as an Accelerator of Cerebral Cryptococcosis. Immunol. Cell Biol. 2006, 84, 551–556. [Google Scholar] [CrossRef]
- Hole, C.; Wormley, F.L. Innate Host Defenses against Cryptococcus Neoformans. J. Microbiol. 2016, 54, 202–211. [Google Scholar] [CrossRef]
- García-Rodas, R.; Zaragoza, O. Catch Me If You Can: Phagocytosis and Killing Avoidance by Cryptococcus Neoformans. FEMS Immunol. Med. Microbiol. 2012, 64, 147–161. [Google Scholar] [CrossRef]
- Furman, D.; Hejblum, B.P.; Simon, N.; Jojic, V.; Dekker, C.L.; Thiebaut, R.; Tibshirani, R.J.; Davis, M.M. Systems Analysis of Sex Differences Reveals an Immunosuppressive Role for Testosterone in the Response to Influenza Vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 869–874. [Google Scholar] [CrossRef]
- Sturdevant, G.L.; Caldwell, H.D. Innate Immunity Is Sufficient for the Clearance of Chlamydia Trachomatis from the Female Mouse Genital Tract. Pathog. Dis. 2014, 72, 70. [Google Scholar] [CrossRef]
- Kadioglu, A.; Cuppone, A.M.; Trappetti, C.; List, T.; Spreafico, A.; Pozzi, G.; Andrew, P.W.; Oggioni, M.R. Sex-Based Differences in Susceptibility to Respiratory and Systemic Pneumococcal Disease in Mice. J. Infect. Dis. 2011, 204, 1971–1979. [Google Scholar] [CrossRef]
- Scriven, J.E.; Graham, L.M.; Schutz, C.; Scriba, T.J.; Wilkinson, K.A.; Wilkinson, R.J.; Boulware, D.R.; Urban, B.C.; Meintjes, G.; Lalloo, D.G. The CSF Immune Response in HIV-1-Associated Cryptococcal Meningitis: Macrophage Activation, Correlates of Disease Severity, and Effect of Antiretroviral Therapy. J. Acquir. Immune Defic. Syndr. 2017, 75, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Portnow, J.; Ahluwalia, M.; Baehring, J.; Brem, H.; Brem, S.; Butowski, N.; Campian, J.L.; Clark, S.W.; Fabiano, A.J.; et al. Central Nervous System Cancers, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 1537–1570. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Wesseling, P. Histologic Classification of Gliomas. Handb. Clin. Neurol 2016, 134, 71–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, R.; Yang, S.; Zhang, J.; Wang, M.; Zhong, D.; Zhang, J.; Han, X. Combining Radiology and Pathology for Automatic Glioma Classification. Front. Bioeng. Biotechnol. 2022, 10, 356. [Google Scholar] [CrossRef]
- Olar, A.; Aldape, K.D. Using the Molecular Classification of Glioblastoma to Inform Personalized Treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef]
- Jiang, H.; Cui, Y.; Wang, J.; Lin, S. Impact of Epidemiological Characteristics of Supratentorial Gliomas in Adults Brought about by the 2016 World Health Organization Classification of Tumors of the Central Nervous System. Oncotarget 2017, 8, 20354. [Google Scholar] [CrossRef]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of Origin for Malignant Gliomas and Its Implication in Therapeutic Development. Cold Spring Harb. Perspect. Biol. 2015, 7, 1–13. [Google Scholar] [CrossRef]
- Kallenberg, K.; Goldmann, T.; Menke, J.; Strik, H.; Bock, H.C.; Stockhammer, F.; Buhk, J.H.; Frahm, J.; Dechent, P.; Knauth, M. Glioma Infiltration of the Corpus Callosum: Early Signs Detected by DTI. J. Neurooncol. 2013, 112, 217. [Google Scholar] [CrossRef]
- Ge, W.P.; Miyawaki, A.; Gage, F.H.; Jan, Y.N.; Jan, L.Y. Local Generation of Glia Is a Major Astrocyte Source in Postnatal Cortex. Nature 2012, 484, 376–380. [Google Scholar] [CrossRef]
- Sayegh, E.T.; Oh, T.; Fakurnejad, S.; Oyon, D.E.; Bloch, O.; Parsa, A.T. Principles of Surgery for Malignant Astrocytomas. Semin. Oncol. 2014, 41, 523–531. [Google Scholar] [CrossRef]
