

Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: A Focus on Insulin Resistance


{kind=link}
{kind=link}
Abstract
1. Introduction
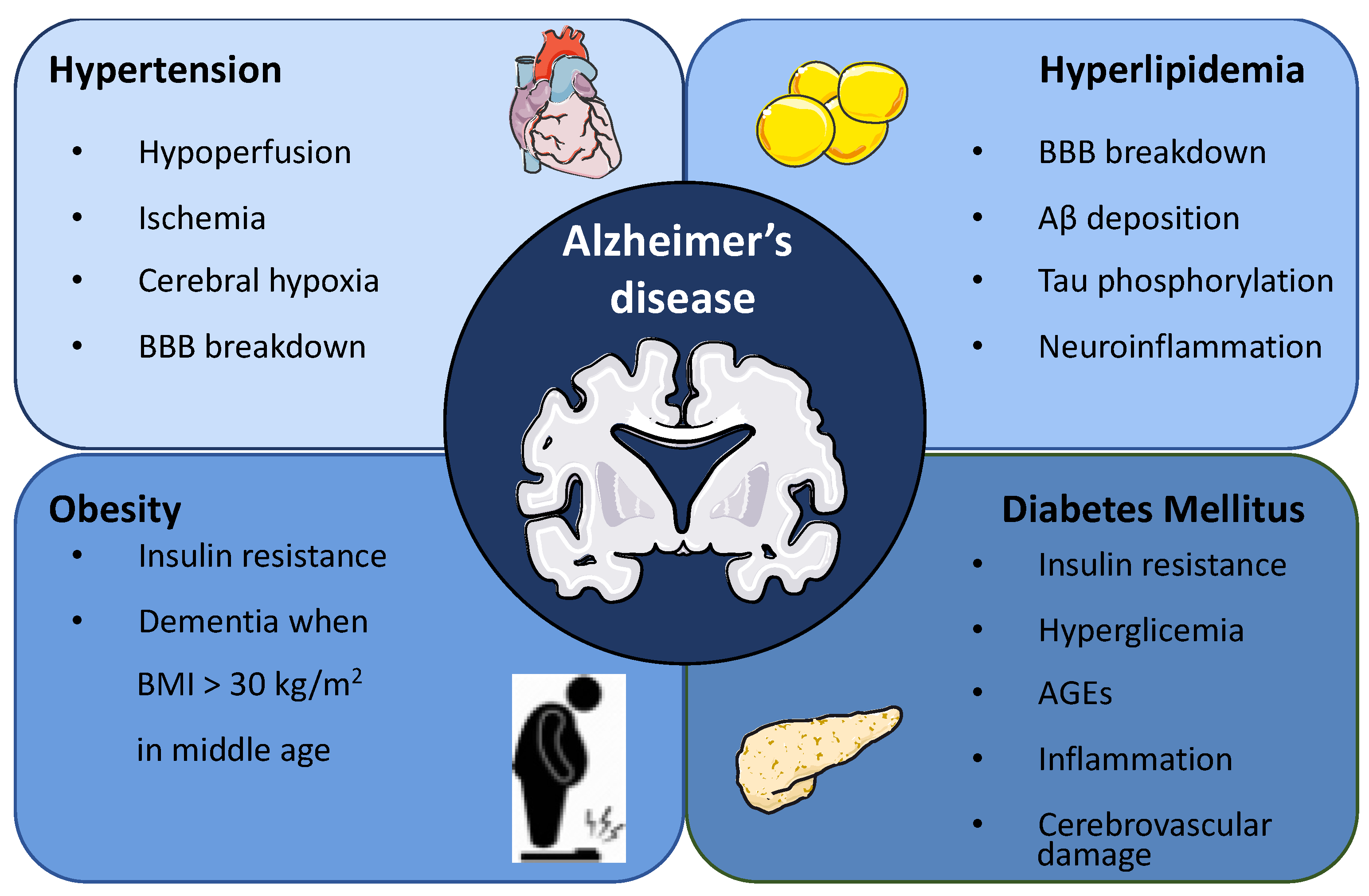
2. Association between Metabolic Syndrome and Alzheimer’s Disease
2.1. Hypertension
2.2. Hyperlipidemia
2.3. Obesity
2.4. Diabetes Mellitus
3. Insulin Action in the Brain
3.1. Insulin Transport and Regional Distribution in the Brain
3.2. Brain Insulin Actions in Peripheral Energy Homeostasis
3.2.1. Energy Homeostasis
3.2.2. Glucose Metabolism
3.3. Brain Insulin Actions in CNS Functions
3.3.1. Neuronal Plasticity
3.3.2. Dendritic Arbor Development
3.3.3. Neuronal Survival
3.3.4. Learning and Memory
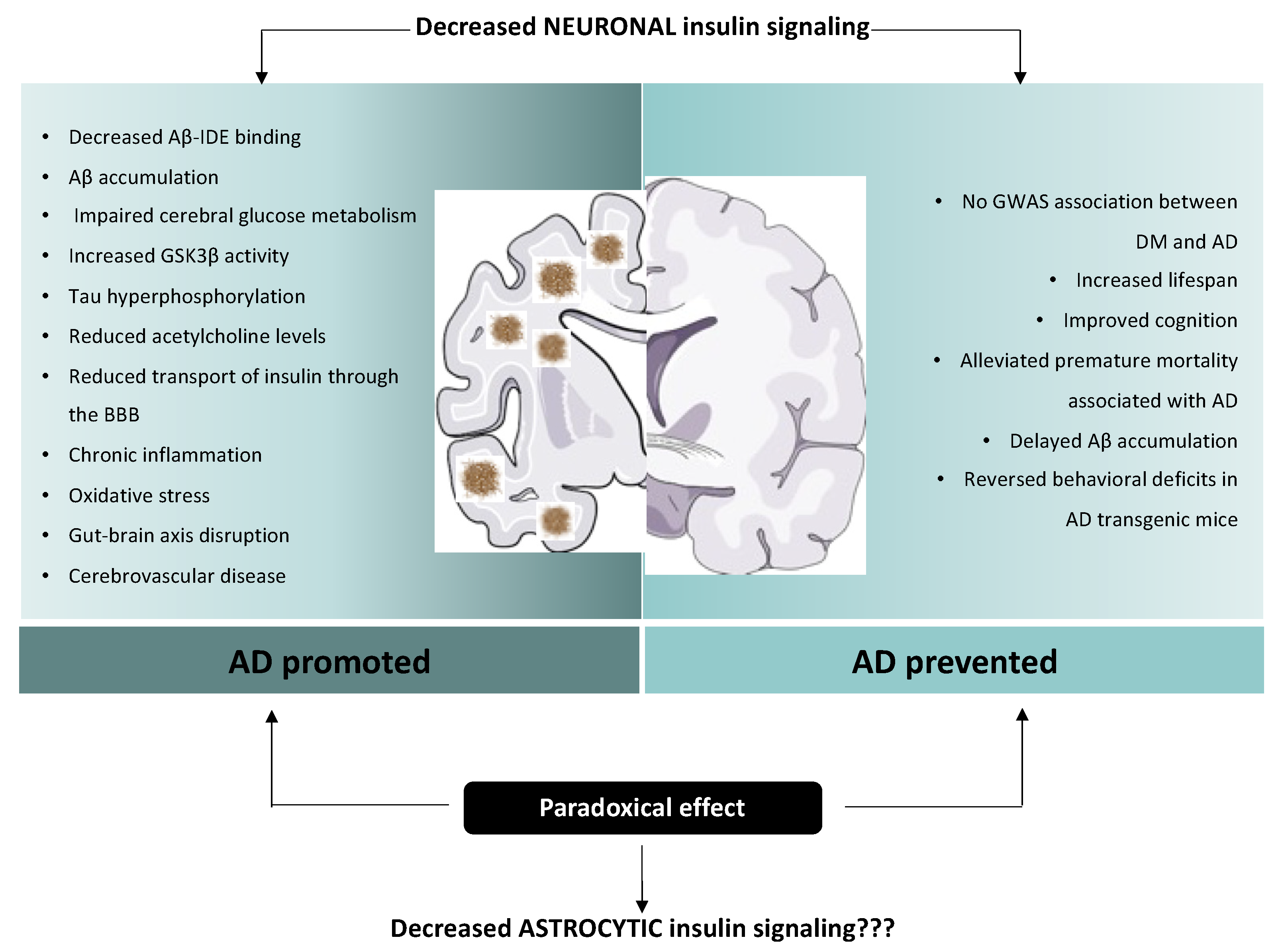
4. Controversial Role of Insulin Alterations in Alzheimer’s Disease
5. Role of Astrocytic Insulin Receptor
5.1. Astrocytic Function in AD
5.2. Key Role of Astrocytic Insulin Signalling in AD
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s Disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Ferreira, D.; Perestelo-Pérez, L.; Westman, E.; Wahlund, L.-O.; Sarría, A.; Serrano-Aguilar, P. Meta-Review of CSF Core Biomarkers in Alzheimer’s Disease: The State-of-the-Art after the New Revised Diagnostic Criteria. Front. Aging Neurosci. 2014, 6, 47. [Google Scholar] [CrossRef]
- Alzheimer Association. 2022 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Kuns, B.; Rosani, A.; Varghese, D. Memantine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7443. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Albarrati, A.; Albratty, M.; Najmi, A.; Meraya, A.M.; Bungau, S. The Road to Precision Medicine: Eliminating the “One Size Fits All” Approach in Alzheimer’s Disease. Biomed. Pharmacother. 2022, 153, 113337. [Google Scholar] [CrossRef]
- d’Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 265. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A Review on Alzheimer’s Disease Pathophysiology and Its Management: An Update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Pini, L.; Pievani, M.; Bocchetta, M.; Altomare, D.; Bosco, P.; Cavedo, E.; Galluzzi, S.; Marizzoni, M.; Frisoni, G.B. Brain Atrophy in Alzheimer’s Disease and Aging. Ageing Res. Rev. 2016, 30, 25–48. [Google Scholar] [CrossRef]
- Francis, P.T. The Interplay of Neurotransmitters in Alzheimer’s Disease. CNS Spectr. 2005, 10, 6–9. [Google Scholar] [CrossRef]
- Pan, X.; Kaminga, A.C.; Jia, P.; Wen, S.W.; Acheampong, K.; Liu, A. Catecholamines in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2020, 12, 184. [Google Scholar] [CrossRef]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How Does Brain Insulin Resistance Develop in Alzheimer’s Disease? Alzheimers Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef]
- de Wilde, M.C.; Vellas, B.; Girault, E.; Yavuz, A.C.; Sijben, J.W. Lower Brain and Blood Nutrient Status in Alzheimer’s Disease: Results from Meta-Analyses. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 416–431. [Google Scholar] [CrossRef]
- Gibas, K.J. The Starving Brain: Overfed Meets Undernourished in the Pathology of Mild Cognitive Impairment (MCI) and Alzheimer’s Disease (AD). Neurochem. Int. 2017, 110, 57–68. [Google Scholar] [CrossRef]
- Atwood, C.S.; Bowen, R.L. A Unified Hypothesis of Early- and Late-Onset Alzheimer’s Disease Pathogenesis. J. Alzheimers Dis. 2015, 47, 33–47. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging Roles of ER Stress in the Etiology and Pathogenesis of Alzheimer’s Disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy Metabolism and Inflammation in Brain Aging and Alzheimer’s Disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Blood-Brain Barrier Dysfunction as a Cause and Consequence of Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef]
- Galvan, V.; Hart, M.J. Vascular MTOR-Dependent Mechanisms Linking the Control of Aging to Alzheimer’s Disease. Biochim. Biophys. Acta 2016, 1862, 992–1007. [Google Scholar] [CrossRef]
- Yassine, H.N.; Anderson, A.; Brinton, R.; Carmichael, O.; Espeland, M.A.; Hoscheidt, S.; Hugenschmidt, C.E.; Keller, J.N.; Peters, A.; Pi-Sunyer, X. Do Menopausal Status and APOE4 Genotype Alter the Long-Term Effects of Intensive Lifestyle Intervention on Cognitive Function in Women with Type 2 Diabetes Mellitus? Neurobiol. Aging 2020, 92, 61–72. [Google Scholar] [CrossRef]
- Neu, S.C.; Pa, J.; Kukull, W.; Beekly, D.; Kuzma, A.; Gangadharan, P.; Wang, L.-S.; Romero, K.; Arneric, S.P.; Redolfi, A.; et al. Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-Analysis. JAMA Neurol. 2017, 74, 1178–1189. [Google Scholar] [CrossRef]
- Kulminski, A.M.; Loika, Y.; Culminskaya, I.; Huang, J.; Arbeev, K.G.; Bagley, O.; Feitosa, M.F.; Zmuda, J.M.; Christensen, K.; Yashin, A.I.; et al. Independent Associations of TOMM40 and APOE Variants with Body Mass Index. Aging Cell 2019, 18, e12869. [Google Scholar] [CrossRef]
- Tao, Q.; Ang, T.F.A.; DeCarli, C.; Auerbach, S.H.; Devine, S.; Stein, T.D.; Zhang, X.; Massaro, J.; Au, R.; Qiu, W.Q. Association of Chronic Low-Grade Inflammation With Risk of Alzheimer Disease in ApoE4 Carriers. JAMA Netw. Open 2018, 1, e183597. [Google Scholar] [CrossRef]
- Malek-Ahmadi, M.; Beach, T.; Obradov, A.; Sue, L.; Belden, C.; Davis, K.; Walker, D.G.; Lue, L.; Adem, A.; Sabbagh, M.N. Increased Alzheimer’s Disease Neuropathology Is Associated with Type 2 Diabetes and ApoE ε.4 Carrier Status. Curr. Alzheimer Res. 2013, 10, 654–659. [Google Scholar] [CrossRef]
