
The Landscape of Point Mutations in Human Protein Coding Genes Leading to Pregnancy Loss

 , , , ,
, , , , 
Abstract
:1. Introduction: Miscarriage and Genetics
2. Genetic Research into Pregnancy Loss: The Evolution of Methods
3. Next Generation Sequencing in the Analysis of Pregnancy Loss Genetics
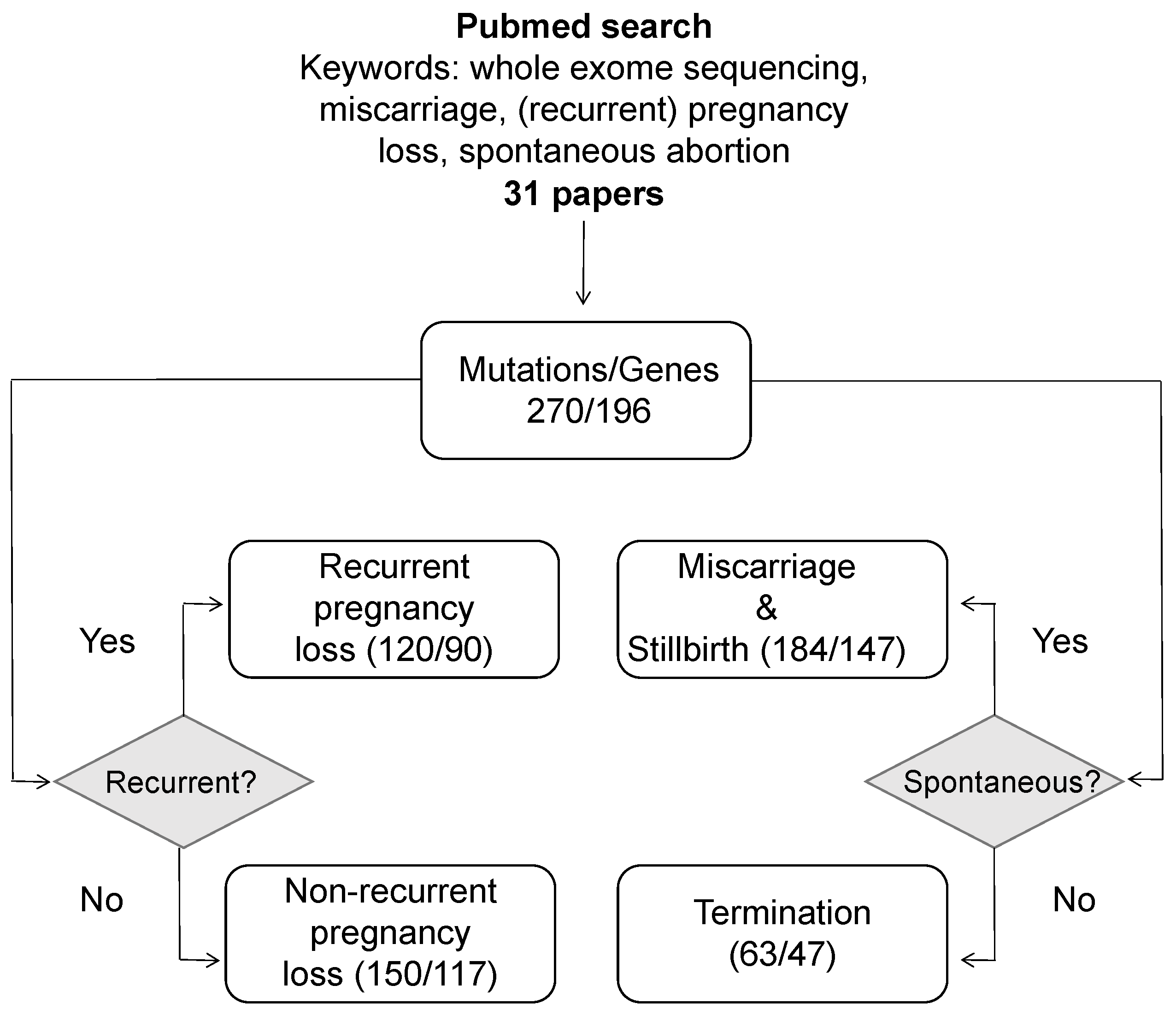
4. A Systematic Review of Pregnancy Loss Genes

{kind=link}
{kind=link}
{kind=link}
| Gene (Locus) | Associated Diseases | Variant | Variant Origin † | Pregnancy Outcome | Reference |
|---|---|---|---|---|---|
| PADI6 chr1p36.13 | OMIM:617234 | NM_207421.4:c.1793A>G (p.Asn598Ser) NM_207421.4:c.2045G>A (p.Arg682Gln) | Inherited | Miscarriage | [55] |
| NM_207421:c.122C>T (p.Ala41Val) | n.a. | Miscarriage | [49] | ||
| STIL chr1p33 | OMIM:612703 | NM_001048166.1:c.1231C>G (p.His411Asp) NM_001048166.1:c.3370A>G (p.Met1124Val) | uncertain | Miscarriage | [65] |
| NM_001048166:c. 1012C>T (p.His338Tyr) | Inherited | Miscarriage | [51] | ||
| DYNC2H1 chr11q22.3 | OMIM:613091 | NM_001080463.1:c.2819-14A4G | Inherited | Termination | [46] |
| NM_001080463.1:c.7577T4G (p. Ile2526Ser) | Inherited | Termination | [46] | ||
| NM_001377.3:c.6047A>G (p.Tyr2016Cys) NM_001377.3:c.6551A>T (p.Asp2184Val) | Inherited | Miscarriage | [33] | ||
| FGFR2 chr10q26.13 | 14 conditions | NM_000141:c.940-1G>A | n.a. | Fetal demise | [48] |
| NM_022970.3:c.764G>A (p.Arg255Gln) | Inherited; | Neonatal death | [34] | ||
| NM_022970.3:c.758C>G (p.Pro253Arg) | de novo | Termination | [34] | ||
| FGFR3 chr4p16.3 | 14 conditions | NM_000142:c.1537G>T (p.Asp513Tyr) | n.a. | Miscarriage | [52] |
| NM_000142:c.742C>T (p.Arg248Cys) | n.a. | Miscarriage | [52] | ||
| NM_000142:c.1118A>G (p.Tyr373Cys) | n.a. | Termination | [26] | ||
| FRAS1 chr4q21.21 | OMIM:219000 | NM_025074:c.8537C>A (p.Ala2846Asp) | Inherited | Miscarriage | [51] |
| NM_025074.7:c.1918C>T (p.Arg640Cys) NM_025074.7:c.5205C>A (p.His1735Gln) | n.a. | Termination | [26] | ||
| GREB1L chr18q11.1-q11.2 | OMIM:619274 | NM_001142966.2:c.5614dupA (p.Thr1872Asnfs*) | de novo | Termination | [34] |
| OMIM:617805 | NM_001142966:c.1305dupA (p.Asp436Argfs*32) | n.a. | Miscarriage | [52] | |
| LZTR1 chr22q11.21 | OMIM:616564 | ENST00000215739.8:c.902G>T (p.Gly301Val) | de novo | Termination | [34] |
| OMIM:605275 | NM_006767:c.2317G>A (p.Val773Met) | n.a. | Miscarriage | [72] | |
| PIEZO1 chr16q24.3 | OMIM:194380 | NM_001142864:c.1264C>T (p.Gln422Ter) | uncertain | Miscarriage | [72] |
| OMIM:616843 | NM_001142864:c.2035G>T (p.Glu679X) | uncertain | Termination | [48] | |
| NM_001142864.3:c.3206G>A (p.Trp1069Ter) NM_001142864.3:c.6208A>C (p.Lys2070Gln) | Inherited | Termination | [73] | ||
| NM_001142864:c.30_31delAC (p.Leu10fs) | uncertain | Miscarriage | [51] | ||
| PIK3R2 chr19p13.11 | OMIM:603387 | NM_005027:c.1117G>A (p.Gly373Arg) | n.a. | Fetal demise | [52] |
| NM_005027:c.1690A>G (p.Lys564Glu) | n.a. | Miscarriage | [48] | ||
| PTPN11 chr12q24.13 | 4 conditions | NM_002834:c.174C>A (p.Asn58Lys) | n.a. | Fetal demise | [48] |
| NM_002834.4:c.218C>T (p.Thr73Ile) | de novo | Neonatal death | [34] | ||
| COL2A1 chr12q13.11 | 15 conditions | NM_001844.5:c.3864_3865delCT (p.Cys1289Pfs*) | Inherited | Termination | [34] |
| NM_001844.5:c.3490G>T (p.Gly1164Cys) | de novo | Miscarriage | [26] | ||
| FOXP3 chrXp11.23 | OMIM:304790 | NM_014009.3:c.1009C>T (p.Arg337Ter) | Inherited | RPL | [74] |
| NM_014009.3:c.906delT (p.Asp303fs*87) | Inherited | Fetal death | [75] | ||
| NM_014009.3:c.1009C>T (p.Arg337X) | Inherited | Miscarriage | [76] | ||
| NM_014009.3:c.1033C>T (p.Leu345Phe) | Inherited | Miscarriage | [77] | ||
| NM_014009.3:c.1189CNT (p.Arg397Trp) | Inherited | Miscarriage | [78] | ||
| NM_014009.3:c.319_320delTC | Inherited | Miscarriage | [78] | ||
| NEB chr2q23.3 | OMIM:619334 | NM_001164507:c.20974delA (p.Val6993Serfs*8) | uncertain | Miscarriage | [72] |
| OMIM:256030 | NM_001271208:c.24094C>T (p.Arg8032Ter) NM_001271208:c.20098C>A (p.Leu6700Ile) | uncertain | Miscarriage | [52] | |
| RYR1 chr19q13.2 | 4 conditions | NM_000540.2:c.14130-2A>G NM_000540.2:c.9221C>T (p.Ser3074Phe) | Inherited | Termination | [46] |
| NM_000540.2:c.6721C>T (p.Arg2241Ter) | Inherited | Miscarriage | [79] | ||
| NM_000540.2:c.2097_2123del (p.Glu699_Gly707del) | Inherited | Termination | [79] | ||