- Krawczyk, M.C.; Haney, J.R.; Pan, L.; Caneda, C.; Khankan, R.R.; Reyes, S.D.; Chang, J.W.; Morselli, M.; Vinters, H.V.; Wang, A.C.; et al. Human Astrocytes Exhibit Tumor Microenvironment-, Age-, and Sex-Related Transcriptomic Signatures. J. Neurosci. 2022, 42, 1587–1603. [Google Scholar] [CrossRef] [PubMed]
- Taal, W.; Bromberg, J.E.C.; van den Bent, M.J. Chemotherapy in Glioma. CNS Oncol. 2015, 4, 179. [Google Scholar] [CrossRef] [PubMed]
- de Gooijer, M.C.; Guillén Navarro, M.; Bernards, R.; Wurdinger, T.; van Tellingen, O. An Experimenter’s Guide to Glioblastoma Invasion Pathways. Trends Mol. Med. 2018, 24, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Nobes, C.D.; Hall, A. Rho, Rac, and Cdc42 GTPases Regulate the Assembly of Multimolecular Focal Complexes Associated with Actin Stress Fibers, Lamellipodia, and Filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Karmur, B.S.; Philteos, J.; Abbasian, A.; Zacharia, B.E.; Lipsman, N.; Levin, V.; Grossman, S.; Mansouri, A. Blood-Brain Barrier Disruption in Neuro-Oncology: Strategies, Failures, and Challenges to Overcome. Front. Oncol. 2020, 10, 563840. [Google Scholar] [CrossRef] [PubMed]
- Finneran, M.; Marotta, D.A.; Altenburger, D.; Nardone, E. Long-Term Survival in a Patient with Butterfly Glioblastoma: A Case Report. Cureus 2020, 12. [Google Scholar] [CrossRef]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. Glioblastoma 2017, 143–153. [Google Scholar] [CrossRef]
- Stabellini, N.; Krebs, H.; Patil, N.; Waite, K.; Barnholtz-Sloan, J.S. Sex Differences in Time to Treat and Outcomes for Gliomas. Front. Oncol. 2021, 11, 1. [Google Scholar] [CrossRef]
- Whitmire, P.; Rickertsen, C.R.; Hawkins-Daarud, A.; Carrasco, E.; Lorence, J.; de Leon, G.; Curtin, L.; Bayless, S.; Clark-Swanson, K.; Peeri, N.C.; et al. Sex-Specific Impact of Patterns of Imageable Tumor Growth on Survival of Primary Glioblastoma Patients. BMC Cancer 2020, 20, 447. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef]
- Pan, I.W.; Ferguson, S.D.; Lam, S. Patient and Treatment Factors Associated with Survival among Adult Glioblastoma Patients: A USA Population-Based Study from 2000-2010. J. Clin. Neurosci. 2015, 22, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, X.; You, Y.; Wang, X.; Liu, Y.; Hu, Q.; Yan, W. Comprehensive Portrait of Recurrent Glioblastoma Multiforme in Molecular and Clinical Characteristics. Oncotarget 2015, 6, 30968–30974. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Warrington, N.M.; Rubin, J.B. Why Does Jack, and Not Jill, Break His Crown? Sex Disparity in Brain Tumors. Biol. Sex Differ. 2012, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- McCrea, H.J.; Bander, E.D.; Venn, R.A.; Reiner, A.S.; Iorgulescu, J.B.; Puchi, L.A.; Schaefer, P.M.; Cederquist, G.; Greenfield, J.P. Sex, Age, Anatomic Location, and Extent of Resection Influence Outcomes in Children With High-Grade Glioma. Neurosurgery 2015, 77, 443–452. [Google Scholar] [CrossRef]
- Franceschi, E.; Tosoni, A.; Minichillo, S.; Depenni, R.; Paccapelo, A.; Bartolini, S.; Michiara, M.; Pavesi, G.; Urbini, B.; Crisi, G.; et al. The Prognostic Roles of Gender and O6-Methylguanine-DNA Methyltransferase Methylation Status in Glioblastoma Patients: The Female Power. World Neurosurg. 2018, 112, e342–e347. [Google Scholar] [CrossRef]
- Yang, W.; Warrington, N.M.; Taylor, S.J.; Whitmire, P.; Carrasco, E.; Singleton, K.W.; Wu, N.; Lathia, J.D.; Berens, M.E.; Kim, A.H.; et al. Sex Differences in GBM Revealed by Analysis of Patient Imaging, Transcriptome, and Survival Data. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Rubin, J.B.; Lathia, J.D.; Berens, M.E.; Barnholtz-Sloan, J.S. Females Have the Survival Advantage in Glioblastoma. Neuro Oncol. 2018, 20, 576. [Google Scholar] [CrossRef]