- Aghajanpour-Mir, M.; Amjadi-Moheb, F.; Dadkhah, T.; Hosseini, S.R.; Ghadami, E.; Assadollahi, E.; Akhavan-Niaki, H.; Ahmadi Ahangar, A. Informative Combination of CLU Rs11136000, Serum HDL Levels, Diabetes, and Age as a New Piece of Puzzle-Picture of Predictive Medicine for Cognitive Disorders. Mol. Biol. Rep. 2019, 46, 1033–1041. [Google Scholar] [CrossRef]
- Šerý, O.; Hlinecká, L.; Balcar, V.J.; Janout, V.; Povova, J. Diabetes, Hypertension and Stroke–Does Alzheimer Protect You? Neuro Endocrinol. Lett. 2014, 35, 691–696. [Google Scholar]
- de Bruijn, R.F.A.G.; Ikram, M.A. Cardiovascular Risk Factors and Future Risk of Alzheimer’s Disease. BMC Med. 2014, 12, 130. [Google Scholar] [CrossRef]
- Leszek, J.; Mikhaylenko, E.V.; Belousov, D.M.; Koutsouraki, E.; Szczechowiak, K.; Kobusiak-Prokopowicz, M.; Mysiak, A.; Diniz, B.S.; Somasundaram, S.G.; Kirkland, C.E.; et al. The Links between Cardiovascular Diseases and Alzheimer’s Disease. Curr. Neuropharmacol. 2021, 19, 152–169. [Google Scholar] [CrossRef]
- Tini, G.; Scagliola, R.; Monacelli, F.; La Malfa, G.; Porto, I.; Brunelli, C.; Rosa, G.M. Alzheimer’s Disease and Cardiovascular Disease: A Particular Association. Cardiol. Res. Pr. 2020, 2020, 2617970. [Google Scholar] [CrossRef]
- Ogama, N.; Sakurai, T.; Kawashima, S.; Tanikawa, T.; Tokuda, H.; Satake, S.; Miura, H.; Shimizu, A.; Kokubo, M.; Niida, S.; et al. Postprandial Hyperglycemia Is Associated With White Matter Hyperintensity and Brain Atrophy in Older Patients With Type 2 Diabetes Mellitus. Front. Aging Neurosci. 2018, 10, 273. [Google Scholar] [CrossRef]
- Rojas-Gutierrez, E.; Muñoz-Arenas, G.; Treviño, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Díaz, A.; Guevara, J. Alzheimer’s Disease and Metabolic Syndrome: A Link from Oxidative Stress and Inflammation to Neurodegeneration. Synapse 2017, 71, e21990. [Google Scholar] [CrossRef]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible Implications of Insulin Resistance and Glucose Metabolism in Alzheimer’s Disease Pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s Disease from Perturbed Cerebral Glucose Metabolism: Implications for Diagnostic and Therapeutic Strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef]
- Craft, S.; Cholerton, B.; Baker, L.D. Insulin and Alzheimer’s Disease: Untangling the Web. J. Alzheimers Dis. 2013, 33 (Suppl. S1), S263–S275. [Google Scholar] [CrossRef]
- Skoog, I.; Gustafson, D. Update on Hypertension and Alzheimer’s Disease. Neurol. Res. 2006, 28, 605–611. [Google Scholar] [CrossRef]
- Skoog, I.; Lernfelt, B.; Landahl, S.; Palmertz, B.; Andreasson, L.A.; Nilsson, L.; Persson, G.; Odén, A.; Svanborg, A. 15-Year Longitudinal Study of Blood Pressure and Dementia. Lancet 1996, 347, 1141–1145. [Google Scholar] [CrossRef]
- Staessen, J.A.; Richart, T.; Birkenhäger, W.H. Less Atherosclerosis and Lower Blood Pressure for a Meaningful Life Perspective with More Brain. Hypertension 2007, 49, 389–400. [Google Scholar] [CrossRef]
- Popp, J.; Meichsner, S.; Kölsch, H.; Lewczuk, P.; Maier, W.; Kornhuber, J.; Jessen, F.; Lütjohann, D. Cerebral and Extracerebral Cholesterol Metabolism and CSF Markers of Alzheimer’s Disease. Biochem. Pharm. 2013, 86, 37–42. [Google Scholar] [CrossRef]
- Xue-Shan, Z.; Juan, P.; Qi, W.; Zhong, R.; Li-Hong, P.; Zhi-Han, T.; Zhi-Sheng, J.; Gui-Xue, W.; Lu-Shan, L. Imbalanced Cholesterol Metabolism in Alzheimer’s Disease. Clin. Chim. Acta 2016, 456, 107–114. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Canepa, E.; Marengo, B.; Marinari, U.M.; Poli, G.; Pronzato, M.A.; Domenicotti, C. Cholesterol and Alzheimer’s Disease: A Still Poorly Understood Correlation. IUBMB Life 2012, 64, 931–935. [Google Scholar] [CrossRef]
- Ullrich, C.; Pirchl, M.; Humpel, C. Hypercholesterolemia in Rats Impairs the Cholinergic System and Leads to Memory Deficits. Mol. Cell.Neurosci. 2010, 45, 408–417. [Google Scholar] [CrossRef]
- Hendrie, H.C.; Hake, A.; Lane, K.; Purnell, C.; Unverzagt, F.; Smith-Gamble, V.; Murrell, J.; Ogunniyi, A.; Baiyewu, O.; Callahan, C.; et al. Statin Use, Incident Dementia and Alzheimer Disease in Elderly African Americans. Ethn. Dis. 2015, 25, 345–354. [Google Scholar] [CrossRef]
- Haag, M.D.M.; Hofman, A.; Koudstaal, P.J.; Stricker, B.H.C.; Breteler, M.M.B. Statins Are Associated with a Reduced Risk of Alzheimer Disease Regardless of Lipophilicity. The Rotterdam Study. J. Neurol. Neurosurg. Psychiatry 2009, 80, 13–17. [Google Scholar] [CrossRef]
- Lin, F.-C.; Chuang, Y.-S.; Hsieh, H.-M.; Lee, T.-C.; Chiu, K.-F.; Liu, C.-K.; Wu, M.-T. Early Statin Use and the Progression of Alzheimer Disease: A Total Population-Based Case-Control Study. Medicine 2015, 94, e2143. [Google Scholar] [CrossRef]
- Simons, M.; Schwärzler, F.; Lütjohann, D.; von Bergmann, K.; Beyreuther, K.; Dichgans, J.; Wormstall, H.; Hartmann, T.; Schulz, J.B. Treatment with Simvastatin in Normocholesterolemic Patients with Alzheimer’s Disease: A 26-Week Randomized, Placebo-Controlled, Double-Blind Trial. Ann. Neurol. 2002, 52, 346–350. [Google Scholar] [CrossRef]
- Sano, M.; Bell, K.L.; Galasko, D.; Galvin, J.E.; Thomas, R.G.; van Dyck, C.H.; Aisen, P.S. A Randomized, Double-Blind, Placebo-Controlled Trial of Simvastatin to Treat Alzheimer Disease. Neurology 2011, 77, 556–563. [Google Scholar] [CrossRef]