| NM_000540.2:c.7043delGA (p.Glu2347del) | Inherited | Termination | [79] | ||
| RYR2 chr1q43 | OMIM:115000 OMIM:115000 | NM_001035.2:c.409C>T (p.Arg137Trp) NM_001035.2:c.4652A>G (p.Asn1551Ser) | Inherited | RPL | [39] |
| NM_001035.2:c.12526G>A (p.Val4176Met) | Inherited | Stillbirth | [34] | ||
| SCN5A chr3p22.2 | 9 conditions | NM_001160161:c.3749C>T (p.Thr1250Met) | Inherited | Miscarriage | [51] |
| NM_198056:c.5393G>A (p.Trp1798Ter) | n.a. | Fetal demise | [52] | ||
| NM_198056.3:c.1663G>T (p.Glu555Ter) | n.a. | Stillbirth | [50] | ||
| NM_198056.3:c.1858C>T (p.Arg620Cys) | n.a. | Stillbirth | [50] | ||
| NM_198056.3:c.5350G>A (p.Glu1784Lys) | n.a. | Stillbirth | [50] | ||
| GBE1 chr3p12.2 | OMIM: 232500 OMIM:263570 | NM_000158:c.467G>A (p.Arg156His) NM_000158:c.-35_-54del | uncertain | Miscarriage | [51] |
| NM_000158:c.1064G>A (p.Arg355His) | Inherited | Fetal demise | [47] | ||
| NM_000158:c.1543C>T (p.Arg515Cys) | Inherited | Fetal demise | [47] |
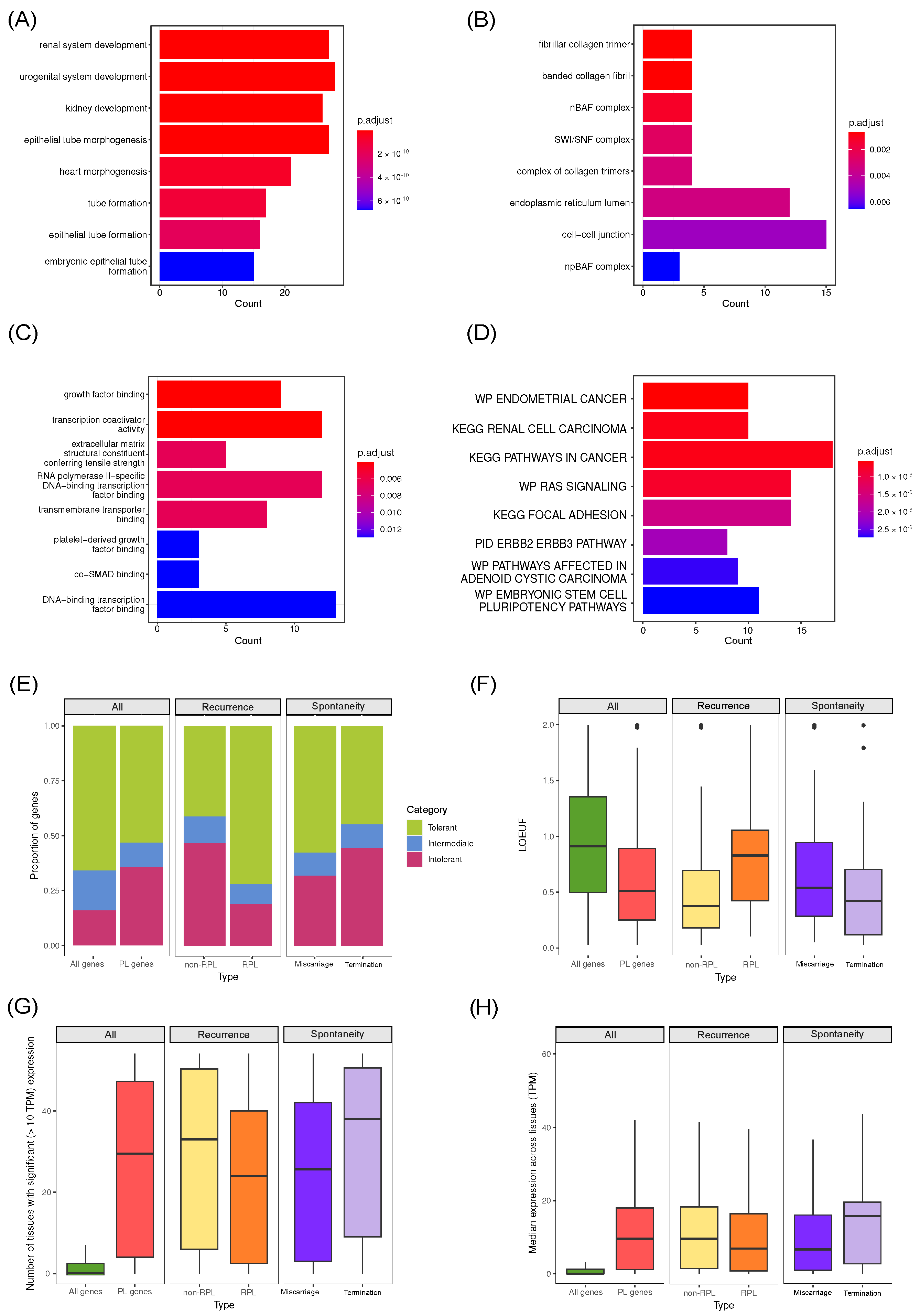
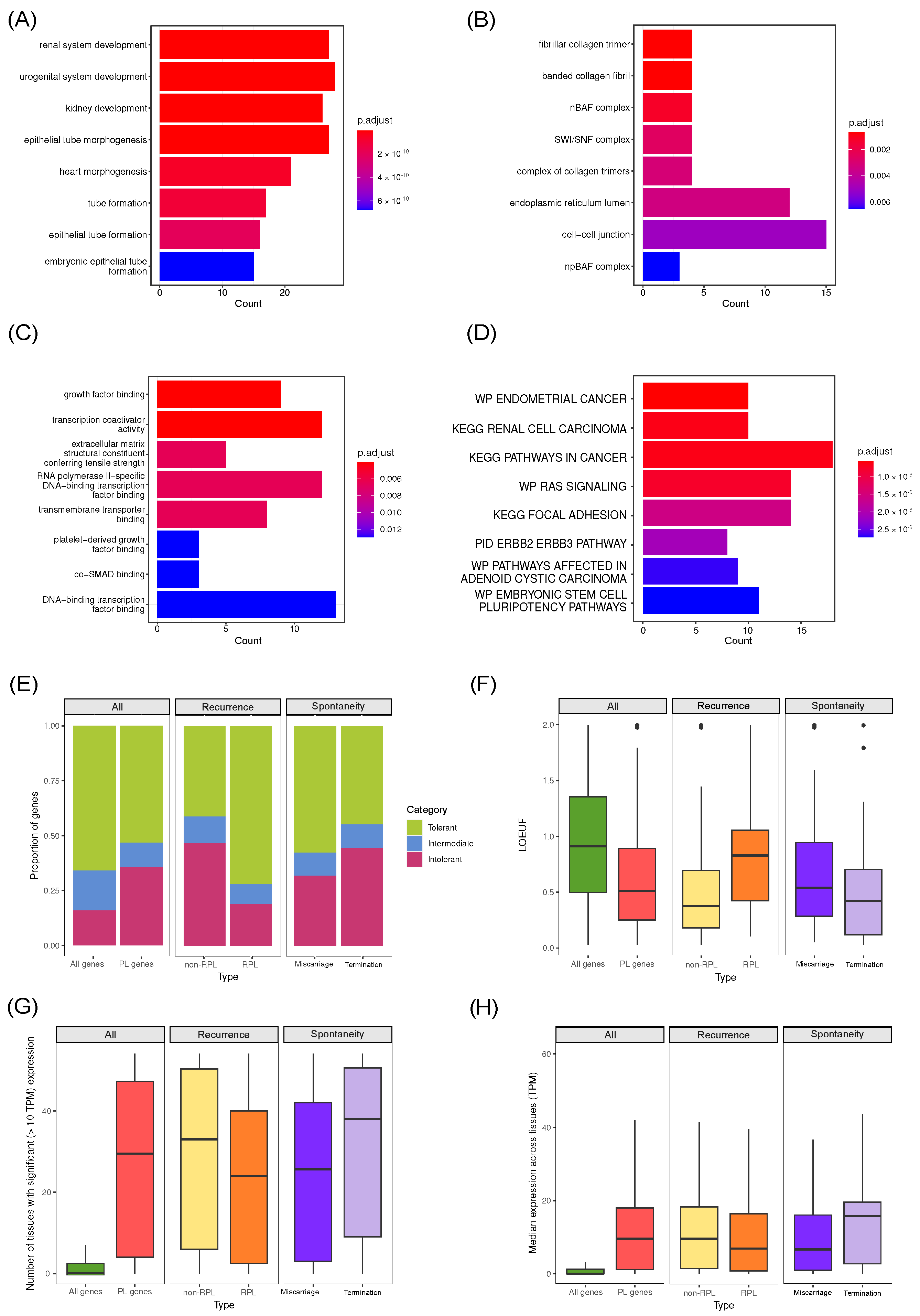
5. Common Properties of Known Pregnancy Loss Genes
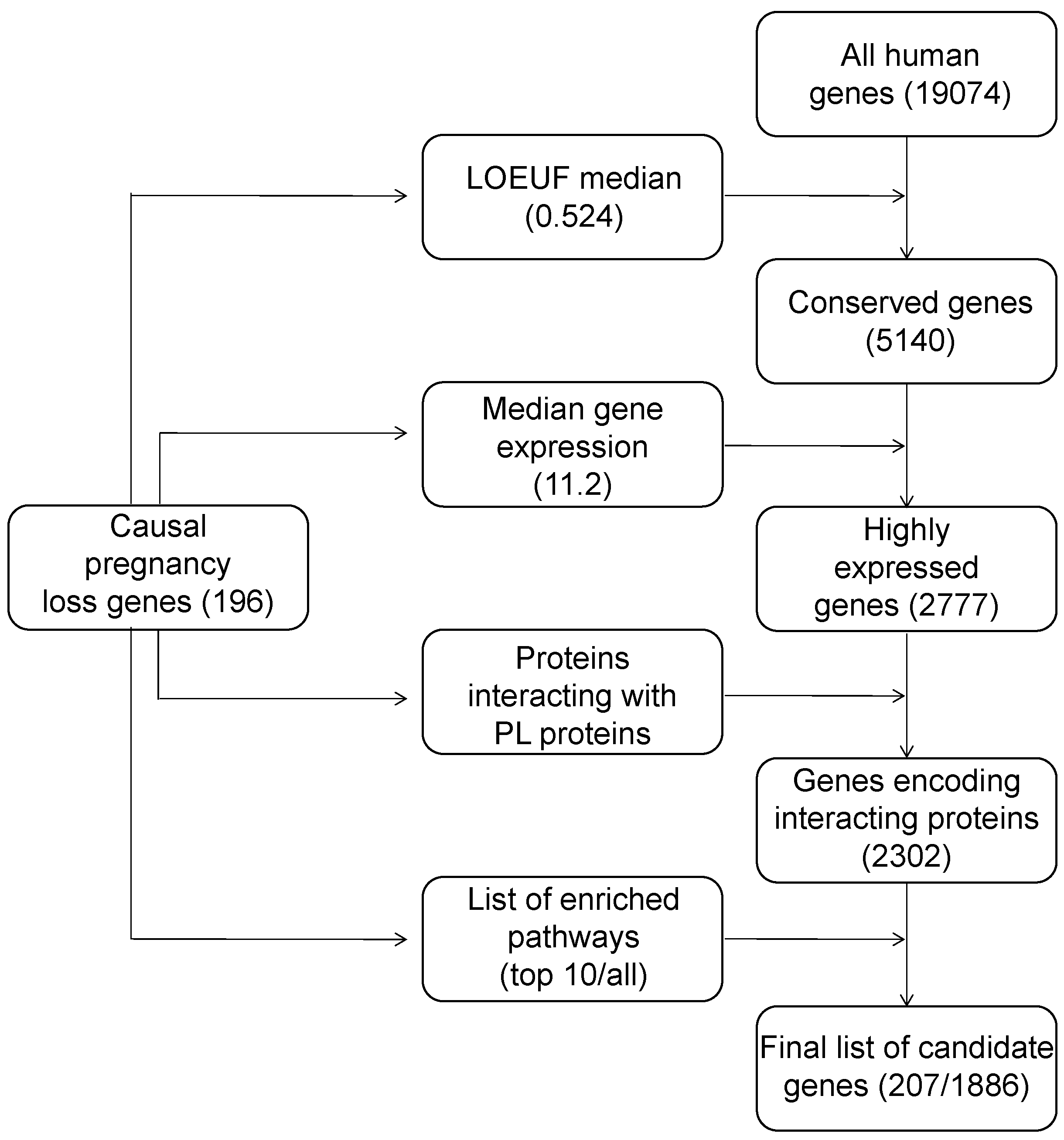
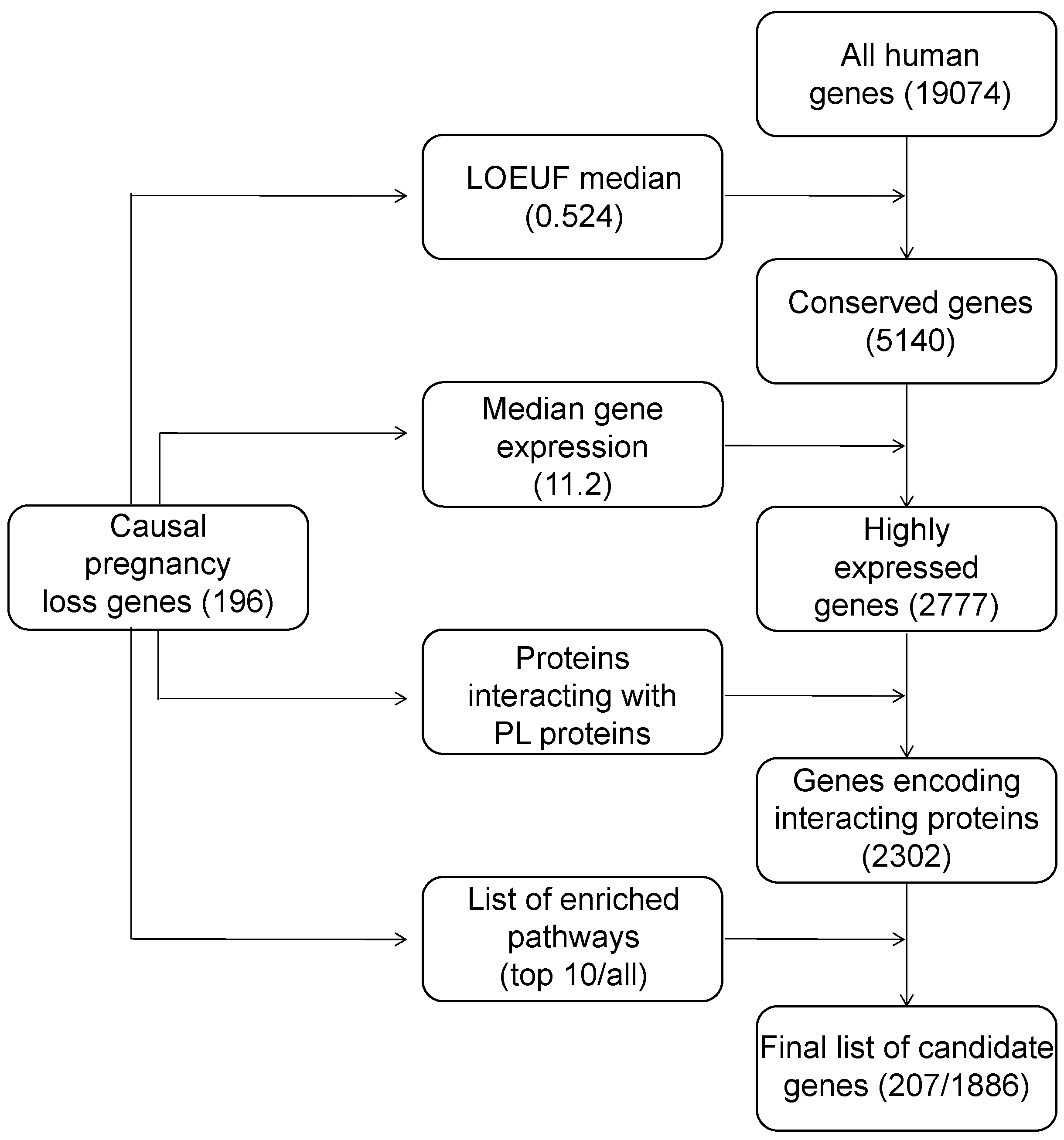
6. Construction of a List of Candidate Pregnancy Loss Genes
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| gnomAD | genome aggregation database |
| pLI | probability of loss-of-function intolerance |
| pLoF | putative loss-of-function variants |
| ACMG | American College of Medical Genetics and Genomics |
| GO | gene ontology |
| GTEx | genotype tissue expression |
| LOEUF | loss-of-function observed-to-expected upper fraction |
| MSigDB | molecular signatures database |
| PL | pregnancy loss |
| POC | product of conception |
| RPL | recurrent pregnancy loss |
| SA | spontaneous abortion |
| TPM | transcripts per million |
| VUS | variant of uncertain significance |
References
- The ESHRE Guideline Group on RPL; Bender Atik, R.; Christiansen, O.B.; Elson, J.; Kolte, A.M.; Lewis, S.; Middeldorp, S.; Nelen, W.; Peramo, B.; Quenby, S.; et al. ESHRE guideline: Recurrent pregnancy loss. Hum. Reprod. Open 2018, 2018, hoy004. [Google Scholar] [CrossRef] [PubMed]
- Tetruashvili, N.; Domar, A.; Bashiri, A. Prevention of Pregnancy Loss: Combining Progestogen Treatment and Psychological Support. J. Clin. Med. 2023, 12, 1827. [Google Scholar] [CrossRef] [PubMed]
- El Hachem, H.; Crepaux, V.; May-Panloup, P.; Descamps, P.; Legendre, G.; Bouet, P.E. Recurrent pregnancy loss: Current perspectives. Int. J. Women Health 2017, 9, 331–345. [Google Scholar] [CrossRef] [PubMed]
- WHO. Recommended definitions, terminology and format for statistical tables related to the perinatal period and use of a new certificate for cause of perinatal deaths. Modifications recommended by FIGO as amended October 14, 1976. Acta Obstet. Gynecol. Scand. 1977, 56, 247–253. [Google Scholar]
- Turesheva, A.; Aimagambetova, G.; Ukybassova, T.; Marat, A.; Kanabekova, P.; Kaldygulova, L.; Amanzholkyzy, A.; Ryzhkova, S.; Nogay, A.; Khamidullina, Z.; et al. Recurrent Pregnancy Loss Etiology, Risk Factors, Diagnosis, and Management. Fresh Look into a Full Box. J. Clin. Med. 2023, 12, 4074. [Google Scholar] [CrossRef]
- Cohain, J.S.; Buxbaum, R.E.; Mankuta, D. Spontaneous first trimester miscarriage rates per woman among parous women with 1 or more pregnancies of 24 weeks or more. BMC Pregnancy Childbirth 2017, 17, 437. [Google Scholar] [CrossRef]
- Linnakaari, R.; Helle, N.; Mentula, M.; Bloigu, A.; Gissler, M.; Heikinheimo, O.; Niinimäki, M. Trends in the incidence, rate and treatment of miscarriage-nationwide register-study in Finland, 1998–2016. Hum. Reprod. 2019, 34, 2120–2128. [Google Scholar] [CrossRef]
- Larsen, E.C.; Christiansen, O.B.; Kolte, A.M.; Macklon, N. New insights into mechanisms behind miscarriage. BMC Med. 2013, 11, 154. [Google Scholar] [CrossRef]
- Qu, S.; Wang, L.; Cai, A.; Cui, S.; Bai, N.; Liu, N.; Kong, X. Exploring the cause of early miscarriage with SNP-array analysis and karyotyping. J. Matern. Fetal Neonatal Med. 2019, 32, 1–10. [Google Scholar] [CrossRef]
- Heaney, S.; Tomlinson, M.; Aventin, Á. Termination of pregnancy for fetal anomaly: A systematic review of the healthcare experiences and needs of parents. BMC Pregnancy Childbirth 2022, 22, 441. [Google Scholar] [CrossRef]