- Tian, M.; Ma, W.; Chen, Y.; Yu, Y.; Zhu, D.; Shi, J.; Zhang, Y. Impact of Gender on the Survival of Patients with Glioblastoma. Biosci. Rep. 2018, 38, BSR20180752. [Google Scholar] [CrossRef]
- Hopewell, J.W. The Effects of Castration on the Induction of Experimental Gliomas in Male Rats. Br. J. Cancer 1970, 24, 187. [Google Scholar] [CrossRef]
- Yu, X.; Jiang, Y.; Wei, W.; Cong, P.; Ding, Y.; Xiang, L.; Wu, K. Androgen Receptor Signaling Regulates Growth of Glioblastoma Multiforme in Men. Tumour Biol. 2015, 36, 967–972. [Google Scholar] [CrossRef]
- Bao, D.; Cheng, C.; Lan, X.; Xing, R.; Chen, Z.; Zhao, H.; Sun, J.; Wang, Y.; Niu, C.; Zhang, B.; et al. Regulation of P53wt Glioma Cell Proliferation by Androgen Receptor-Mediated Inhibition of Small VCP/P97-Interacting Protein Expression. Oncotarget 2017, 8, 23142–23154. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, N.; Canello, T.; Ovadia, H.; Charbit, H.; Zelikovitch, B.; Mordechai, A.; Fellig, Y.; Rabani, S.; Shahar, T.; Lossos, A.; et al. Androgen Receptor: A Potential Therapeutic Target for Glioblastoma. Oncotarget 2018, 9, 19980. [Google Scholar] [CrossRef] [PubMed]
- Atallah, A.; Mhaouty-Kodja, S.; Grange-Messent, V. Chronic Depletion of Gonadal Testosterone Leads to Blood-Brain Barrier Dysfunction and Inflammation in Male Mice. J. Cereb. Blood Flow Metab. 2017, 37, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lozano, D.C.; Piña-Medina, A.G.; Hansberg-Pastor, V.; Bello-Alvarez, C.; Camacho-Arroyo, I. Testosterone Promotes Glioblastoma Cell Proliferation, Migration, and Invasion Through Androgen Receptor Activation. Front. Endocrinol. 2019, 10, 16. [Google Scholar] [CrossRef]
- Barone, T.A.; Gorski, J.W.; Greenberg, S.J.; Plunkett, R.J. Estrogen Increases Survival in an Orthotopic Model of Glioblastoma. J. Neurooncol. 2009, 95, 37–48. [Google Scholar] [CrossRef]
- McKinley, B.P.; Michalek, A.M.; Fenstermarker, R.A.; Plunkett, R.J. The Impact of Age and Sex on the Incidence of Glial Tumors in New York State from 1976 to 1995. J. Neurosurg. 2000, 93, 932–939. [Google Scholar] [CrossRef]
- Kabat, G.C.; Park, Y.; Hollenbeck, A.R.; Schatzkin, A.; Rohan, T.E. Reproductive Factors and Exogenous Hormone Use and Risk of Adult Glioma in Women in the NIH-AARP Diet and Health Study. Int. J. Cancer 2011, 128, 944–950. [Google Scholar] [CrossRef]
- Broestl, L.; Warrington, N.M.; Grandison, L.; Abou-Antoun, T.; Tung, O.; Shenoy, S.; Tallman, M.M.; Rhee, G.; Yang, W.; Sponagel, J.; et al. Gonadal Sex Patterns P21-Induced Cellular Senescence in Mouse and Human Glioblastoma. Commun. Biol. 2022, 5, 781. [Google Scholar] [CrossRef]
- Locascio, C.; Kupp, R.; Singh, S.; Szeto, E.; Loo, V.; Rauf, F.; Derogatis, A.; Labaer, J.; Sanai, N.; Mehta, S.; et al. CSIG-13. Androgen receptor is involved in glioblastoma and presents a potential therapeutic target. Neuro Oncol. 2016, 18, vi43. [Google Scholar] [CrossRef]
- Roved, J.; Westerdahl, H.; Hasselquist, D. Sex Differences in Immune Responses: Hormonal Effects, Antagonistic Selection, and Evolutionary Consequences. Horm. Behav. 2017, 88, 95–105. [Google Scholar] [CrossRef]
- Lin, G.L.; Nagaraja, S.; Filbin, M.G.; Suvà, M.L.; Vogel, H.; Monje, M. Non-Inflammatory Tumor Microenvironment of Diffuse Intrinsic Pontine Glioma. Acta Neuropathol. Commun. 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Ley, K. M1 and M2 Macrophages: The Chicken and the Egg of Immunity. J. Innate Immun. 2014, 6, 716–726. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage Plasticity and Polarization in Tissue Repair and Remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-Associated Microglia/Macrophages Predict Poor Prognosis in High-Grade Gliomas and Correlate with an Aggressive Tumour Subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma Intrinsic Gene Expression Subtype Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42. [Google Scholar] [CrossRef]