- Feldman, H.H.; Doody, R.S.; Kivipelto, M.; Sparks, D.L.; Waters, D.D.; Jones, R.W.; Schwam, E.; Schindler, R.; Hey-Hadavi, J.; DeMicco, D.A.; et al. Randomized Controlled Trial of Atorvastatin in Mild to Moderate Alzheimer Disease: LEADe. Neurology 2010, 74, 956–964. [Google Scholar] [CrossRef]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.E.M.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al. Pravastatin in Elderly Individuals at Risk of Vascular Disease (PROSPER): A Randomised Controlled Trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of Cholesterol Lowering with Simvastatin in 20 536 High-Risk Individuals: A Randomised Placebocontrolled Trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Song, Y.; Nie, H.; Xu, Y.; Zhang, L.; Wu, Y. Association of Statin Use with Risk of Dementia: A Meta-Analysis of Prospective Cohort Studies. Geriatr. Gerontol. Int. 2013, 13, 817–824. [Google Scholar] [CrossRef]
- Purdon, D.; Arai, T.; Rapoport, S. No Evidence for Direct Incorporation of Esterified Palmitic Acid from Plasma into Brain Lipids of Awake Adult Rat. J. Lipid Res. 1997, 38, 526–530. [Google Scholar] [CrossRef]
- Karmi, A.; Iozzo, P.; Viljanen, A.; Hirvonen, J.; Fielding, B.A.; Virtanen, K.; Oikonen, V.; Kemppainen, J.; Viljanen, T.; Guiducci, L.; et al. Increased Brain Fatty Acid Uptake in Metabolic Syndrome. Diabetes 2010, 59, 2171–2177. [Google Scholar] [CrossRef]
- Cole, G.M.; Ma, Q.-L.; Frautschy, S.A. Dietary Fatty Acids and the Aging Brain. Nutr. Rev. 2010, 68 (Suppl. S2), S102–S111. [Google Scholar] [CrossRef]
- Gupta, S.; Knight, A.G.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. Saturated Long-Chain Fatty Acids Activate Inflammatory Signaling in Astrocytes. J. Neurochem. 2012, 120, 1060–1071. [Google Scholar] [CrossRef]
- Wilson, D.M.; Binder, L.I. Free Fatty Acids Stimulate the Polymerization of Tau and Amyloid Beta Peptides. In Vitro Evidence for a Common Effector of Pathogenesis in Alzheimer’s Disease. Am. J. Pathol. 1997, 150, 2181–2195. [Google Scholar]
- Kelly, T.; Yang, W.; Chen, C.-S.; Reynolds, K.; He, J. Global Burden of Obesity in 2005 and Projections to 2030. Int. J. Obes. (Lond.) 2008, 32, 1431–1437. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central Obesity and Increased Risk of Dementia More than Three Decades Later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef]
- Jagust, W.; Harvey, D.; Mungas, D.; Haan, M. Central Obesity and the Aging Brain. Arch. Neurol. 2005, 62, 1545–1548. [Google Scholar] [CrossRef]
- Hassing, L.B.; Dahl, A.K.; Thorvaldsson, V.; Berg, S.; Gatz, M.; Pedersen, N.L.; Johansson, B. Overweight in Midlife and Risk of Dementia: A 40-Year Follow-up Study. Int. J. Obes. 2009, 33, 893–898. [Google Scholar] [CrossRef]
- Hassing, L.B.; Dahl, A.K.; Pedersen, N.L.; Johansson, B. Overweight in Midlife Is Related to Lower Cognitive Function 30 Years Later: A Prospective Study with Longitudinal Assessments. Dement. Geriatr. Cogn. Disord. 2010, 29, 543–552. [Google Scholar] [CrossRef]
- Anstey, K.J.; Cherbuin, N.; Budge, M.; Young, J. Body Mass Index in Midlife and Late-Life as a Risk Factor for Dementia: A Meta-Analysis of Prospective Studies. Obes. Rev. 2011, 12, e426–e437. [Google Scholar] [CrossRef]
- Fitzpatrick, A.L.; Kuller, L.H.; Lopez, O.L.; Diehr, P.; O’Meara, E.S.; Longstreth, W.T.; Luchsinger, J.A. Midlife and Late-Life Obesity and the Risk of Dementia: Cardiovascular Health Study. Arch. Neurol. 2009, 66, 336–342. [Google Scholar] [CrossRef]
- Xu, W.L.; Atti, A.R.; Gatz, M.; Pedersen, N.L.; Johansson, B.; Fratiglioni, L. Midlife Overweight and Obesity Increase Late-Life Dementia Risk: A Population-Based Twin Study. Neurology 2011, 76, 1568–1574. [Google Scholar] [CrossRef]
- Wotton, C.J.; Goldacre, M.J. Age at Obesity and Association with Subsequent Dementia: Record Linkage Study. Postgrad. Med. J. 2014, 90, 547–551. [Google Scholar] [CrossRef]
- Nepal, B.; Brown, L.J.; Anstey, K.J. Rising Midlife Obesity Will Worsen Future Prevalence of Dementia. PLoS ONE 2014, 9, e99305. [Google Scholar] [CrossRef]
- Qizilbash, N.; Gregson, J.; Johnson, M.E.; Pearce, N.; Douglas, I.; Wing, K.; Evans, S.J.W.; Pocock, S.J. BMI and Risk of Dementia in Two Million People over Two Decades: A Retrospective Cohort Study. Lancet Diabetes Endocrinol. 2015, 3, 431–436. [Google Scholar] [CrossRef]
- Li, X.; Song, D.; Leng, S.X. Link between Type 2 Diabetes and Alzheimer’s Disease: From Epidemiology to Mechanism and Treatment. Clin. Interv. Aging 2015, 10, 549–560. [Google Scholar] [CrossRef]
- Em, B.; Jw, G.; Kc, C.; Nm, P.; Wr, M.; Pw, L. Incipient Alzheimer’s Disease: Microarray Correlation Analyses Reveal Major Transcriptional and Tumor Suppressor Responses. Proc. Natl. Acad. Sci. USA 2004, 101, 2173–2178. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired Insulin and Insulin-like Growth Factor Expression and Signaling Mechanisms in Alzheimer’s Disease—Is This Type 3 Diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, Increased Phosphorylation of Akt Substrates and Loss and Altered Distribution of Akt and PTEN Are Features of Alzheimer’s Disease Pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef]