- Cai, M.; Lin, N.; Xu, L.; Huang, H. Comparative clinical genetic testing in spontaneous miscarriage: Insights from a study in Southern Chinese women. J. Cell. Mol. Med. 2021, 25, 5721–5728. [Google Scholar] [CrossRef]
- Manning, M.; Hudgins, L.; Professional Practice and Guidelines Committee. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet. Med. 2010, 12, 742–745. [Google Scholar] [CrossRef]
- Suzumori, N.; Sugiura-Ogasawara, M. Genetic factors as a cause of miscarriage. Curr. Med. Chem. 2010, 17, 3431–3437. [Google Scholar] [CrossRef]
- Bowen, P.; Lee, C.S. Spontaneous abortion. Chromosome studies on 41 cases and an analysis of maternal age and duration of pregnancy in relation to karyotype. Am. J. Obstet. Gynecol. 1969, 104, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Boué, J.; Boué, A.; Lazar, P. Retrospective and prospective epidemiological studies of 1500 karyotyped spontaneous human abortions. Teratology 1975, 12, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Menasha, J.; Levy, B.; Hirschhorn, K.; Kardon, N.B. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: New insights from a 12-year study. Genet. Med. 2005, 7, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Goddijn, M.; Leschot, N. Genetic aspects of miscarriage. Best Pract. Res. Clin. Obstet. Gynaecol. 2000, 14, 855–865. [Google Scholar] [CrossRef]
- Griffin, D.K.; Millie, E.A.; Redline, R.W.; Hassold, T.J.; Zaragoza, M.V. Cytogenetic analysis of spontaneous abortions: Comparison of techniques and assessment of the incidence of confined placental mosaicism. Am. J. Med Genet. 1997, 72, 297–301. [Google Scholar] [CrossRef]
- Lebedev, I.N.; Ostroverkhova, N.V.; Nikitina, T.V.; Sukhanova, N.N.; Nazarenko, S.A. Features of chromosomal abnormalities in spontaneous abortion cell culture failures detected by interphase FISH analysis. Eur. J. Hum. Genet. EJHG 2004, 12, 513–520. [Google Scholar] [CrossRef]
- Kallioniemi, A.; Kallioniemi, O.P.; Sudar, D.; Rutovitz, D.; Gray, J.W.; Waldman, F.; Pinkel, D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258, 818–821. [Google Scholar] [CrossRef]
- Kallioniemi, O.P.; Kallioniemi, A.; Piper, J.; Isola, J.; Waldman, F.M.; Gray, J.W.; Pinkel, D. Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes Chromosom. Cancer 1994, 10, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Lomax, B.; Tang, S.; Separovic, E.; Phillips, D.; Hillard, E.; Thomson, T.; Kalousek, D.K. Comparative genomic hybridization in combination with flow cytometry improves results of cytogenetic analysis of spontaneous abortions. Am. J. Hum. Genet. 2000, 66, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Lestou, V.S.; Desilets, V.; Lomax, B.L.; Barrett, I.J.; Wilson, R.D.; Langlois, S.; Kalousek, D.K. Comparative genomic hybridization: A new approach to screening for intrauterine complete or mosaic aneuploidy. Am. J. Med Genet. 2000, 92, 281–284. [Google Scholar] [CrossRef]
- Dória, S.; Carvalho, F.; Ramalho, C.; Lima, V.; Francisco, T.; Machado, A.P.; Brandão, O.; Sousa, M.; Matias, A.; Barros, A. An efficient protocol for the detection of chromosomal abnormalities in spontaneous miscarriages or foetal deaths. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 147, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, M.; Liu, Q.Y.; Hu, S.Q.; Li, L.R.; Li, J.; Ma, R.M. Detecting trisomy in products of conception from first-trimester spontaneous miscarriages by next-generation sequencing (NGS). Medicine 2020, 99, e18731. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Hillman, S.C.; Parthiban, V.; McMullan, D.J.; Maher, E.R.; Kilby, M.D.; Hurles, M.E. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum. Mol. Genet. 2014, 23, 3269–3277. [Google Scholar] [CrossRef]
- Van den Veyver, I.B.; Patel, A.; Shaw, C.A.; Pursley, A.N.; Kang, S.L.; Simovich, M.J.; Ward, P.A.; Darilek, S.; Johnson, A.; Neill, S.E.; et al. Clinical use of array comparative genomic hybridization (aCGH) for prenatal diagnosis in 300 cases. Prenat. Diagn. 2009, 29, 29–39. [Google Scholar] [CrossRef]
- Hillman, S.C.; McMullan, D.J.; Hall, G.; Togneri, F.S.; James, N.; Maher, E.J.; Meller, C.H.; Williams, D.; Wapner, R.J.; Maher, E.R.; et al. Use of prenatal chromosomal microarray: Prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2013, 41, 610–620. [Google Scholar] [CrossRef]
