

The Role of Bacteria–Mitochondria Communication in the Activation of Neuronal Innate Immunity: Implications to Parkinson’s Disease
 ,
, 

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Bacteria-Mediated Neuronal Innate Immunity Activation: The Role of α-Syn
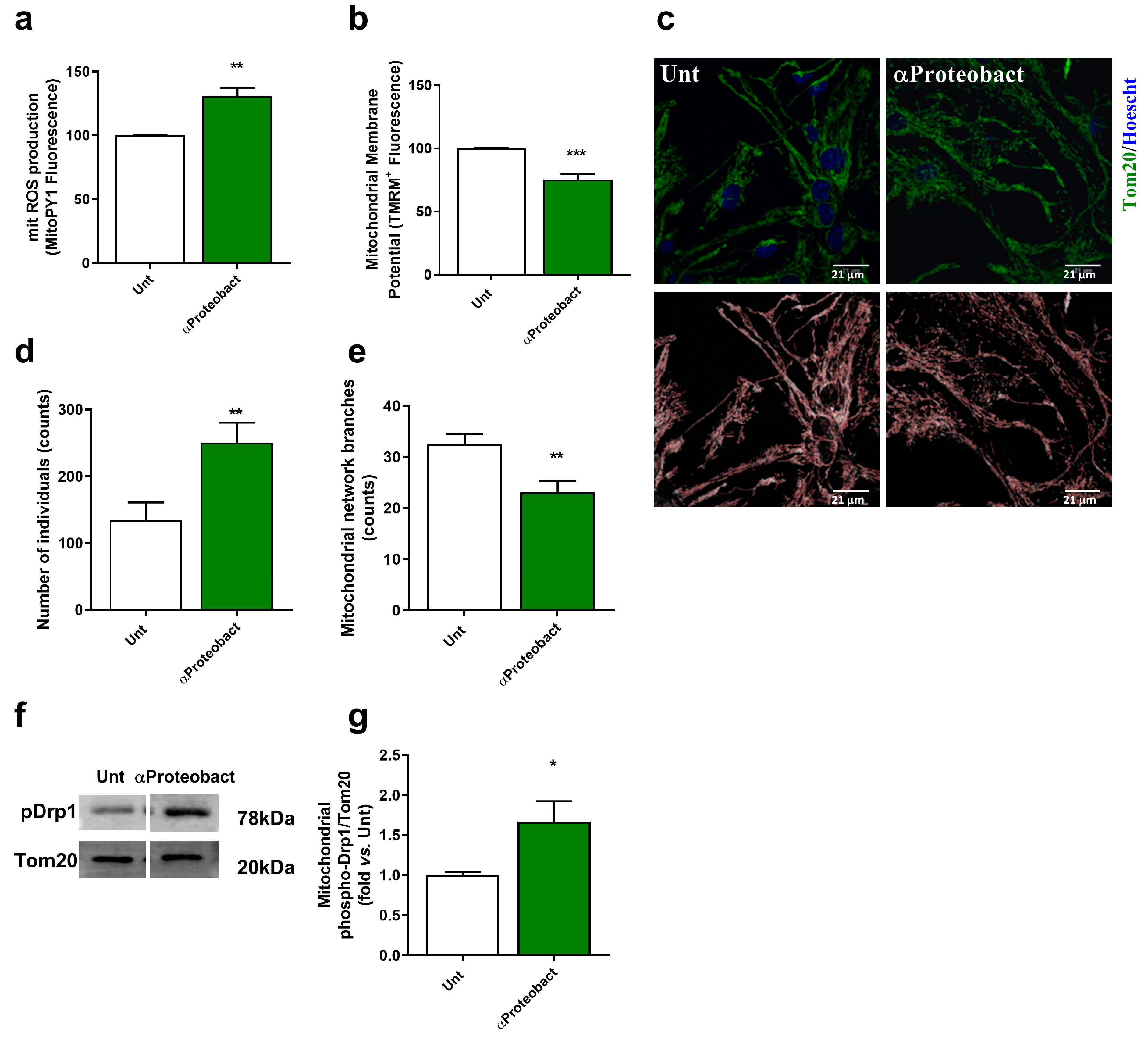
2.2. Mitochondrial Dysfunction: A Positive Feedback Loop to Potentiate Innate Immunity Activation
3. Discussions
4. Materials and Methods
4.1. Primary Mesencephalic Cultures Preparation and Treatments
4.2. Bacterial Strain, Culture Conditions and Treatment
4.3. Cellular Extracts Preparation
4.4. Western Blotting
4.5. Immunocytochemistry and Confocal Microscopy Analysis
4.6. Evaluation of Mitochondrial Membrane Potential (Δψm)
4.7. Determination of Mitochondrial-Derived Reactive Oxygen Species
4.8. Caspase-1 Activity Assay
4.9. Inflammatory Markers and α-Syn Oligomers Determination by ELISA
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Menšíková, K.; Matěj, R.; Colosimo, C.; Rosales, R.; Tučková, L.; Ehrmann, J.; Hraboš, D.; Kolaříková, K.; Vodička, R.; Vrtěl, R.; et al. Lewy body disease or diseases with Lewy bodies? NPJ Park. Dis. 2022, 8, 3. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M. The Mitochondrial Cascade Hypothesis for Parkinsons Disease. Curr. Pharm. Des. 2011, 17, 3390–3397. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; McGeer, E.G.; McGeer, P.L. Therapeutic approaches to inflammation in neurodegenerative disease. Curr. Opin. Neurol. 2007, 20, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Empadinhas, N. The Microbiome-Mitochondria Dance in Prodromal Parkinson’s Disease. Front. Physiol. 2018, 9, 471. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s Disease Brain Mitochondrial Complex I Has Oxidatively Damaged Subunits and Is Functionally Impaired and Misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef]
- Li, J.-L.; Lin, T.-Y.; Chen, P.-L.; Guo, T.-N.; Huang, S.-Y.; Chen, C.-H.; Lin, C.-H.; Chan, C.-C. Mitochondrial Function and Parkinson’s Disease: From the Perspective of the Electron Transport Chain. Front. Mol. Neurosci. 2021, 14, 797833. [Google Scholar] [CrossRef]
- Esteves, A.R.; Munoz-Pinto, M.F.; Nunes-Costa, D.; Candeias, E.; Silva, D.F.; Magalhães, J.D.; Pereira-Santos, A.R.; Ferreira, I.L.; Alarico, S.; Tiago, I.; et al. Footprints of a microbial toxin from the gut microbiome to mesencephalic mitochondria. Gut 2021, 72, 73–89. [Google Scholar] [CrossRef]
- Won, J.-H.; Park, S.; Hong, S.; Son, S.; Yu, J.-W. Rotenone-induced Impairment of Mitochondrial Electron Transport Chain Confers a Selective Priming Signal for NLRP3 Inflammasome Activation. J. Biol. Chem. 2015, 290, 27425–27437. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduíno, D.M.; Silva, D.F.; Viana, S.D.; Pereira, F.C.; Cardoso, S.M. Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 1440–1462. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Chen, M.; Du, T.; Duan, C.; Gao, G.; Yang, H. The novel mechanism of rotenone-induced α-synuclein phosphorylation via reduced protein phosphatase 2A activity. Int. J. Biochem. Cell Biol. 2016, 75, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Lach, B.; Grimes, D.; Benoit, B.; Minkiewicz-Janda, A. Caudate nucleus pathology in Parkinson’s disease: Ultrastructural and biochemical findings in biopsy material. Acta Neuropathol. 