
dCas9-BE3 and dCas12a-BE3 Systems Mediated Base Editing in Kiwifruit Canker Causal Agent Pseudomonas syringae pv. actinidiae

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Result
2.1. Genome Information of PSA.AH.01
2.2. Successfully Knockout Flic in Psa
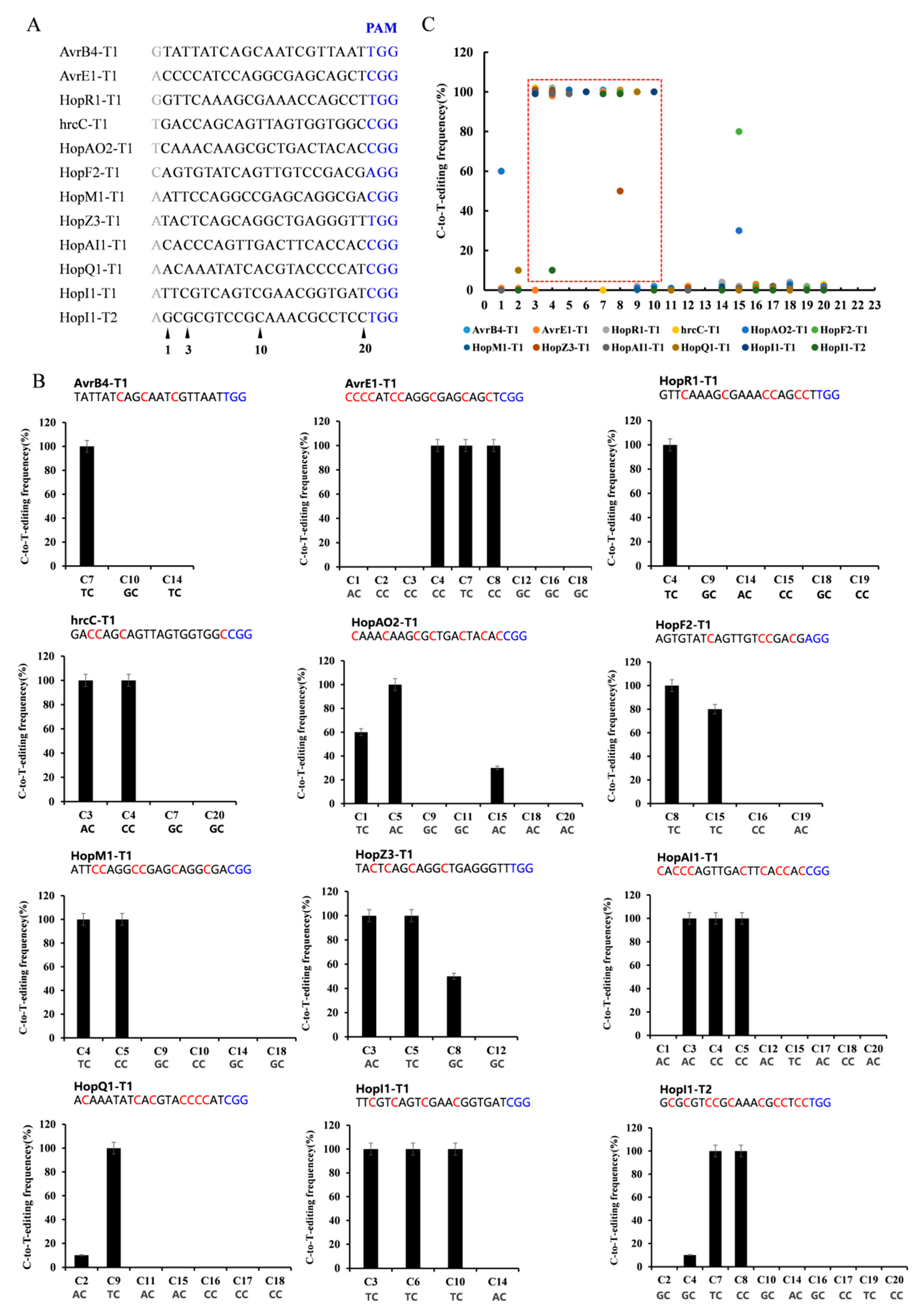
2.3. Editing Rule of dCas9-BE3 in Psa
2.4. Multi-Site Knockout by Using the dCas9-BE3 System
2.5. pdCas12a-BE Knockout System
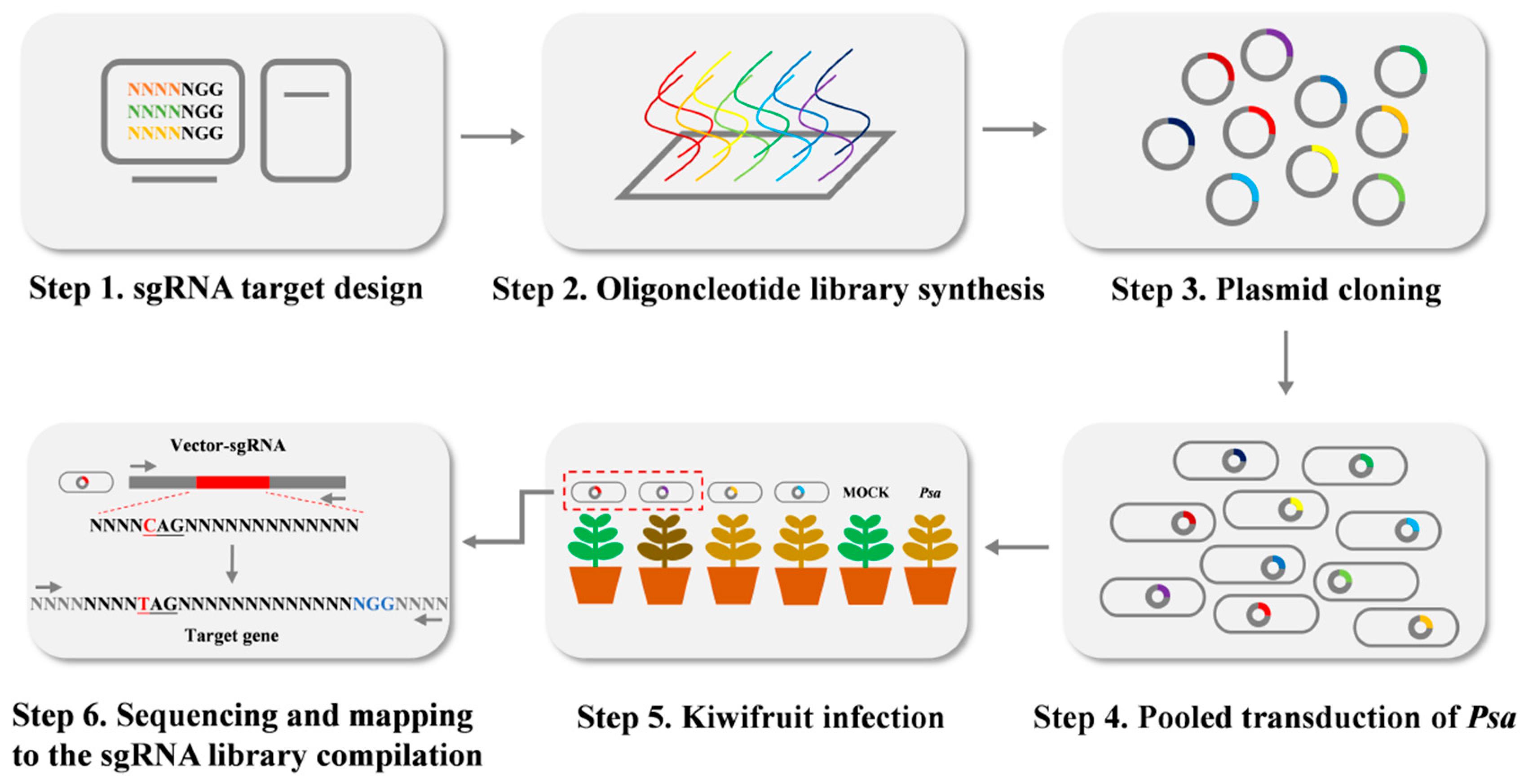
2.6. Construction of sgRNA Libraries Targeting Genome-Wide Editing
2.7. Pathogenicity Assays of Psa Mutant Strains
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Plant Growth Conditions
4.2. Genome Sequencing and Annotation
4.3. Construction of the Base-Editing System pdCas12a-BE
4.4. Target Design Procedure
4.5. Knockout Vector Construction
4.6. Construction of a Genome-Wide Mutant Library
4.7. Plasmid Transformations into Psa
4.8. Pathogenicity Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef]
- Terns, M.P.; Terns, R.M. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 2011, 14, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da, C.M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Paul, B.; Montoya, G. CRISPR-Cas12a: Functional overview and applications. Biomed. J. 2020, 43, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ren, K.; Qiu, X.; Zheng, J.; Guo, M.; Guan, X.; Liu, H.; Li, N.; Zhang, B.; Yang, D.; et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 2016, 532, 522–526. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yang, J.; Yang, S.; Ye, R.D.; Chen, D.; Jiang, Y. A CRISPR-Cpf1-Assisted Non-Homologous End Joining Genome Editing System of Mycobacterium smegmatis. Biotechnol. J. 2018, 13, e1700588. [Google Scholar] [CrossRef]
- Schatoff, E.M.; Zafra, M.P.; Dow, L.E. Base editing the mammalian genome. Methods 2019, 164–165, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, B.; Chen, L.; Xie, L.; Yu, W.; Wang, Y.; Li, L.; Yin, S.; Yang, L.; Hu, H.; et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat. Biotechnol. 2020, 38, 856–860. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Scortichini, M.; Marcelletti, S.; Ferrante, P.; Petriccione, M.; Firrao, G. Pseudomonas syringae pv. actinidiae: A re-emerging, multi-faceted, pandemic pathogen. Mol. Plant Pathol. 