- Rajcan-Separovic, E.; Qiao, Y.; Tyson, C.; Harvard, C.; Fawcett, C.; Kalousek, D.; Stephenson, M.; Philipp, T. Genomic changes detected by array CGH in human embryos with developmental defects. Mol. Hum. Reprod. 2010, 16, 125–134. [Google Scholar] [CrossRef]
- Rajcan-Separovic, E.; Diego-Alvarez, D.; Robinson, W.P.; Tyson, C.; Qiao, Y.; Harvard, C.; Fawcett, C.; Kalousek, D.; Philipp, T.; Somerville, M.J.; et al. Identification of copy number variants in miscarriages from couples with idiopathic recurrent pregnancy loss. Hum. Reprod. 2010, 25, 2913–2922. [Google Scholar] [CrossRef]
- Bagheri, H.; Mercier, E.; Qiao, Y.; Stephenson, M.D.; Rajcan-Separovic, E. Genomic characteristics of miscarriage copy number variants. Mol. Hum. Reprod. 2015, 21, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Sigurjonsson, S.; Pettersen, B.; Maisenbacher, M.K.; Hall, M.P.; Demko, Z.; Lathi, R.B.; Tao, R.; Aggarwal, V.; Rabinowitz, M. Genomic imbalance in products of conception: Single-nucleotide polymorphism chromosomal microarray analysis. Obstet. Gynecol. 2014, 124, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Wen, J.; Tang, F.; Martell, S.; Shomer, N.; Leung, P.C.K.; Stephenson, M.D.; Rajcan-Separovic, E. Whole exome sequencing in recurrent early pregnancy loss. Mol. Hum. Reprod. 2016, 22, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.B.; Arts, P.; Ha, T.T.; Kassahn, K.S.; Pais, L.S.; O’Donnell-Luria, A.; Broad Institute Center for Mendelian Genomics; Babic, M.; Frank, M.S.B.; Feng, J.; et al. Genomic autopsy to identify underlying causes of pregnancy loss and perinatal death. Nat. Med. 2023, 29, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Normand, E.A.; Braxton, A.; Nassef, S.; Ward, P.A.; Vetrini, F.; He, W.; Patel, V.; Qu, C.; Westerfield, L.E.; Stover, S.; et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med. 2018, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Colley, E.; Hamilton, S.; Smith, P.; Morgan, N.V.; Coomarasamy, A.; Allen, S. Potential genetic causes of miscarriage in euploid pregnancies: A systematic review. Hum. Reprod. Update 2019, 25, 452–472. [Google Scholar] [CrossRef] [PubMed]
- Talkowski, M.E.; Ordulu, Z.; Pillalamarri, V.; Benson, C.B.; Blumenthal, I.; Connolly, S.; Hanscom, C.; Hussain, N.; Pereira, S.; Picker, J.; et al. Clinical diagnosis by whole-genome sequencing of a prenatal sample. N. Engl. J. Med. 2012, 367, 2226–2232. [Google Scholar] [CrossRef]
- Quintero-Ronderos, P.; Laissue, P. Genetic Variants Contributing to Early Recurrent Pregnancy Loss Etiology Identified by Sequencing Approaches. Reprod. Sci. 2020, 27, 1541–1552. [Google Scholar] [CrossRef]
- Wang, X.; Shi, W.; Zhao, S.; Gong, D.; Li, S.; Hu, C.; Chen, Z.J.; Li, Y.; Yan, J. Whole exome sequencing in unexplained recurrent miscarriage families identified novel pathogenic genetic causes of euploid miscarriage. Hum. Reprod. 2023, 38, 1003–1018. [Google Scholar] [CrossRef]
- Xiang, H.; Wang, C.; Pan, H.; Hu, Q.; Wang, R.; Xu, Z.; Li, T.; Su, Y.; Ma, X.; Cao, Y.; et al. Exome-Sequencing Identifies Novel Genes Associated with Recurrent Pregnancy Loss in a Chinese Cohort. Front. Genet. 2021, 12, 746082. [Google Scholar] [CrossRef]
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Polev, D.E.; Glotov, A.S.; Serebryakova, E.A.; Shcherbakova, I.V.; Kiselev, A.M.; Kostareva, A.A.; Glotov, O.S.; Predeus, A.V. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Sci. Rep. 2020, 10, 2057. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.; Choi, M. Application of whole exome sequencing to identify disease-causing variants in inherited human diseases. Genom. Inform. 2012, 10, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Li, Z.; Lu, J. Advances of Genetic Testing Technology in Etiology Diagnosis of Recurrent Spontaneous Abortion. Yangtze Med. 2023, 7, 76–86. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Swaid, A.; Alkuraya, F.S. Lifting the lid on unborn lethal Mendelian phenotypes through exome sequencing. Genet. Med. 2013, 15, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Kivuva, E.; Turnpenny, P.; Stals, K.; Johnson, M.; Xie, W.; Caswell, R.; Lango Allen, H. An exome sequencing strategy to diagnose lethal autosomal recessive disorders. Eur. J. Hum. Genet. EJHG 2015, 23, 401–404. [Google Scholar] [CrossRef]