- Sun, T.; Plutynski, A.; Ward, S.; Rubin, J.B. An Integrative View on Sex Differences in Brain Tumors. Cell Mol. Life Sci. 2015, 72, 3323–3342. [Google Scholar] [CrossRef]
- Sarkar, S.; Döring, A.; Zemp, F.J.; Silva, C.; Lun, X.; Wang, X.; Kelly, J.; Hader, W.; Hamilton, M.; Mercier, P.; et al. Therapeutic Activation of Macrophages and Microglia to Suppress Brain Tumor-Initiating Cells. Nat. Neurosci. 2013, 17, 46–55. [Google Scholar] [CrossRef]
- Müller, A.; Brandenburg, S.; Turkowski, K.; Müller, S.; Vajkoczy, P. Resident Microglia, and Not Peripheral Macrophages, Are the Main Source of Brain Tumor Mononuclear Cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, W.; Yang, P.; Wang, Y.; Li, M.; Zhang, C.; Wang, Z.; Hu, H.; Liu, Y.; Li, Q.; et al. Identification of a 6-Cytokine Prognostic Signature in Patients with Primary Glioblastoma Harboring M2 Microglia/Macrophage Phenotype Relevance. PLoS ONE 2015, 10, e0126022. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Bocian, K.; Dabrowski, M.; Rajan, W.D.; Kloss, M.; Mieczkowski, J.; Kaminska, B. Immune Microenvironment of Experimental Rat C6 Gliomas Resembles Human Glioblastomas. Sci. Rep. 2017, 7, 17556. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.C.; Selwood, D.L.; Tsirka, S.E. Tuftsin Signals through Its Receptor Neuropilin-1 via the Transforming Growth Factor Beta Pathway. J. Neurochem. 2013, 127, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, J.T.; Chen, D.; Choi, M.; Nissen, J.C.; Shroyer, K.R.; Djordevic, S.; Zachary, I.C.; Selwood, D.; Tsirka, S.E. Ablation of Neuropilin 1 from Glioma-Associated Microglia and Macrophages Slows Tumor Progression. Oncotarget 2016, 7, 9801–9814. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, J.T.; Caponegro, M.D.; Chen, D.; Choi, M.K.; Melvin, L.I.; Tsirka, S.E. Deletion of Neuropilin 1 from Microglia or Bone Marrow–Derived Macrophages Slows Glioma Progression. Cancer Res. 2018, 78, 685–694. [Google Scholar] [CrossRef]
- Lisi, L.; Ciotti, G.M.P.; Braun, D.; Kalinin, S.; Currò, D.; dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of INOS, CD163 and ARG-1 Taken as M1 and M2 Markers of Microglial Polarization in Human Glioblastoma and the Surrounding Normal Parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Kuntzel, T.; Bagnard, D. Manipulating Macrophage/Microglia Polarization to Treat Glioblastoma or Multiple Sclerosis. Pharmaceutics 2022, 14, 344. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Model | Species | Evidence Supporting Female M1 Predominance | Reference |
|---|---|---|---|
| Immunohistochemistry for Iba1 | Rat | Increased numbers of activated microglia in P60 females | [115] |
| Whole transcriptome profiling of primary microglial cells | Mouse | Female microglia have a more transcriptionally mature profile, increased expression of inflammatory transcripts in adulthood | [119] |
| Whole transcriptome profiling of primary microglial cells | Mouse | Increase expression of MHC I, complement pathway, and inflammatory genes in aged females | [120] |
| Transwell migration assays | Rat | Increased motility in male microglia | [122] |
| Renal ischemia/reperfusion | Rat | Increased IL-10 production following testosterone treatment | [128] |
| Western blotting | Mouse | Increased expression of inflammation-associated CCR5 following estrogen treatment | [133] |