2012, 13, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Purahong, W.; Orru, L.; Donati, I.; Perpetuini, G.; Cellini, A.; Lamontanara, A.; Michelotti, V.; Tacconi, G.; Spinelli, F. Plant Microbiome and Its Link to Plant Health: Host Species, Organs and Pseudomonas syringae pv. actinidiae Infection Shaping Bacterial Phyllosphere Communities of Kiwifruit Plants. Front. Plant Sci. 2018, 9, 1563. [Google Scholar] [CrossRef] [PubMed]
- Donati, I.; Cellini, A.; Buriani, G.; Mauri, S.; Kay, C.; Tacconi, G.; Spinelli, F. Pathways of flower infection and pollen-mediated dispersion of Pseudomonas syringae pv. actinidiae, the causal agent of kiwifruit bacterial canker. Hortic. Res.-Engl. 2018, 5, 56. [Google Scholar] [CrossRef]
- Portaliou, A.G.; Tsolis, K.C.; Loos, M.S.; Zorzini, V.; Economou, A. Type III Secretion: Building and Operating a Remarkable Nanomachine. Trends Biochem. Sci. 2016, 41, 175–189. [Google Scholar] [CrossRef]
- Wilton, M.; Subramaniam, R.; Elmore, J.; Felsensteiner, C.; Coaker, G.; Desveaux, D. The type III effector HopF2Pto targets Arabidopsis RIN4 protein to promote Pseudomonas syringae virulence. Proc. Natl. Acad. Sci. USA 2010, 107, 2349–2354. [Google Scholar] [CrossRef]
- Bretz, J.R.; Mock, N.M.; Charity, J.C.; Zeyad, S.; Baker, C.J.; Hutcheson, S.W. A translocated protein tyrosine phosphatase of Pseudomonas syringae pv. tomato DC3000 modulates plant defence response to infection. Mol. Microbiol. 2003, 49, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, J.; Yoon, M.; Applegate, E.R.; Stroud, E.A.; Templeton, M.D. AvrE1 and HopR1 from Pseudomonas syringae pv. actinidiae are additively required for full virulence on kiwifruit. Mol. Plant Pathol. 2020, 21, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Han, N.; Tian, R.; Chen, J.; Gao, X.; Wu, Z.; Liu, Y.; Huang, L. Role of the Type VI Secretion System in the Pathogenicity of Pseudomonas syringae pv. actinidiae, the Causative Agent of Kiwifruit Bacterial Canker. Front. Microbiol. 2021, 12, 627785. [Google Scholar] [CrossRef] [PubMed]
- Hemara, L.M.; Jayaraman, J.; Sutherland, P.W.; Montefiori, M.; Arshed, S.; Chatterjee, A.; Chen, R.; Andersen, M.T.; Mesarich, C.H.; van der Linden, O.; et al. Effector loss drives adaptation of Pseudomonas syringae pv. actinidiae biovar 3 to Actinidia arguta. PLoS Pathog. 2022, 18, e1010542. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Y.; Zhang, Y.; Pi, Y.; Gu, T.; Song, L.; Wang, Y.; Ji, Q. CRISPR/Cas9-based Genome Editing in Pseudomonas aeruginosa and Cytidine Deaminase-Mediated Base Editing in Pseudomonas Species. iScience 2018, 6, 222–231. [Google Scholar] [CrossRef]