- Alamillo, C.L.; Powis, Z.; Farwell, K.; Shahmirzadi, L.; Weltmer, E.C.; Turocy, J.; Lowe, T.; Kobelka, C.; Chen, E.; Basel, D.; et al. Exome sequencing positively identified relevant alterations in more than half of cases with an indication of prenatal ultrasound anomalies. Prenat. Diagn. 2015, 35, 1073–1078. [Google Scholar] [CrossRef]
- Yates, C.L.; Monaghan, K.G.; Copenheaver, D.; Retterer, K.; Scuffins, J.; Kucera, C.R.; Friedman, B.; Richard, G.; Juusola, J. Whole-exome sequencing on deceased fetuses with ultrasound anomalies: Expanding our knowledge of genetic disease during fetal development. Genet. Med. 2017, 19, 1171–1178. [Google Scholar] [CrossRef]
- Fu, M.; Mu, S.; Wen, C.; Jiang, S.; Li, L.; Meng, Y.; Peng, H. Whole-exome sequencing analysis of products of conception identifies novel mutations associated with missed abortion. Mol. Med. Rep. 2018, 18, 2027–2032. [Google Scholar] [CrossRef]
- Sahlin, E.; Gréen, A.; Gustavsson, P.; Liedén, A.; Nordenskjöld, M.; Papadogiannakis, N.; Pettersson, K.; Nilsson, D.; Jonasson, J.; Iwarsson, E. Identification of putative pathogenic single nucleotide variants (SNVs) in genes associated with heart disease in 290 cases of stillbirth. PLoS ONE 2019, 14, e0210017. [Google Scholar] [CrossRef]
- Najafi, K.; Mehrjoo, Z.; Ardalani, F.; Ghaderi-Sohi, S.; Kariminejad, A.; Kariminejad, R.; Najmabadi, H. Identifying the causes of recurrent pregnancy loss in consanguineous couples using whole exome sequencing on the products of miscarriage with no chromosomal abnormalities. Sci. Rep. 2021, 11, 6952. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chai, H.; Zhou, Q.; Wen, J.; Reddy, U.M.; Kastury, R.; Jiang, Y.; Mak, W.; Bale, A.E.; Zhang, H.; et al. Exome sequencing analysis on products of conception: A cohort study to evaluate clinical utility and genetic etiology for pregnancy loss. Genet. Med. 2021, 23, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shi, Y.; Fu, J.; Yu, M.; Feng, R.; Sang, Q.; Liang, B.; Chen, B.; Qu, R.; Li, B.; et al. Mutations in PADI6 Cause Female Infertility Characterized by Early Embryonic Arrest. Am. J. Hum. Genet. 2016, 99, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Nguyen, N.M.P.; Rezaei, M.; Huang, B.; Tao, Y.; Zhang, X.; Cheng, Q.; Yang, H.; Asangla, A.; Majewski, J.; et al. Biallelic PADI6 variants linking infertility, miscarriages, and hydatidiform moles. Eur. J. Hum. Genet. EJHG 2018, 26, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Morency, E.; Li, T.; Qin, H.; Zhang, X.; Zhang, X.; Coonrod, S. Role for PADI6 in securing the mRNA-MSY2 complex to the oocyte cytoplasmic lattices. Cell Cycle 2017, 16, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Yurttas, P.; Vitale, A.M.; Fitzhenry, R.J.; Cohen-Gould, L.; Wu, W.; Gossen, J.A.; Coonrod, S.A. Role for PADI6 and the cytoplasmic lattices in ribosomal storage in oocytes and translational control in the early mouse embryo. Development 2008, 135, 2627–2636. [Google Scholar] [CrossRef]
- Eggermann, T. Maternal Effect Mutations: A Novel Cause for Human Reproductive Failure. Geburtshilfe Frauenheilkd. 2021, 81, 780–788. [Google Scholar] [CrossRef]
- Begemann, M.; Rezwan, F.I.; Beygo, J.; Docherty, L.E.; Kolarova, J.; Schroeder, C.; Buiting, K.; Chokkalingam, K.; Degenhardt, F.; Wakeling, E.L.; et al. Maternal variants in NLRP and other maternal effect proteins are associated with multilocus imprinting disturbance in offspring. J. Med Genet. 2018, 55, 497–504. [Google Scholar] [CrossRef]
- Cubellis, M.V.; Pignata, L.; Verma, A.; Sparago, A.; Del Prete, R.; Monticelli, M.; Calzari, L.; Antona, V.; Melis, D.; Tenconi, R.; et al. Loss-of-function maternal-effect mutations of PADI6 are associated with familial and sporadic Beckwith-Wiedemann syndrome with multi-locus imprinting disturbance. Clin. Epigenetics 2020, 12, 139. [Google Scholar] [CrossRef]