| RT-PCR | Rat | E2 diminished the inflammatory release of IL-1β following LPS stimulation in male microglia, but enhanced the inflammatory response in female microglia. | [134] |
| RNASeq | Mouse | Microglia from female mice had higher mRNA levels for TNFα, IL-1β, and IL-6 than those from males | [137] |
| Brain stab wound model, MHC II immunostaining | Rat | Decreased microglial reactivity with testosterone treatment following brain injury | [126] |
| IL6 stimulation | Rat | Diminished stress responses with reduced corticosterone and ACTH release in rats with increased testosterone | [138] |
| Whole blood samples | Human | Higher endogenous testosterone associated with lower serum cytokine levels | [139] |
| Various clinical studies of human disease | Human | Suppressed TNFα, IL-6, IL-1β and increased IL-10 with high levels of testosterone | [129,140] |
| Disease | Species | Evidence Supporting Female Predominance | Reference |
|---|---|---|---|
| Multiple Sclerosis | Human | The female to male ratio was reported as 2.35:1. | [151] |
| Multiple Sclerosis | Human | Female incidence of MS in all races is about three times of their male counterparts | [152] |
| Multiple Sclerosis | Human | 3:1 ratio of MS diagnosis between females and males | [169] |
| Multiple Sclerosis | Human | Women with MS had more inflammatory MS lesions than men. | [157] |
| Neuromyelitis Optica Spectrum Disorders | Human | Female patients were more likely to be positive for susceptibility-associated AQP4-abs (92% vs. 55%; p < 0.001) | [168] |
| Neuromyelitis Optica Spectrum Disorders | Human | 4.6:1 ratio between females and males for NMOSD diagnosis; among AQP4-ab seropositive groups the ratio is 12:1 | [170] |
| Neuromyelitis Optica Spectrum Disorders | Human | High AQP4-antibody seropositivity prevalence in females compared to males (9:1) | [171] |
| Neuromyelitis Optica Spectrum Disorders | Human | Seropositivity for AQP4-IgG is 8:1 between females and males, and particularly in people ages 18 years and older. | [172] |
| Neuromyelitis Optica Spectrum Disorders | Human | Reduced early-onset optic neuritis in male patients | [173] |
| Model | Species | Evidence Supporting M1-Associated Pathology | Reference |
|---|---|---|---|
| EAE | Mouse | Microglial transcriptomes show increased neuroinflammatory and immune signaling pathways during EAE progression | [175] |
| EAE | Mouse | Polarization of microglial cells to M2 substantially reduces EAE symptoms and progression | [181,182,183] |
| EAE | Mouse | Ablation of microglia improves EAE symptoms | [184] |
| EAE | Mouse | Increased inflammatory cytokine production and Th17 activation in microglial repopulated mice during EAE | [185] |
| EAE | Mouse | Deletion of M1-associated MAPK pathway proteins in microglia is protective in EAE. | [194] |
| EAE–optic neuritis (EAEON) | Mouse | TNFα was significantly upregulated in the optic nerve of EAE animals with a peak at 35 dpi. | [224] |
| Lysolecithin-mediated demyelination, primary microglial culture | Mouse | Microglial cells switch from M1 to M2 phenotypes at the initiation of remyelination, M2 cells promote oligodendrocyte differentiation. | [179] |
| EAE | Mouse | Infiltrating T cells promote inflammatory microglial activation, which recruit additional peripheral immune cells | [186] |
| Postmortem MS autopsy samples | Human | Increased myelin phagocytosis and T cell activation by microglia in areas of active inflammation | [187] |
| Postmortem MS autopsy samples | Human | Active and chronic active MS lesions showed abundant expression of M1 markers, with M2 markers lacking overall | [196] |