- Busquets, O.; Ettcheto, M.; Pallàs, M.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C.; Folch, J.; Camins, A. Long-Term Exposition to a High Fat Diet Favors the Appearance of β-Amyloid Depositions in the Brain of C57BL/6J Mice. A Potential Model of Sporadic Alzheimer’s Disease. Mech. Ageing Dev. 2017, 162, 38–45. [Google Scholar] [CrossRef]
- Salas, I.H.; Weerasekera, A.; Ahmed, T.; Callaerts-Vegh, Z.; Himmelreich, U.; D’Hooge, R.; Balschun, D.; Saido, T.C.; De Strooper, B.; Dotti, C.G. High Fat Diet Treatment Impairs Hippocampal Long-Term Potentiation without Alterations of the Core Neuropathological Features of Alzheimer Disease. Neurobiol. Dis. 2018, 113, 82–96. [Google Scholar] [CrossRef]
- Carvalho, C.; Cardoso, S.; Correia, S.C.; Santos, R.X.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Metabolic Alterations Induced by Sucrose Intake and Alzheimer’s Disease Promote Similar Brain Mitochondrial Abnormalities. Diabetes 2012, 61, 1234–1242. [Google Scholar] [CrossRef]
- Hiltunen, M.; Khandelwal, V.K.M.; Yaluri, N.; Tiilikainen, T.; Tusa, M.; Koivisto, H.; Krzisch, M.; Vepsäläinen, S.; Mäkinen, P.; Kemppainen, S.; et al. Contribution of Genetic and Dietary Insulin Resistance to Alzheimer Phenotype in APP/PS1 Transgenic Mice. J. Cell. Mol. Med. 2012, 16, 1206–1222. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Spires-Jones, T.; Pooler, A.M.; Lechuga-Sancho, A.M.; Bacskai, B.J.; Garcia-Alloza, M. Progressive Neuronal Pathology and Synaptic Loss Induced by Prediabetes and Type 2 Diabetes in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 3428–3438. [Google Scholar] [CrossRef]
- Alagiakrishnan, K.; Sankaralingam, S.; Ghosh, M.; Mereu, L.; Senior, P. Antidiabetic Drugs and Their Potential Role in Treating Mild Cognitive Impairment and Alzheimer’s Disease. Discov. Med. 2013, 16, 277–286. [Google Scholar]
- Hölscher, C. Novel Dual GLP-1/GIP Receptor Agonists Show Neuroprotective Effects in Alzheimer’s and Parkinson’s Disease Models. Neuropharmacology 2018, 136, 251–259. [Google Scholar] [CrossRef]
- Grieb, P. Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer’s Disease: In Search of a Relevant Mechanism. Mol. Neurobiol. 2016, 53, 1741–1752. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Rai, S.; Tota, S.K.; Kumar, A.; Ahmad, A.S. Streptozotocin Intracerebroventricular-Induced Neurotoxicity and Brain Insulin Resistance: A Therapeutic Intervention for Treatment of Sporadic Alzheimer’s Disease (SAD)-Like Pathology. Mol. Neurobiol. 2016, 53, 4548–4562. [Google Scholar] [CrossRef]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the Brain: Sources, Localization and Functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef]
- Agrawal, R.; Reno, C.M.; Sharma, S.; Christensen, C.; Huang, Y.; Fisher, S.J. Insulin Action in the Brain Regulates Both Central and Peripheral Functions. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E156–E163. [Google Scholar] [CrossRef]
- Devaskar, S.U.; Giddings, S.J.; Rajakumar, P.A.; Carnaghi, L.R.; Menon, R.K.; Zahm, D.S. Insulin Gene Expression and Insulin Synthesis in Mammalian Neuronal Cells. J. Biol. Chem. 1994, 269, 8445–8454. [Google Scholar] [CrossRef]
- Giddings, S.J.; Chirgwin, J.; Permutt, M.A. Evaluation of Rat Insulin Messenger RNA in Pancreatic and Extrapancreatic Tissues. Diabetologia 1985, 28, 343–347. [Google Scholar] [CrossRef]
- Young, W.S. Periventricular Hypothalamic Cells in the Rat Brain Contain Insulin MRNA. Neuropeptides 1986, 8, 93–97. [Google Scholar] [CrossRef]
- Mehran, A.E.; Templeman, N.M.; Brigidi, G.S.; Lim, G.E.; Chu, K.-Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.G.; Kieffer, T.J.; et al. Hyperinsulinemia Drives Diet-Induced Obesity Independently of Brain Insulin Production. Cell Metab. 2012, 16, 723–737. [Google Scholar] [CrossRef]
- Dorn, A.; Rinne, A.; Hahn, H.J.; Bernstein, H.G.; Ziegler, M. C-Peptide Immunoreactive Neurons in Human Brain. Acta Histochem. 1982, 70, 326–330. [Google Scholar] [CrossRef]
- Frölich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Türk, A.; Hoyer, S.; et al. Brain Insulin and Insulin Receptors in Aging and Sporadic Alzheimer’s Disease. J. Neural. Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef]
- Milstein, J.L.; Ferris, H.A. The Brain as an Insulin-Sensitive Metabolic Organ. Mol. Metab. 2021, 52, 101234. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain Insulin Resistance in Type 2 Diabetes and Alzheimer Disease: Concepts and Conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Wallum, B.J.; Taborsky, G.J.; Porte, D.; Figlewicz, D.P.; Jacobson, L.; Beard, J.C.; Ward, W.K.; Dorsa, D. Cerebrospinal Fluid Insulin Levels Increase during Intravenous Insulin Infusions in Man. J. Clin. Endocrinol. Metab. 1987, 64, 190–194. [Google Scholar] [CrossRef]
- Hill, J.M.; Lesniak, M.A.; Pert, C.B.; Roth, J. Autoradiographic Localization of Insulin Receptors in Rat Brain: Prominence in Olfactory and Limbic Areas. Neuroscience 1986, 17, 1127–1138. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin Receptors Are Widely Distributed in the Central Nervous System of the Rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef]