- Eggermann, T.; Kadgien, G.; Begemann, M.; Elbracht, M. Biallelic PADI6 variants cause multilocus imprinting disturbances and miscarriages in the same family. Eur. J. Hum. Genet. EJHG 2021, 29, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Vulprecht, J.; David, A.; Tibelius, A.; Castiel, A.; Konotop, G.; Liu, F.; Bestvater, F.; Raab, M.S.; Zentgraf, H.; Izraeli, S.; et al. STIL is required for centriole duplication in human cells. J. Cell Sci. 2012, 125, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Izraeli, S.; Colaizzo-Anas, T.; Bertness, V.L.; Mani, K.; Aplan, P.D.; Kirsch, I.R. Expression of the SIL gene is correlated with growth induction and cellular proliferation. Cell Growth Differ. 1997, 8, 1171–1179. [Google Scholar] [PubMed]
- Izraeli, S.; Lowe, L.A.; Bertness, V.L.; Good, D.J.; Dorward, D.W.; Kirsch, I.R.; Kuehn, M.R. The SIL gene is required for mouse embryonic axial development and left-right specification. Nature 1999, 399, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Cristofoli, F.; De Keersmaecker, B.; De Catte, L.; Vermeesch, J.R.; Van Esch, H. Novel STIL Compound Heterozygous Mutations Cause Severe Fetal Microcephaly and Centriolar Lengthening. Mol. Syndromol. 2017, 8, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, M.; Arts, H.H.; Bongers, E.M.H.F.; Yap, Z.; Oud, M.M.; Antony, D.; Duijkers, L.; Emes, R.D.; Stalker, J.; Yntema, J.B.L.; et al. Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J. Med. Genet. 2013, 50, 309–323. [Google Scholar] [CrossRef]
- Dagoneau, N.; Goulet, M.; Geneviève, D.; Sznajer, Y.; Martinovic, J.; Smithson, S.; Huber, C.; Baujat, G.; Flori, E.; Tecco, L.; et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 2009, 84, 706–711. [Google Scholar] [CrossRef]
- Pitera, J.E.; Scambler, P.J.; Woolf, A.S. Fras1, a basement membrane-associated protein mutated in Fraser syndrome, mediates both the initiation of the mammalian kidney and the integrity of renal glomeruli. Hum. Mol. Genet. 2008, 17, 3953–3964. [Google Scholar] [CrossRef]
- Vrontou, S.; Petrou, P.; Meyer, B.I.; Galanopoulos, V.K.; Imai, K.; Yanagi, M.; Chowdhury, K.; Scambler, P.J.; Chalepakis, G. Fras1 deficiency results in cryptophthalmos, renal agenesis and blebbed phenotype in mice. Nat. Genet. 2003, 34, 209–214. [Google Scholar] [CrossRef]
- Talbot, J.C.; Walker, M.B.; Carney, T.J.; Huycke, T.R.; Yan, Y.L.; BreMiller, R.A.; Gai, L.; Delaurier, A.; Postlethwait, J.H.; Hammerschmidt, M.; et al. fras1 shapes endodermal pouch 1 and stabilizes zebrafish pharyngeal skeletal development. Development 2012, 139, 2804–2813. [Google Scholar] [CrossRef]
- Rivière, J.B.; Mirzaa, G.M.; O’Roak, B.J.; Beddaoui, M.; Alcantara, D.; Conway, R.L.; St-Onge, J.; Schwartzentruber, J.A.; Gripp, K.W.; Nikkel, S.M.; et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012, 44, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Kurdi, W.; Almusafri, F.; Alnemer, M.; Alkaff, A.; Babay, Z.; Alhashem, A.; Tulbah, M.; Alsahan, N.; Khan, R.; et al. Molecular autopsy in maternal-fetal medicine. Genet. Med. 2018, 20, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Datkhaeva, I.; Arboleda, V.A.; Senaratne, T.N.; Nikpour, G.; Meyerson, C.; Geng, Y.; Afshar, Y.; Scibetta, E.; Goldstein, J.; Quintero-Rivera, F.; et al. Identification of novel PIEZO1 variants using prenatal exome sequencing and correlation to ultrasound and autopsy findings of recurrent hydrops fetalis. Am. J. Med Genet. Part A 2018, 176, 2829–2834. [Google Scholar] [CrossRef] [PubMed]
- Rae, W.; Gao, Y.; Bunyan, D.; Holden, S.; Gilmour, K.; Patel, S.; Wellesley, D.; Williams, A. A novel FOXP3 mutation causing fetal akinesia and recurrent male miscarriages. Clin. Immunol. 2015, 161, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Shehab, O.; Tester, D.J.; Ackerman, N.C.; Cowchock, F.S.; Ackerman, M.J. Whole genome sequencing identifies etiology of recurrent male intrauterine fetal death. Prenat. Diagn. 2017, 37, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Reichert, S.L.; McKay, E.M.; Moldenhauer, J.S. Identification of a novel nonsense mutation in the FOXP3 gene in a fetus with hydrops–Expanding the phenotype of IPEX syndrome. Am. J. Med Genet. Part A 2016, 170A, 226–232. [Google Scholar] [CrossRef]
- Vasiljevic, A.; Poreau, B.; Bouvier, R.; Lachaux, A.; Arnoult, C.; Fauré, J.; Cordier, M.P.; Ray, P.F. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome and recurrent intrauterine fetal death. Lancet 2015, 385, 2120. [Google Scholar] [CrossRef]