- Ho, J.; Zhao, M.; Wojcik, S.; Taiaroa, G.; Butler, M.; Poulter, R. The application of the CRISPR-Cas9 system in Pseudomonas syringae pv. actinidiae. J. Med. Microbiol. 2020, 69, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Wang, Y.; Li, N.; Jiang, F.F.; Wu, C.X.; Liu, F.; Chen, H.C.; Liu, Z.F. Highly efficient base editing in bacteria using a Cas9-cytidine deaminase fusion. Commun. Biol. 2018, 1, 32. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, L.B.; Liang, T.X.; Yang, L.R.; Wu, J.P. CRISPR-Assisted Multiplex Base Editing System in Pseudomonas putida KT2440. Front. Bioeng. Biotechnol. 2020, 8, 905. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef]
- Manghwar, H.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas System: Recent Advances and Future Prospects for Genome Editing. Trends Plant Sci. 2019, 24, 1102–1125. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Liu, Y.; Yang, B.; Wang, X.; Wei, J.; Lu, Z.; Zhang, Y.; Wu, J.; Huang, X.; et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat. Biotechnol. 2018, 36, 324–327. [Google Scholar] [CrossRef]
- Mesarich, C.H.; Rees-George, J.; Gardner, P.P.; Ghomi, F.A.; Gerth, M.L.; Andersen, M.T.; Rikkerink, E.H.; Fineran, P.C.; Templeton, M.D. Transposon insertion libraries for the characterization of mutants from the kiwifruit pathogen Pseudomonas syringae pv. actinidiae. PLoS ONE 2017, 12, e0172790. [Google Scholar] [CrossRef] [PubMed]
- Kolisnychenko, V.; Plunkett, G.; Herring, C.D., 3rd; Fehér, T.; Pósfai, J.; Blattner, F.R.; Pósfai, G. Engineering a reduced Escherichia coli genome. Genome Res. 2002, 12, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Hou, S.; Wang, X.; Li, Y.; Ren, D.; Chen, S.; Tang, X.; Zhou, J.M. A Pseudomonas syringae ADP-ribosyltransferase inhibits Arabidopsis mitogen-activated protein kinase kinases. Plant Cell 2010, 22, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, S.; Chen, X.; Liu, C.; Sheen, J.; Shan, L.; He, P. The Pseudomonas syringae effector HopF2 suppresses Arabidopsis immunity by targeting BAK1. Plant J. 2014, 77, 235–245. [Google Scholar] [CrossRef]