| Injection of patient-derived NMO-IgG | Rat | High levels of IL-1β positive microglia found in active NMO lesions | [210] |
| Primary microglial culture | Rat | NMO-IgG promotes release of M1-polarizing, inflammatory CCL5 and C3 from astrocytes | [212] |
| Infusion of patient-derived serum NMO-IgG or AQP4-IgG | Mouse | Direct interaction between astrocyte and microglial soma following antibody infusion. Increased inflammatory microglial activation and C1q production in NMO lesions, elimination of NMO-IgG motor impairment following microglial ablation | [199] |
| Disease | Species | Evidence Supporting Female Predominance | Reference |
|---|---|---|---|
| Cryptococcus neoformans infection | Human | 2:1 ratio between male and female patients prior to the HIV epidemic | [236] |
| Cryptococcus neoformans infection | Human | 3:1 ratio between male and female HIV negative patients, 8:1 ratio between male and female HIV positive patients | [238] |
| Cryptococcus neoformans infection | Human | 7:3 ratio between male and female patients | [239] |
| Cryptococcus neoformans infection | Human | 97.3% of HIV+ Cn patients were male; 62.7% of HIV- Cn patients were male | [240] |
| Candida albicans infection | Mouse | Testosterone functions in an immunosuppressive manner, infusion of testosterone into female mice increases infections | [256] |
| Nippostrongylus brasiliensis infection | Mouse | Parasite infection commonly recurred in male animals and rarely in females; this recurrence in males was eliminated following orchiectomy | [258] |
| Cryptococcus neoformans infection | In vitro | Synthetic estrogen compounds have potent antifungal activity | [260] |
| Cryptococcus neoformans infection | Human | High levels of testosterone were associated with greater Cn pathology, male-derived strains had shorter doubling times, greater fungal burden and increased splenic cell death rate | [261] |
| Vibrio vulnificus infection | Rat | Gonadectomy in females increases mortality; estrogen treatment decreases mortality in males and females | [263] |
| Enterococcus faecalis infection | Rat | Estradiol treatment reduces bacterial counts | [264] |
| Helicobacter pylori infection | Mouse | Reduced gastric lesions in estradiol treated mice | [265] |
| Coxiella burnetti infection | Mouse | Bacterial load and granuloma numbers were reduced in female mice, estradiol treatment in ovariectomized mice had an analogous effect | [268] |
| Model | Species | Evidence Supporting M2-Associated Pathology | References |
|---|---|---|---|
| Infection with Cryptococcus neoformans | Mouse | Inflammatory IFNγ is critical for microglial anticryptococcal efficacy | [278] |
| Infection with Cryptococcus neoformans | Mouse | Microglial proliferation is suppressed by Cn infection, reduction of IFNγ and delayed microglial activation associated with worse disease symptoms | [276] |
| Infection with Cryptococcus neoformans | Mouse | Increased microglial numbers in the brains of immune mice | [283] |
| Microglial cell culture | Human | Increased M1-associated MIP-1α, MCP1, and IL-8 in the presence of opsonized Cn | [284] |
| Infection with Cryptococcus neoformans | Mouse | M2-associated CCL2 production correlates to increased Cn susceptibility | [287] |
| Cryptococcus gatti infection | Mouse | Loss of M1-associated TLR9 results in a higher mortality rate and a greater number of fungal CFUs in the brain. Reduced inflammatory IFNγ and IL-17 cytokines were correlated with higher fungal burden. | [272] |
| Intracerebral Cryptococcus gatti infection | Mouse | Administration of M1-polarizing IL-2 activated microglia and significantly prolonged survival time. | [278] |