- Steculorum, S.M.; Solas, M.; Brüning, J.C. The Paradox of Neuronal Insulin Action and Resistance in the Development of Aging-Associated Diseases. Alzheimers Dement. 2014, 10, S3–S11. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin Action in Brain Regulates Systemic Metabolism and Brain Function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Vogt, M.C.; Brüning, J.C. CNS Insulin Signaling in the Control of Energy Homeostasis and Glucose Metabolism–From Embryo to Old Age. Trends Endocrinol. Metab. 2013, 24, 76–84. [Google Scholar] [CrossRef]
- Plum, L.; Schubert, M.; Brüning, J.C. The Role of Insulin Receptor Signaling in the Brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef]
- Kitamura, T.; Feng, Y.; Kitamura, Y.I.; Chua, S.C.; Xu, A.W.; Barsh, G.S.; Rossetti, L.; Accili, D. Forkhead Protein FoxO1 Mediates Agrp-Dependent Effects of Leptin on Food Intake. Nat. Med. 2006, 12, 534–540. [Google Scholar] [CrossRef]
- Könner, A.C.; Klöckener, T.; Brüning, J.C. Control of Energy Homeostasis by Insulin and Leptin: Targeting the Arcuate Nucleus and Beyond. Physiol. Behav. 2009, 97, 632–638. [Google Scholar] [CrossRef]
- Könner, A.C.; Janoschek, R.; Plum, L.; Jordan, S.D.; Rother, E.; Ma, X.; Xu, C.; Enriori, P.; Hampel, B.; Barsh, G.S.; et al. Insulin Action in AgRP-Expressing Neurons Is Required for Suppression of Hepatic Glucose Production. Cell Metab. 2007, 5, 438–449. [Google Scholar] [CrossRef]
- Pocai, A.; Lam, T.K.T.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic K(ATP) Channels Control Hepatic Glucose Production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef]
- Ren, H.; Orozco, I.J.; Su, Y.; Suyama, S.; Gutiérrez-Juárez, R.; Horvath, T.L.; Wardlaw, S.L.; Plum, L.; Arancio, O.; Accili, D. FoxO1 Target Gpr17 Activates AgRP Neurons to Regulate Food Intake. Cell 2012, 149, 1314–1326. [Google Scholar] [CrossRef]
- Zhao, W.-Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the Insulin Receptor in Experimental Models of Learning and Memory. Eur. J. Pharm. 2004, 490, 71–81. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Alemán, I. The Many Faces of Insulin-like Peptide Signalling in the Brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Artola, A.; Singer, W. Long-Term Depression of Excitatory Synaptic Transmission and Its Relationship to Long-Term Potentiation. Trends Neurosci. 1993, 16, 480–487. [Google Scholar] [CrossRef]
- Xing, C.; Yin, Y.; Chang, R.; Gong, X.; He, X.; Xie, Z. Effects of Insulin-like Growth Factor 1 on Synaptic Excitability in Cultured Rat Hippocampal Neurons. Exp. Neurol. 2007, 205, 222–229. [Google Scholar] [CrossRef]
- Wan, Q.; Xiong, Z.G.; Man, H.Y.; Ackerley, C.A.; Braunton, J.; Lu, W.Y.; Becker, L.E.; MacDonald, J.F.; Wang, Y.T. Recruitment of Functional GABA(A) Receptors to Postsynaptic Domains by Insulin. Nature 1997, 388, 686–690. [Google Scholar] [CrossRef]
- Zhao, W.-Q.; Townsend, M. Insulin Resistance and Amyloidogenesis as Common Molecular Foundation for Type 2 Diabetes and Alzheimer’s Disease. Biochim. Biophys. Acta 2009, 1792, 482–496. [Google Scholar] [CrossRef]
- Man, H.Y.; Lin, J.W.; Ju, W.H.; Ahmadian, G.; Liu, L.; Becker, L.E.; Sheng, M.; Wang, Y.T. Regulation of AMPA Receptor-Mediated Synaptic Transmission by Clathrin-Dependent Receptor Internalization. Neuron 2000, 25, 649–662. [Google Scholar] [CrossRef]
- Dalton, G.L.; Wang, Y.T.; Floresco, S.B.; Phillips, A.G. Disruption of AMPA Receptor Endocytosis Impairs the Extinction, but Not Acquisition of Learned Fear. Neuropsychopharmacology 2008, 33, 2416–2426. [Google Scholar] [CrossRef]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef]
- Skeberdis, V.A.; Lan, J.; Zheng, X.; Zukin, R.S.; Bennett, M.V. Insulin Promotes Rapid Delivery of N-Methyl-D- Aspartate Receptors to the Cell Surface by Exocytosis. Proc. Natl. Acad. Sci. USA 2001, 98, 3561–3566. [Google Scholar] [CrossRef]
- Christie, J.M.; Wenthold, R.J.; Monaghan, D.T. Insulin Causes a Transient Tyrosine Phosphorylation of NR2A and NR2B NMDA Receptor Subunits in Rat Hippocampus. J. Neurochem. 1999, 72, 1523–1528. [Google Scholar] [CrossRef]
- Chiu, S.-L.; Cline, H.T. Insulin Receptor Signaling in the Development of Neuronal Structure and Function. Neural Dev. 2010, 5, 7. [Google Scholar] [CrossRef]
- Govind, S.; Kozma, R.; Monfries, C.; Lim, L.; Ahmed, S. Cdc42Hs Facilitates Cytoskeletal Reorganization and Neurite Outgrowth by Localizing the 58-KD Insulin Receptor Substrate to Filamentous Actin. J. Cell Biol. 2001, 152, 579–594. [Google Scholar] [CrossRef]
- Choi, J.; Ko, J.; Racz, B.; Burette, A.; Lee, J.-R.; Kim, S.; Na, M.; Lee, H.W.; Kim, K.; Weinberg, R.J.; et al. Regulation of Dendritic Spine Morphogenesis by Insulin Receptor Substrate 53, a Downstream Effector of Rac1 and Cdc42 Small GTPases. J. Neurosci. 2005, 25, 869–879. [Google Scholar] [CrossRef]
- Mielke, J.G.; Taghibiglou, C.; Wang, Y.T. Endogenous Insulin Signaling Protects Cultured Neurons from Oxygen-Glucose Deprivation-Induced Cell Death. Neuroscience 2006, 143, 165–173. [Google Scholar] [CrossRef]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal Insulin Improves Memory in Humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. [Google Scholar] [CrossRef]
- Benedict, C.; Hallschmid, M.; Schmitz, K.; Schultes, B.; Ratter, F.; Fehm, H.L.; Born, J.; Kern, W. Intranasal Insulin Improves Memory in Humans: Superiority of Insulin Aspart. Neuropsychopharmacology 2007, 32, 239–243. [Google Scholar] [CrossRef]