- Espinosa, A.; Guo, M.; Tam, V.C.; Fu, Z.Q.; Alfano, J.R. The Pseudomonas syringae type III-secreted protein HopPtoD2 possesses protein tyrosine phosphatase activity and suppresses programmed cell death in plants. Mol. Microbiol. 2003, 49, 377–387. [Google Scholar] [CrossRef]
- Macho, A.P.; Schwessinger, B.; Ntoukakis, V.; Brutus, A.; Segonzac, C.; Roy, S.; Kadota, Y.; Oh, M.H.; Sklenar, J.; Derbyshire, P.; et al. A bacterial tyrosine phosphatase inhibits plant pattern recognition receptor activation. Science 2014, 343, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Ojeda, M.P.; Moreno-Pérez, A.; Ramos, C.; López-Solanilla, E. Suppression of Plant Immune Responses by the Pseudomonas savastanoi pv. savastanoi NCPPB 3335 Type III Effector Tyrosine Phosphatases HopAO1 and HopAO2. Front. Plant Sci. 2017, 8, 680. [Google Scholar] [CrossRef]
- Denu, J.M.; Stuckey, J.A.; Saper, M.A.; Dixon, J.E. Form and function in protein dephosphorylation. Cell 1996, 87, 361–364. [Google Scholar] [CrossRef]
- Kvitko, B.H.; Park, D.H.; Velásquez, A.C.; Wei, C.F.; Russell, A.B.; Martin, G.B.; Schneider, D.J.; Collmer, A. Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog. 2009, 5, e1000388. [Google Scholar] [CrossRef]
- Ishiga, T.; Sakata, N.; Nguyen, V.T.; Ishiga, Y. Flood inoculation of seedlings on culture medium to study interactions between Pseudomonas syringae pv. actinidiae and kiwifruit. J. Gen. Plant Pathol. 2020, 86, 257–265. [Google Scholar] [CrossRef]
- McAtee, P.A.; Brian, L.; Curran, B.; van der Linden, O.; Nieuwenhuizen, N.J.; Chen, X.; Henry-Kirk, R.A.; Stroud, E.A.; Nardozza, S.; Jayaraman, J.; et al. Re-programming of Pseudomonas syringae pv. actinidiae gene expression during early stages of infection of kiwifruit. BMC Genom. 2018, 19, 822. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yadeta, K.A.; Elmore, J.M.; Coaker, G. The Pseudomonas syringae effector HopQ1 promotes bacterial virulence and interacts with tomato 14-3-3 proteins in a phosphorylation-dependent manner. Plant Physiol. 2013, 161, 2062–2074. [Google Scholar] [CrossRef]
- Zhang, J.; Shao, F.; Li, Y.; Cui, H.; Chen, L.; Li, H.; Zou, Y.; Long, C.; Lan, L.; Chai, J.; et al. A Pseudomonas syringae effector inactivates MAPKs to suppress PAMP-induced immunity in plants. Cell Host. Microbe. 