| Analysis of neutralizing antibody responses to the trivalent seasonal influenza vaccine | Human | Female serum samples showed an elevated expression of inflammatory cytokines, STAT3 proteins in monocytes, and antibody responses to the vaccine compared to males | [290] |
| Model | Species | Evidence Supporting Male Predominance | References |
|---|---|---|---|
| Central Brain Tumor Registry of the United States, 2004–2016 | Human | 57.5% of GBM patients were male, higher risk of death in male patients | [310] |
| Multi- institutional repository of clinical data from 1,400 GBM patients | Human | 60.5% of patients were male | [311] |
| Incidence of primary brain and other CNS tumors in the US | Human | Glioblastoma is 1.57 times more common in males. | [312] |
| SEER database from 2000–2010 | Human | Of 14,675 GBM patients, 59.4% were male. | [313] |
| GBM tumor samples from the Chinese Glioma Genome Atlas | Human | Male to female ratio of primary GBM is 1.67, recurrent GBM is 1.75. | [314] |
| Retrospective Analysis of all patients ≤21 years with high-grade glioma | Human | Female patients demonstrated enhanced survival following gross total resection of tumors compared to male patients, with a mean of 8.1 years in females and 2.4 years in males. Females had increased progression-free survival. | [316] |
| Multivariate Analysis of the Project of Emilia Romagna on Neuro-Oncology (PERNO) registry | Human | 58.6% of patients were male. In those that had hypermethylated O6 Methylguanine-DNA-Methyltransferase (MGMT), methylated females had longer survival compared to methylated males. | [317] |
| Analysis of serial Magnetic Resonance Images | Human | Quantitative imaging-based measure of response revealed standard therapy is more effective in female GBM patients. | [318] |
| SEER database from 2000–2008 | Human | 5-year cancer-specific survival rate was 6.8% in males and 8.3% in females | [320] |
| OBTS database from 2007–2017 | Human | Male median survival was 15.9 months post-diagnosis, 22.6 months in females. | [319] |
| 3,4- benzopyrene induced glial tumor formation | Rat | 77.8% of intact male rats developed glial tumors, which dropped to 50% following castration | [321] |
| GBM cell lines | Human | Upregulation of androgen receptor in all eight examined GBM cell lines, DHT administration prevents TGF β -mediated inhibition of GBM. | [322] |
| Glioma patients, cell lines | Human | Elevated androgen receptor and serum testosterone in GBM patients relative to healthy controls, androgen receptor antagonists inhibit proliferation. | [323] |
| GBM female patient samples | Human | Increased androgen receptor mRNA in 93% of samples | [331] |
| GBM patient specimens, cell lines | Human | Increased androgen receptor protein in 56% of GBM samples, 30% are constitutively active. Androgen receptor siRNA silencing induced GBM cell death. | [324] |
| GBM cell culture | Human | Testosterone increased GBM proliferation, migration, and invasion. | [326] |
| Intracerebral implantation of cultured U87MG cells | Rat | The apoptotic index for U87 tumor cells was significantly higher in females than males. Females survived longer than males and ovariectomy negated the advantage. | [327] |
| Incidence Rate Calculation from New York State Cancer Registry Data | Human | The female population had a lower risk of developing GBM than males, with the protective effect of sex most evident at the approximate age of menopause. Protective effects decreased in postmenopausal age strata. | [328] |
| Histologically confirmed cases of glioma identified from population- based cancer registries | Human | Risk of glioma increased among women with a relatively older age at menarche and among women with a history of hysterectomy. | [329] |