1992, 83, 352–360. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, C.; Huang, J.; Liu, W.; Lai, W.; Leng, F.; Tang, Q.; Liu, Y.; Wang, Q.; Zhou, M.; et al. ROCK1 induces dopaminergic nerve cell apoptosis via the activation of Drp1-mediated aberrant mitochondrial fission in Parkinson’s disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Gegg, M.E.; Cooper, J.M.; Chau, K.-Y.; Rojo, M.; Schapira, A.H.; Taanman, J.-W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Gegg, M.E.; Schapira, A.H. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson disease pathogenesis. Autophagy 2011, 7, 243–245. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.M.; Pereira-Santos, A.R.; Esteves, A.R.; Cardoso, S.M.; Empadinhas, N. The Mitochondrial Ribosome: A World of Opportunities for Mitochondrial Dysfunction Toward Parkinson’s Disease. Antioxidants Redox Signal. 2021, 34, 694–711. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.; Marzetti, E. Cell Death and Inflammation: The Role of Mitochondria in Health and Disease. Cells 2021, 10, 537. [Google Scholar] [CrossRef]
- Martijn, J.; Vosseberg, J.; Guy, L.; Offre, P.; Ettema, T.J.G. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature 2018, 557, 101–105. [Google Scholar] [CrossRef]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef]
- Behzadi, P.; García-Perdomo, H.A.; Karpiński, T.M. Toll-Like Receptors: General Molecular and Structural Biology. J. Immunol. Res. 2021, 2021, 9914854. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Dzamko, N.; Gysbers, A.; Perera, G.; Bahar, A.; Shankar, A.; Gao, J.; Fu, Y.; Halliday, G.M. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017, 133, 303–319. [Google Scholar] [CrossRef]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef] [PubMed]
- Rietdijk, C.D.; Van Wezel, R.J.A.; Garssen, J.; Kraneveld, A.D. Neuronal toll-like receptors and neuro-immunity in Parkinson’s disease, Alzheimer’s disease and stroke. Neuroimmunol. Neuroinflammation 2016, 3, 27–37. [Google Scholar] [CrossRef]
- Campolo, M.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain, Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef]
- Otani, K.; Shichita, T. Cerebral sterile inflammation in neurodegenerative diseases. Inflamm. Regen. 2020, 40, 28. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.F.; Candeias, E.; Esteves, A.R.; Magalhães, J.D.; Ferreira, I.L.; Nunes-Costa, D.; Rego, A.C.; Empadinhas, N.; Cardoso, S.M. Microbial BMAA elicits mitochondrial dysfunction, innate immunity activation, and Alzheimer’s disease features in cortical neurons. J. Neuroinflamm. 2020, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Clayton, R.; Clark, J.B.; Sharpe, M. Cytochrome c release from rat brain mitochondria is proportional to the mitochondrial functional deficit: Implications for apoptosis and neurodegenerative disease. J. Neurochem. 2005, 92, 840–849. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Bajwa, E.; Klegeris, A. Cytochrome c can be released into extracellular space and modulate functions of human astrocytes in a toll-like receptor 4-dependent manner. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 129400. [Google Scholar] [CrossRef]