- Kern, W.; Peters, A.; Fruehwald-Schultes, B.; Deininger, E.; Born, J.; Fehm, H.L. Improving Influence of Insulin on Cognitive Functions in Humans. Neuroendocrinology 2001, 74, 270–280. [Google Scholar] [CrossRef]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal Memory Processes Are Modulated by Insulin and High-Fat-Induced Insulin Resistance. Neurobiol. Learn Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Küstermann, E.; et al. Role for Neuronal Insulin Resistance in Neurodegenerative Diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Kim, B.; Feldman, E.L. Chapter 13–Diabetes and Cognitive Dysfunction. In Neurobiology of Brain Disorders; Zigmond, M.J., Rowland, L.P., Coyle, J.T., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 189–201. ISBN 978-0-12-398270-4. [Google Scholar]
- Imamura, T.; Yanagihara, Y.T.; Ohyagi, Y.; Nakamura, N.; Iinuma, K.M.; Yamasaki, R.; Asai, H.; Maeda, M.; Murakami, K.; Irie, K.; et al. Insulin Deficiency Promotes Formation of Toxic Amyloid-Β42 Conformer Co-Aggregating with Hyper-Phosphorylated Tau Oligomer in an Alzheimer’s Disease Model. Neurobiol. Dis. 2020, 137, 104739. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is Hyperinsulinemia the Cart or the Horse? Diabetes Care 2008, 31 (Suppl. S2), S262–S268. [Google Scholar] [CrossRef]
- Kim, S.H.; Reaven, G.M. Insulin Resistance and Hyperinsulinemia: You Can’t Have One without the Other. Diabetes Care 2008, 31, 1433–1438. [Google Scholar] [CrossRef]
- Anand, S.S.; Friedrich, M.G.; Desai, D.; Schulze, K.M.; Awadalla, P.; Busseuil, D.; Dummer, T.J.B.; Jacquemont, S.; Dick, A.; Kelton, D.; et al. Reduced Cognitive Assessment Scores Among Individuals With Magnetic Resonance Imaging-Detected Vascular Brain Injury. Stroke 2020, 51, 1158–1165. [Google Scholar] [CrossRef]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between Insulin Resistance and the Development of Cardiovascular Disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 118. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Folstein, M.F. Insulin, Insulin-Degrading Enzyme and Amyloid-Beta Peptide in Alzheimer’s Disease: Review and Hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; Van Bronswijk, W.; Martins, R. Alzheimer’s Beta-Amyloid Peptides Compete for Insulin Binding to the Insulin Receptor. J. Neurosci. 2002, 22, RC221. [Google Scholar] [CrossRef]
- Toledo, J.B.; Arnold, M.; Kastenmüller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. Metabolic Network Failures in Alzheimer’s Disease: A Biochemical Road Map. Alzheimers Dement. 2017, 13, 965–984. [Google Scholar] [CrossRef]
- Bressler, J.; Yu, B.; Mosley, T.H.; Knopman, D.S.; Gottesman, R.F.; Alonso, A.; Sharrett, A.R.; Wruck, L.M.; Boerwinkle, E. Metabolomics and Cognition in African American Adults in Midlife: The Atherosclerosis Risk in Communities Study. Transl. Psychiatry 2017, 7, e1173. [Google Scholar] [CrossRef]
- Xiang, Q.; Zhang, J.; Li, C.-Y.; Wang, Y.; Zeng, M.-J.; Cai, Z.-X.; Tian, R.-B.; Jia, W.; Li, X.-H. Insulin Resistance-Induced Hyperglycemia Decreased the Activation of Akt/CREB in Hippocampus Neurons: Molecular Evidence for Mechanism of Diabetes-Induced Cognitive Dysfunction. Neuropeptides 2015, 54, 9–15. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ambegaokar, S.S.; Jackson, G.R.; Mudher, A. Insulin-Mediated Changes in Tau Hyperphosphorylation and Autophagy in a Drosophila Model of Tauopathy and Neuroblastoma Cells. Front. Neurosci. 2019, 13, 801. [Google Scholar] [CrossRef]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormö, M.; Nilsson, Y.; Radesäter, A.-C.; Jerning, E.; Markgren, P.-O.; Borgegård, T.; et al. Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-Specific Inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and Insulin-like Growth Factor Expression and Function Deteriorate with Progression of Alzheimer’s Disease: Link to Brain Reductions in Acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Rizzo, M.R.; Di Meo, I.; Polito, R.; Auriemma, M.C.; Gambardella, A.; di Mauro, G.; Capuano, A.; Paolisso, G. Cognitive Impairment and Type 2 Diabetes Mellitus: Focus of SGLT2 Inhibitors Treatment. Pharm. Res. 2022, 176, 106062. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, W.; Xie, J.-W.; Wang, T.; Wang, S.-L.; Teng, W.-P.; Wang, Z.-Y. Insulin Deficiency Exacerbates Cerebral Amyloidosis and Behavioral Deficits in an Alzheimer Transgenic Mouse Model. Mol. Neurodegener. 2010, 5, 46. [Google Scholar] [CrossRef]
- Sędzikowska, A.; Szablewski, L. Insulin and Insulin Resistance in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9987. [Google Scholar] [CrossRef]
- de la Monte, S.M. Contributions of Brain Insulin Resistance and Deficiency in Amyloid-Related Neurodegeneration in Alzheimer’s Disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef]
- Park, J.-C.; Han, S.-H.; Mook-Jung, I. Peripheral Inflammatory Biomarkers in Alzheimer’s Disease: A Brief Review. BMB Rep. 2020, 53, 10–19. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, Insulin Resistance, and the Metabolic Syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef]
- Xie, B.; Waters, M.J.; Schirra, H.J. Investigating Potential Mechanisms of Obesity by Metabolomics. J. Biomed. Biotechnol. 2012, 2012, 805683. [Google Scholar] [CrossRef]
- Shan, Z.; Sun, T.; Huang, H.; Chen, S.; Chen, L.; Luo, C.; Yang, W.; Yang, X.; Yao, P.; Cheng, J.; et al. Association between Microbiota-Dependent Metabolite Trimethylamine-N-Oxide and Type 2 Diabetes. Am. J. Clin. Nutr. 2017, 106, 888–894. [Google Scholar] [CrossRef]