| Irradiation of mouse astrocytes | Mouse | Significantly decreased proliferation in female astrocytes compared to male post-irradiation, increased markers of cellular senescence in females | [330] |
| Model | Species | Evidence Supporting Male M2 Predominance | References |
|---|---|---|---|
| Primary glioma patient samples | Human | The amount of M2 GAMs increases with malignancy grade and is associated with shorter survival. | [336] |
| Patient-derived glioma stem cells | Human | Short-term relapse following radiation therapy was correlated with high M2 GAM frequency. The frequency of M2 macrophage/microglia was increased in recurrent mesenchymal GBM compared to primary non-mesenchymal GBM. | [337] |
| Mouse- and patient-derived cells | Mouse, Human | Microglial/macrophage activation by amphotericin B upregulates the M1 markers MCP-1 and IL-8, and inhibits brain tumor initiating cells (BTICs). Cells cultured from the tumor microenvironment could not inhibit BTICs | [339] |
| Chimeric mice to track origins of GAM populations | Mouse | Mouse chimeras showed that macrophages infiltrated at late stage GBM, and comprise only ¼ of the total myeloid-cell fraction in experimental gliomas. Microglia were found to up-regulate CD45 expression levels and represent up to 40% of the CD45high cell population. | [340] |
| Genome sequencing profile from the Chinese Glioma Genome Atlas (CGGA) database | Human | In analysis of human GBM genomes, the high-risk group was characterized by upregulated mRNA expression of M2 microglia/macrophage markers and higher levels of IL-10 and TGFβ1. | [341] |
| Transcriptomic profile analysis of experimental C6 gliomas | Rat | In microglia from rat gliomas, genes characteristic of M1 activation such as Tlr2, Tlr4 and Il18, Cd80, Il12a, Il15 were downregulated, and gene expression characteristic for M2 activation such as Tgm2, Il1rn, Cxcl10, Cxcl16, Ccl2, Ccl17, Ccl22, Vegfa, Stat3, Arg1 and Mmp14 were upregulated. | [342] |
| Implantation of GL261 GBM cells | Mouse | Pharmacological blockade or conditional knockout of Nrp1 from microglia/macrophages polarizes GAMs towards an M1 phenotype, reduces tumor size and vascularity and increases survival | [345] |
| Implantation of GL261 GBM cells in chimeric mice | Mouse | Conditional knockout of Nrp1 from microglia/macrophages polarizes GAMs towards an M1 phenotype and increases antitumor cytotoxic T cell immune responses | [346] |
| GBM tissue specimens of from the Neurosurgery Unit of the Catholic University Medical School | Human | Increased M2-specific CD163 expression was observed within the tumor than in the surrounding periphery. | [347] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O'Connor, J.L.; Nissen, J.C. The Pathological Activation of Microglia Is Modulated by Sexually Dimorphic Pathways. Int. J. Mol. Sci. 2023, 24, 4739. https://doi.org/10.3390/ijms24054739
O'Connor JL, Nissen JC. The Pathological Activation of Microglia Is Modulated by Sexually Dimorphic Pathways. International Journal of Molecular Sciences. 2023; 24(5):4739. https://doi.org/10.3390/ijms24054739
Chicago/Turabian StyleO'Connor, Jennifer L., and Jillian C. Nissen. 2023. "The Pathological Activation of Microglia Is Modulated by Sexually Dimorphic Pathways" International Journal of Molecular Sciences 24, no. 5: 4739. https://doi.org/10.3390/ijms24054739
APA StyleO'Connor, J. L., & Nissen, J. C. (2023). The Pathological Activation of Microglia Is Modulated by Sexually Dimorphic Pathways. International Journal of Molecular Sciences, 24(5), 4739. https://doi.org/10.3390/ijms24054739