- Hertz, C.J.; Wu, Q.; Porter, E.M.; Zhang, Y.J.; Weismüller, K.-H.; Godowski, P.J.; Ganz, T.; Randell, S.H.; Modlin, R.L. Activation of Toll-Like Receptor 2 on Human Tracheobronchial Epithelial Cells Induces the Antimicrobial Peptide Human β Defensin-2. J. Immunol. 2003, 171, 6820–6826. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Schenk, M.; Walker, V.P.; Dempsey, P.W.; Kanchanapoomi, M.; Wheelwright, M.; Vazirnia, A.; Zhang, X.; Steinmeyer, A.; Zügel, U.; et al. Convergence of IL-1β and VDR Activation Pathways in Human TLR2/1-Induced Antimicrobial Responses. PLoS ONE 2009, 4, e5810. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 2019, 68, 829–843. [Google Scholar] [CrossRef]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Koppel, S.J.; Weidling, I.W.; Roy, N.; Ryan, L.N.; Stanford, J.A.; Swerdlow, R.H. Extracellular Mitochondria and Mitochondrial Components Act as Damage-Associated Molecular Pattern Molecules in the Mouse Brain. J. Neuroimmune Pharmacol. 2016, 11, 622–628. [Google Scholar] [CrossRef]
- Kouli, A.; Horne, C.; Williams-Gray, C. Toll-like receptors and their therapeutic potential in Parkinson’s disease and α-synucleinopathies. Brain Behav. Immun. 2019, 81, 41–51. [Google Scholar] [CrossRef]
- Cain, M.D.; Salimi, H.; Diamond, M.S.; Klein, R.S. Mechanisms of Pathogen Invasion into the Central Nervous System. Neuron 2019, 103, 771–783. [Google Scholar] [CrossRef]
- Munoz-Pinto, M.F.; Empadinhas, N.; Cardoso, S.M. The neuromicrobiology of Parkinson’s disease: A unifying theory. Ageing Res. Rev. 2021, 70, 101396. [Google Scholar] [CrossRef]
- Bi, D.; Wang, Y.; Gao, Y.; Li, X.; Chu, Q.; Cui, J.; Xu, T. Recognition of Lipopolysaccharide and Activation of NF-κB by Cytosolic Sensor NOD1 in Teleost Fish. Front. Immunol. 2018, 9, 1413. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.W.; Zhang, Y.; Herrup, K. Non-Neuronal Cells Are Required to Mediate the Effects of Neuroinflammation: Results from a Neuron-Enriched Culture System. PLoS ONE 2016, 11, e0147134. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Alam, M.; Yang, D.; Li, X.-Q.; Liu, J.; Back, T.C.; Trivett, A.; Karim, B.; Barbut, D.; Zasloff, M.; Oppenheim, J.J. Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep. 2022, 38, 110090. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduíno, D.M.; Silva, D.F.F.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial Dysfunction: The Road to Alpha-Synuclein Oligomerization in PD. Park. Dis. 2011, 2011, 693761. [Google Scholar] [CrossRef]
- Park, J.-H.; Burgess, J.D.; Faroqi, A.H.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 2020, 15, 5. [Google Scholar] [CrossRef]
- Minakaki, G.; Krainc, D.; Burbulla, L.F. The Convergence of Alpha-Synuclein, Mitochondrial, and Lysosomal Pathways in Vulnerability of Midbrain Dopaminergic Neurons in Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 580634. [Google Scholar] [CrossRef]
- Magalhães, J.D.; Fão, L.; Vilaça, R.; Cardoso, S.M.; Rego, A.C. Macroautophagy and Mitophagy in Neurodegenerative Disorders: Focus on Therapeutic Interventions. Biomedicines 2021, 9, 1625. [Google Scholar] [CrossRef]