- Xavier-da Silva, M.M.; Moreira-Filho, C.A.; Suzuki, E.; Patricio, F.; Coutinho, A.; Carneiro-Sampaio, M. Fetal-onset IPEX: Report of two families and review of literature. Clin. Immunol. 2015, 156, 131–140. [Google Scholar] [CrossRef]
- McKie, A.B.; Alsaedi, A.; Vogt, J.; Stuurman, K.E.; Weiss, M.M.; Shakeel, H.; Tee, L.; Morgan, N.V.; Nikkels, P.G.J.; van Haaften, G.; et al. Germline mutations in RYR1 are associated with foetal akinesia deformation sequence/lethal multiple pterygium syndrome. Acta Neuropathol. Commun. 2014, 2, 148. [Google Scholar] [CrossRef]
- De Tomasi, L.; David, P.; Humbert, C.; Silbermann, F.; Arrondel, C.; Tores, F.; Fouquet, S.; Desgrange, A.; Niel, O.; Bole-Feysot, C.; et al. Mutations in GREB1L Cause Bilateral Kidney Agenesis in Humans and Mice. Am. J. Hum. Genet. 2017, 101, 803–814. [Google Scholar] [CrossRef]
- Schrauwen, I.; Liaqat, K.; Schatteman, I.; Bharadwaj, T.; Nasir, A.; Acharya, A.; Ahmad, W.; Van Camp, G.; Leal, S.M. Autosomal Dominantly Inherited GREB1L Variants in Individuals with Profound Sensorineural Hearing Impairment. Genes 2020, 11, 687. [Google Scholar] [CrossRef] [PubMed]
- Karamatic Crew, V.; Tilley, L.A.; Satchwell, T.J.; AlSubhi, S.A.; Jones, B.; Spring, F.A.; Walser, P.J.; Martins Freire, C.; Murciano, N.; Rotordam, M.G.; et al. Missense mutations in PIEZO1, which encodes the Piezo1 mechanosensor protein, define Er red blood cell antigens. Blood 2023, 141, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Sewduth, R.N.; Pandolfi, S.; Steklov, M.; Sheryazdanova, A.; Zhao, P.; Criem, N.; Baietti, M.F.; Lechat, B.; Quarck, R.; Impens, F.; et al. The Noonan Syndrome Gene Lztr1 Controls Cardiovascular Function by Regulating Vesicular Trafficking. Circ. Res. 2020, 126, 1379–1393. [Google Scholar] [CrossRef] [PubMed]
- El Bouchikhi, I.; Belhassan, K.; Moufid, F.Z.; Iraqui Houssaini, M.; Bouguenouch, L.; Samri, I.; Atmani, S.; Ouldim, K. Noonan syndrome-causing genes: Molecular update and an assessment of the mutation rate. Int. J. Pediatr. Adolesc. Med. 2016, 3, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.; Ottenheijm, C.A.C. Nebulin: Big protein with big responsibilities. J. Muscle Res. Cell Motil. 2020, 41, 103–124. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Tulbah, M.; Kurdi, W.; Nemer, M.; Alsahan, N.; Al Mardawi, E.; Khalifa, O.; Hashem, A.; Kurdi, A.; Babay, Z.; et al. Identification of embryonic lethal genes in humans by autozygosity mapping and exome sequencing in consanguineous families. Genome Biol. 2015, 16, 116. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Barzaghi, F.; Passerini, L.; Bacchetta, R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: A paradigm of immunodeficiency with autoimmunity. Front. Immunol. 2012, 3, 211. [Google Scholar] [CrossRef]
- Zhuang, J.; Luo, Q.; Xie, M.; Chen, Y.; Jiang, Y.; Zeng, S.; Wang, Y.; Xie, Y.; Chen, C. Etiological identification of recurrent male fatality due to a novel NSDHL gene mutation using trio whole-exome sequencing: A rare case report and literature review. Mol. Genet. Genom. Med. 2023, 11, e2121. [Google Scholar] [CrossRef]
- König, A.; Happle, R.; Bornholdt, D.; Engel, H.; Grzeschik, K.H. Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am. J. Med Genet. 2000, 90, 339–346. [Google Scholar] [CrossRef]
- Jamieson, S.E.; de Roubaix, L.A.; Cortina-Borja, M.; Tan, H.K.; Mui, E.J.; Cordell, H.J.; Kirisits, M.J.; Miller, E.N.; Peacock, C.S.; Hargrave, A.C.; et al. Genetic and epigenetic factors at COL2A1 and ABCA4 influence clinical outcome in congenital toxoplasmosis. PloS ONE 2008, 3, e2285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Brockington, M.; Jungbluth, H.; Monk, D.; Stanier, P.; Sewry, C.A.; Moore, G.E.; Muntoni, F. Epigenetic allele silencing unveils recessive RYR1 mutations in core myopathies. Am. J. Hum. Genet. 2006, 79, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Hopton, C.; Tijsen, A.J.; Maizels, L.; Arbel, G.; Gepstein, A.; Bates, N.; Brown, B.; Huber, I.; Kimber, S.J.; Newman, W.G.; et al. Characterization of the mechanism by which a nonsense variant in RYR2 leads to disordered calcium handling. Physiol. Rep. 2022, 10, e15265. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, C.; Huang, S.; Zhang, W.; Liutang, Q.; Wang, Y.; Ma, G.; Chen, R. Case report: Familial glycogen storage disease type IV caused by novel compound heterozygous mutations in a glycogen branching enzyme 1 gene. Front. Genet. 2022, 13, 1033944. [Google Scholar] [CrossRef]