2007, 1, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Jelenska, J.; Yao, N.; Vinatzer, B.A.; Wright, C.M.; Brodsky, J.L.; Greenberg, J.T. A J domain virulence effector of Pseudomonas syringae remodels host chloroplasts and suppresses defenses. Curr. Biol. 2007, 17, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Badel, J.L.; Shimizu, R.; Oh, H.S.; Collmer, A. A Pseudomonas syringae pv. tomato avrE1/hopM1 mutant is severely reduced in growth and lesion formation in tomato. Mol. Plant Microbe. Interact. 2006, 19, 99–111. [Google Scholar] [CrossRef]
- Nomura, K.; Debroy, S.; Lee, Y.H.; Pumplin, N.; Jones, J.; He, S.Y. A bacterial virulence protein suppresses host innate immunity to cause plant disease. Science 2006, 313, 220–223. [Google Scholar] [CrossRef]
- Zumaquero, A.; Macho, A.P.; Rufián, J.S.; Beuzón, C.R. Analysis of the role of the type III effector inventory of Pseudomonas syringae pv. phaseolicola 1448a in interaction with the plant. J. Bacteriol. 2010, 192, 4474–4488. [Google Scholar] [CrossRef]
- Lee, J.; Teitzel, G.M.; Greenberg, J.T. SGT1b is required for HopZ3-mediated suppression of the epiphytic growth of Pseudomonas syringae on N. benthamiana. Plant Signal Behav. 2012, 7, 1129–1131. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Song, W.; Wang, L.; Wu, Y.; Xu, X.; Niu, X.; Huang, S.; Liu, Y.; Tang, W. dCas9-BE3 and dCas12a-BE3 Systems Mediated Base Editing in Kiwifruit Canker Causal Agent Pseudomonas syringae pv. actinidiae. Int. J. Mol. Sci. 2023, 24, 4597. https://doi.org/10.3390/ijms24054597
Liu B, Song W, Wang L, Wu Y, Xu X, Niu X, Huang S, Liu Y, Tang W. dCas9-BE3 and dCas12a-BE3 Systems Mediated Base Editing in Kiwifruit Canker Causal Agent Pseudomonas syringae pv. actinidiae. International Journal of Molecular Sciences. 2023; 24(5):4597. https://doi.org/10.3390/ijms24054597
Chicago/Turabian StyleLiu, Bo, Wenpeng Song, Linchao Wang, Yantao Wu, Xiaoting Xu, Xiangli Niu, Shengxiong Huang, Yongsheng Liu, and Wei Tang. 2023. "dCas9-BE3 and dCas12a-BE3 Systems Mediated Base Editing in Kiwifruit Canker Causal Agent Pseudomonas syringae pv. actinidiae" International Journal of Molecular Sciences 24, no. 5: 4597. https://doi.org/10.3390/ijms24054597
APA StyleLiu, B., Song, W., Wang, L., Wu, Y., Xu, X., Niu, X., Huang, S., Liu, Y., & Tang, W. (2023). dCas9-BE3 and dCas12a-BE3 Systems Mediated Base Editing in Kiwifruit Canker Causal Agent Pseudomonas syringae pv. actinidiae. International Journal of Molecular Sciences, 24(5), 4597. https://doi.org/10.3390/ijms24054597