- Ettema, T.J.; Andersson, S.G. The α-proteobacteria: The Darwin finches of the bacterial world. Biol. Lett. 2009, 5, 429–432. [Google Scholar] [CrossRef]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef]
- Rongvaux, A. Innate immunity and tolerance toward mitochondria. Mitochondrion 2017, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Kaul, D.; Habbel, P.; Derkow, K.; Krüger, C.; Franzoni, E.; Wulczyn, F.G.; Bereswill, S.; Nitsch, R.; Schott, E.; Veh, R.; et al. Expression of Toll-Like Receptors in the Developing Brain. PLoS ONE 2012, 7, e37767. [Google Scholar] [CrossRef]
- Bowyer, J.F.; Sarkar, S.; Burks, S.M.; Hess, J.N.; Tolani, S.; O’Callaghan, J.P.; Hanig, J.P. Microglial activation and responses to vasculature that result from an acute LPS exposure. Neurotoxicology 2020, 77, 181–192. [Google Scholar] [CrossRef]
- Heidari, A.; Yazdanpanah, N.; Rezaei, N. The role of Toll-like receptors and neuroinflammation in Parkinson’s disease. J. Neuroinflamm. 2022, 19, 135. [Google Scholar] [CrossRef]
- Kim, R.; Kim, H.-J.; Kim, A.; Jang, M.; Kim, A.; Kim, Y.; Yoo, D.; Im, J.H.; Choi, J.-H.; Jeon, B. Peripheral blood inflammatory markers in early Parkinson’s disease. J. Clin. Neurosci. 2018, 58, 30–33. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chen, C.-C.; Chiang, H.-L.; Liou, J.-M.; Chang, C.-M.; Lu, T.-P.; Chuang, E.Y.; Tai, Y.-C.; Cheng, C.; Lin, H.-Y.; et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflamm. 2019, 16, 129. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Hunot, S.; Dugas, N.; Faucheux, B.; Hartmann, A.; Tardieu, M.; Debré, P.; Agid, Y.; Dugas, B.; Hirsch, E.C. FcεRII/CD23 Is Expressed in Parkinson’s Disease and Induces, In Vitro, Production of Nitric Oxide and Tumor Necrosis Factor-α in Glial Cells. J. Neurosci. 1999, 19, 3440–3447. [Google Scholar] [CrossRef]
- Zimmermann, M.; Brockmann, K. Blood and Cerebrospinal Fluid Biomarkers of Inflammation in Parkinson’s Disease. J. Park. Dis. 2022, 12, S183–S200. [Google Scholar] [CrossRef]
- Arena, G.; Sharma, K.; Agyeah, G.; Krüger, R.; Grünewald, A.; Fitzgerald, J.C. Neurodegeneration and Neuroinflammation in Parkinson’s Disease: A Self-Sustained Loop. Curr. Neurol. Neurosci. Rep. 2022, 22, 427–440. [Google Scholar] [CrossRef]
- Rajendran, M.; Queralt-Martín, M.; Gurnev, P.A.; Rosencrans, W.M.; Rovini, A.; Jacobs, D.; Abrantes, K.; Hoogerheide, D.P.; Bezrukov, S.M.; Rostovtseva, T.K. Restricting α-synuclein transport into mitochondria by inhibition of α-synuclein–VDAC complexation as a potential therapeutic target for Parkinson’s disease treatment. Cell Mol. Life Sci. 2022, 79, 368. [Google Scholar] [CrossRef]
- Flønes, I.H.; Nyland, H.; Sandnes, D.-A.; Alves, G.W.; Tysnes, O.-B.; Tzoulis, C. Early Forms of α-Synuclein Pathology Are Associated with Neuronal Complex I Deficiency in the Substantia Nigra of Individuals with Parkinson’s Disease. Biomolecules 2022, 12, 747. [Google Scholar] [CrossRef]
- Santos, M.C.J.; Esteves, A.R.; Silva, D.F.; Januário, C.; Cardoso, S.M. The Impact of Mitochondrial Fusion and Fission Modulation in Sporadic Parkinson’s Disease. Mol. Neurobiol. 2015, 52, 573–586. [Google Scholar] [CrossRef]