- McEntyre, C.J.; Lever, M.; Chambers, S.T.; George, P.M.; Slow, S.; Elmslie, J.L.; Florkowski, C.M.; Lunt, H.; Krebs, J.D. Variation of Betaine, N,N-Dimethylglycine, Choline, Glycerophosphorylcholine, Taurine and Trimethylamine-N-Oxide in the Plasma and Urine of Overweight People with Type 2 Diabetes over a Two-Year Period. Ann. Clin. Biochem. 2015, 52, 352–360. [Google Scholar] [CrossRef]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through MTORC1. Cell 2018, 175, 947–961.e17. [Google Scholar] [CrossRef]
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Di Somma, C.; Laudisio, D.; Maisto, M.; de Alteriis, G.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine-N-Oxide (TMAO) as Novel Potential Biomarker of Early Predictors of Metabolic Syndrome. Nutrients 2018, 10, 1971. [Google Scholar] [CrossRef]
- Lanz, M.; Janeiro, M.H.; Milagro, F.I.; Puerta, E.; Ludwig, I.A.; Pineda-Lucena, A.; Ramírez, M.J.; Solas, M. Trimethylamine N-Oxide (TMAO) Drives Insulin Resistance and Cognitive Deficiencies in a Senescence Accelerated Mouse Model. Mech. Ageing Dev. 2022, 204, 111668. [Google Scholar] [CrossRef]
- Fujii, Y.; Khasnobish, A.; Morita, H. Relationship between Alzheimer’s Disease and the Human Microbiome. In Alzheimer’s Disease; Wisniewski, T., Ed.; Codon Publications: Brisbane, Australia, 2019; ISBN 978-0-646-80968-7. [Google Scholar]
- Marizzoni, M.; Cattaneo, A.; Mirabelli, P.; Festari, C.; Lopizzo, N.; Nicolosi, V.; Mombelli, E.; Mazzelli, M.; Luongo, D.; Naviglio, D.; et al. Short-Chain Fatty Acids and Lipopolysaccharide as Mediators Between Gut Dysbiosis and Amyloid Pathology in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 78, 683–697. [Google Scholar] [CrossRef]
- Kitta, Y.; Nakamura, T.; Uematsu, M.; Sugamata, W.; Deyama, J.; Fujioka, D.; Saito, Y.; Kawabata, K.; Obata, J.; Kugiyama, K. Insulin Resistance Negatively Affects Long-Term Outcome in Non-Diabetic Patients with Coronary Artery Disease after Therapies to Reduce Atherosclerotic Risk Factors. J. Cardiol. 2013, 62, 348–353. [Google Scholar] [CrossRef]
- Reaven, G.M. Insulin Resistance and Compensatory Hyperinsulinemia: Role in Hypertension, Dyslipidemia, and Coronary Heart Disease. Am. Heart J. 1991, 121, 1283–1288. [Google Scholar] [CrossRef]
- Salonen, J.T.; Lakka, T.A.; Lakka, H.M.; Valkonen, V.P.; Everson, S.A.; Kaplan, G.A. Hyperinsulinemia Is Associated with the Incidence of Hypertension and Dyslipidemia in Middle-Aged Men. Diabetes 1998, 47, 270–275. [Google Scholar] [CrossRef]
- Stern, M.P.; Haffner, S.M. Body Fat Distribution and Hyperinsulinemia as Risk Factors for Diabetes and Cardiovascular Disease. Arteriosclerosis 1986, 6, 123–130. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s Disease Is Type 3 Diabetes-Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- Hardy, J.; de Strooper, B.; Escott-Price, V. Diabetes and Alzheimer’s Disease: Shared Genetic Susceptibility? Lancet Neurol. 2022, 21, 962–964. [Google Scholar] [CrossRef]
- Ewald, C.Y.; Castillo-Quan, J.I.; Blackwell, T.K. Untangling Longevity, Dauer, and Healthspan in Caenorhabditis Elegans Insulin/IGF-1-Signalling. Gerontology 2018, 64, 96–104. [Google Scholar] [CrossRef]
- Dillin, A.; Crawford, D.K.; Kenyon, C. Timing Requirements for Insulin/IGF-1 Signaling in C. Elegans. Science 2002, 298, 830–834. [Google Scholar] [CrossRef]
- Venz, R.; Pekec, T.; Katic, I.; Ciosk, R.; Ewald, C.Y. End-of-Life Targeted Degradation of DAF-2 Insulin/IGF-1 Receptor Promotes Longevity Free from Growth-Related Pathologies. Elife 2021, 10, e71335. [Google Scholar] [CrossRef]
- Mao, K.; Quipildor, G.F.; Tabrizian, T.; Novaj, A.; Guan, F.; Walters, R.O.; Delahaye, F.; Hubbard, G.B.; Ikeno, Y.; Ejima, K.; et al. Late-Life Targeting of the IGF-1 Receptor Improves Healthspan and Lifespan in Female Mice. Nat. Commun. 2018, 9, 2394. [Google Scholar] [CrossRef]
- Selman, C.; Lingard, S.; Choudhury, A.I.; Batterham, R.L.; Claret, M.; Clements, M.; Ramadani, F.; Okkenhaug, K.; Schuster, E.; Blanc, E.; et al. Evidence for Lifespan Extension and Delayed Age-Related Biomarkers in Insulin Receptor Substrate 1 Null Mice. FASEB J. 2008, 22, 807–818. [Google Scholar] [CrossRef]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stöhr, O.; Koch, L.; Köhler, C.; Udelhoven, M.; Leeser, U.; Müller, M.; Kubota, N.; et al. Neuronal IGF-1 Resistance Reduces Abeta Accumulation and Protects against Premature Death in a Model of Alzheimer’s Disease. FASEB J. 2009, 23, 3315–3324. [Google Scholar] [CrossRef]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 Receptor Regulates Lifespan and Resistance to Oxidative Stress in Mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Killick, R.; Scales, G.; Leroy, K.; Causevic, M.; Hooper, C.; Irvine, E.E.; Choudhury, A.I.; Drinkwater, L.; Kerr, F.; Al-Qassab, H.; et al. Deletion of Irs2 Reduces Amyloid Deposition and Rescues Behavioural Deficits in APP Transgenic Mice. Biochem. Biophys. Res. Commun. 2009, 386, 257–262. [Google Scholar] [CrossRef]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte Contribution to Dysfunction, Risk and Progression in Neurodegenerative Disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.-X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Rodriguez, R.D.; Suemoto, C.K.; et al. Molecular Characterization of Selectively Vulnerable Neurons in Alzheimer’s Disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-Associated Astrocytes in Alzheimer’s Disease and Aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J. Neurosci. 2016, 36, 3453–3470. [Google Scholar] [CrossRef]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic Alterations in Parkinson’s Disease Astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef]
- Beach, T.G.; McGeer, E.G. Lamina-Specific Arrangement of Astrocytic Gliosis and Senile Plaques in Alzheimer’s Disease Visual Cortex. Brain Res. 1988, 463, 357–361. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Hoshi, A.; Yamamoto, T.; Shimizu, K.; Ugawa, Y.; Nishizawa, M.; Takahashi, H.; Kakita, A. Characteristics of Aquaporin Expression Surrounding Senile Plaques and Cerebral Amyloid Angiopathy in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2012, 71, 750–759. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of Aquaporin-4 in APP/PS1 Mice Exacerbates Brain Aβ Accumulation and Memory Deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Ungerer, N. Cholesterol Metabolism in Neurons and Astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Bindocci, E.; Savtchouk, I.; Liaudet, N.; Becker, D.; Carriero, G.; Volterra, A. Three-Dimensional Ca2+ Imaging Advances Understanding of Astrocyte Biology. Science 2017, 356, eaai8185. [Google Scholar] [CrossRef]
- Paumier, A.; Boisseau, S.; Jacquier-Sarlin, M.; Pernet-Gallay, K.; Buisson, A.; Albrieux, M. Astrocyte-Neuron Interplay Is Critical for Alzheimer’s Disease Pathogenesis and Is Rescued by TRPA1 Channel Blockade. Brain 2022, 145, 388–405. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate Uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-Mediated Excitotoxicity and Neurodegeneration in Alzheimer’s Disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ Induces Astrocytic Glutamate Release, Extrasynaptic NMDA Receptor Activation, and Synaptic Loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus Extrasynaptic NMDA Receptor Signalling: Implications for Neurodegenerative Disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Kowalewski, J.M.; Renner, M.; Bousset, L.; Koulakoff, A.; Melki, R.; Giaume, C.; Triller, A. β-Amyloid and ATP-Induced Diffusional Trapping of Astrocyte and Neuronal Metabotropic Glutamate Type-5 Receptors. Glia 2013, 61, 1673–1686. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from Reactive Astrocytes Impairs Memory in Mouse Models of Alzheimer’s Disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Cai, W.; Xue, C.; Sakaguchi, M.; Konishi, M.; Shirazian, A.; Ferris, H.A.; Li, M.E.; Yu, R.; Kleinridders, A.; Pothos, E.N.; et al. Insulin Regulates Astrocyte Gliotransmission and Modulates Behavior. J. Clin. Investig. 2018, 128, 2914–2926. [Google Scholar] [CrossRef]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin Resistance in Brain Alters Dopamine Turnover and Causes Behavioral Disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef]
- Shin, A.C.; Filatova, N.; Lindtner, C.; Chi, T.; Degann, S.; Oberlin, D.; Buettner, C. Insulin Receptor Signaling in POMC, but Not AgRP, Neurons Controls Adipose Tissue Insulin Action. Diabetes 2017, 66, 1560–1571. [Google Scholar] [CrossRef]
- Hascup, E.R.; Broderick, S.O.; Russell, M.K.; Fang, Y.; Bartke, A.; Boger, H.A.; Hascup, K.N. Diet-Induced Insulin Resistance Elevates Hippocampal Glutamate as Well as VGLUT1 and GFAP Expression in AβPP/PS1 Mice. J. Neurochem. 2019, 148, 219–237. [Google Scholar] [CrossRef]
- Rajasekar, N.; Dwivedi, S.; Nath, C.; Hanif, K.; Shukla, R. Protection of Streptozotocin Induced Insulin Receptor Dysfunction, Neuroinflammation and Amyloidogenesis in Astrocytes by Insulin. Neuropharmacology 2014, 86, 337–352. [Google Scholar] [CrossRef]
- Pitt, J.; Wilcox, K.C.; Tortelli, V.; Diniz, L.P.; Oliveira, M.S.; Dobbins, C.; Yu, X.-W.; Nandamuri, S.; Gomes, F.C.A.; DiNunno, N.; et al. Neuroprotective Astrocyte-Derived Insulin/Insulin-like Growth Factor 1 Stimulates Endocytic Processing and Extracellular Release of Neuron-Bound Aβ Oligomers. Mol. Biol. Cell 2017, 28, 2623–2636. [Google Scholar] [CrossRef]
- González-García, I.; Gruber, T.; García-Cáceres, C. Insulin Action on Astrocytes: From Energy Homeostasis to Behaviour. J. Neuroendocr. 2021, 33, e12953. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezkurdia, A.; Ramírez, M.J.; Solas, M. Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: A Focus on Insulin Resistance. Int. J. Mol. Sci. 2023, 24, 4354. https://doi.org/10.3390/ijms24054354
Ezkurdia A, Ramírez MJ, Solas M. Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: A Focus on Insulin Resistance. International Journal of Molecular Sciences. 2023; 24(5):4354. https://doi.org/10.3390/ijms24054354
Chicago/Turabian StyleEzkurdia, Amaia, María J. Ramírez, and Maite Solas. 2023. "Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: A Focus on Insulin Resistance" International Journal of Molecular Sciences 24, no. 5: 4354. https://doi.org/10.3390/ijms24054354
APA StyleEzkurdia, A., Ramírez, M. J., & Solas, M. (2023). Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: A Focus on Insulin Resistance. International Journal of Molecular Sciences, 24(5), 4354. https://doi.org/10.3390/ijms24054354