- Arduíno, D.M.; Esteves, A.R.; Cardoso, S.M. Mitochondria drive autophagy pathology via microtubule disassembly: A new hypothesis for Parkinson disease. Autophagy 2013, 9, 112–114. [Google Scholar] [CrossRef]
- Chung, L.Y.-R.; Lin, Y.-T.; Liu, C.; Tai, Y.-C.; Lin, H.-Y.; Lin, C.-H.; Chen, C.-C. Neuroinflammation Upregulated Neuronal Toll-Like Receptors 2 and 4 to Drive Synucleinopathy in Neurodegeneration. Front. Pharmacol. 2022, 13, 845930. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef]
- Scheiblich, H.; Dansokho, C.; Mercan, D.; Schmidt, S.V.; Bousset, L.; Wischhof, L.; Eikens, F.; Odainic, A.; Spitzer, J.; Griep, A.; et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 2021, 184, 5089–5106.e21. [Google Scholar] [CrossRef]
- Liang, Y.; Cui, L.; Gao, J.; Zhu, M.; Zhang, Y.; Zhang, H.-L. Gut Microbial Metabolites in Parkinson’s Disease: Implications of Mitochondrial Dysfunction in the Pathogenesis and Treatment. Mol. Neurobiol. 2021, 58, 3745–3758. [Google Scholar] [CrossRef]
- Elkjaer, M.L.; Simon, L.; Frisch, T.; Bente, L.-M.; Kacprowski, T.; Thomassen, M.; Reynolds, R.; Baumbach, J.; Röttger, R.; Illes, Z. Hypothesis of a potential BrainBiota and its relation to CNS autoimmune inflammation. Front. Immunol. 2022, 13, 1043579. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-M.; Liu, B.; Zhang, W.; Hong, J.-S. Synergistic dopaminergic neurotoxicity of MPTP and inflammogen lipopolysaccharide: Relevance to the etiology of Parkinson’s disease. FASEB J. 2003, 17, 1957–1959. [Google Scholar] [CrossRef] [PubMed]
- Arduíno, D.M.; Esteves, A.R.; Cortes, L.; Silva, D.F.; Patel, B.; Grazina, M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial metabolism in Parkinson’s disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum. Mol. Genet. 2012, 21, 4680–4702. [Google Scholar] [CrossRef] [PubMed]
- Scaduto, R.C.; Grotyohann, L.W. Measurement of Mitochondrial Membrane Potential Using Fluorescent Rhodamine Derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhães, J.D.; Esteves, A.R.; Candeias, E.; Silva, D.F.; Empadinhas, N.; Cardoso, S.M. The Role of Bacteria–Mitochondria Communication in the Activation of Neuronal Innate Immunity: Implications to Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 4339. https://doi.org/10.3390/ijms24054339
Magalhães JD, Esteves AR, Candeias E, Silva DF, Empadinhas N, Cardoso SM. The Role of Bacteria–Mitochondria Communication in the Activation of Neuronal Innate Immunity: Implications to Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(5):4339. https://doi.org/10.3390/ijms24054339
Chicago/Turabian StyleMagalhães, João D., Ana Raquel Esteves, Emanuel Candeias, Diana F. Silva, Nuno Empadinhas, and Sandra Morais Cardoso. 2023. "The Role of Bacteria–Mitochondria Communication in the Activation of Neuronal Innate Immunity: Implications to Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 5: 4339. https://doi.org/10.3390/ijms24054339
APA StyleMagalhães, J. D., Esteves, A. R., Candeias, E., Silva, D. F., Empadinhas, N., & Cardoso, S. M. (2023). The Role of Bacteria–Mitochondria Communication in the Activation of Neuronal Innate Immunity: Implications to Parkinson’s Disease. International Journal of Molecular Sciences, 24(5), 4339. https://doi.org/10.3390/ijms24054339