- Dainese, L.; Adam, N.; Boudjemaa, S.; Hadid, K.; Rosenblatt, J.; Jouannic, J.M.; Heron, D.; Froissart, R.; Coulomb, A. Glycogen Storage Disease Type IV and Early Implantation Defect: Early Trophoblastic Involvement Associated with a New GBE1 Mutation. Pediatr. Dev. Pathol. 2016, 19, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Phelan, M.L.; Sif, S.; Narlikar, G.J.; Kingston, R.E. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 1999, 3, 247–253. [Google Scholar] [CrossRef]
- Tseng, Y.C.; Cabot, B.; Cabot, R.A. ARID1A, a component of SWI/SNF chromatin remodeling complexes, is required for porcine embryo development. Mol. Reprod. Dev. 2017, 84, 1250–1256. [Google Scholar] [CrossRef]
- Zheng, P.; Patel, B.; McMenamin, M.; Paprocki, A.M.; Schramm, R.D.; Nagl, N.G.; Wilsker, D.; Wang, X.; Moran, E.; Latham, K.E. Expression of genes encoding chromatin regulatory factors in developing rhesus monkey oocytes and preimplantation stage embryos: Possible roles in genome activation. Biol. Reprod. 2004, 70, 1419–1427. [Google Scholar] [CrossRef]
- Böttcher, R.T.; Niehrs, C. Fibroblast growth factor signaling during early vertebrate development. Endocr. Rev. 2005, 26, 63–77. [Google Scholar] [CrossRef]
- McIntosh, I.; Bellus, G.A.; Jab, E.W. The pleiotropic effects of fibroblast growth factor receptors in mammalian development. CEll Struct. Funct. 2000, 25, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Dorey, K.; Amaya, E. FGF signalling: Diverse roles during early vertebrate embryogenesis. Development 2010, 137, 3731–3742. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhou, Y. Identification of novel biomarkers and immune infiltration features of recurrent pregnancy loss by machine learning. Sci. Rep. 2023, 13, 10751. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Balasubramanian, S.; Frankish, A.; Huang, N.; Morris, J.; Walter, K.; Jostins, L.; Habegger, L.; Pickrell, J.K.; Montgomery, S.B.; et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012, 335, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, I.; Fuller, Z.L.; Myers, S.R.; Przeworski, M. Relating pathogenic loss-of-function mutations in humans to their evolutionary fitness costs. eLife 2023, 12, e83172. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Oved, J.H.; Babushok, D.V.; Lambert, M.P.; Wolfset, N.; Kowalska, M.A.; Poncz, M.; Karczewski, K.J.; Olson, T.S. Human mutational constraint as a tool to understand biology of rare and emerging bone marrow failure syndromes. Blood Adv. 2020, 4, 5232–5245. [Google Scholar] [CrossRef]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Schwerdtfeger, K.L.; Shreffler, K.M. Trauma of pregnancy loss and infertility among mothers and involuntarily childless women in the United States. J. Loss Trauma 2009, 14, 211–227. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maksiutenko, E.M.; Barbitoff, Y.A.; Nasykhova, Y.A.; Pachuliia, O.V.; Lazareva, T.E.; Bespalova, O.N.; Glotov, A.S. The Landscape of Point Mutations in Human Protein Coding Genes Leading to Pregnancy Loss. Int. J. Mol. Sci. 2023, 24, 17572. https://doi.org/10.3390/ijms242417572
Maksiutenko EM, Barbitoff YA, Nasykhova YA, Pachuliia OV, Lazareva TE, Bespalova ON, Glotov AS. The Landscape of Point Mutations in Human Protein Coding Genes Leading to Pregnancy Loss. International Journal of Molecular Sciences. 2023; 24(24):17572. https://doi.org/10.3390/ijms242417572
Chicago/Turabian StyleMaksiutenko, Evgeniia M., Yury A. Barbitoff, Yulia A. Nasykhova, Olga V. Pachuliia, Tatyana E. Lazareva, Olesya N. Bespalova, and Andrey S. Glotov. 2023. "The Landscape of Point Mutations in Human Protein Coding Genes Leading to Pregnancy Loss" International Journal of Molecular Sciences 24, no. 24: 17572. https://doi.org/10.3390/ijms242417572
APA StyleMaksiutenko, E. M., Barbitoff, Y. A., Nasykhova, Y. A., Pachuliia, O. V., Lazareva, T. E., Bespalova, O. N., & Glotov, A. S. (2023). The Landscape of Point Mutations in Human Protein Coding Genes Leading to Pregnancy Loss. International Journal of Molecular Sciences, 24(24), 17572. https://doi.org/10.3390/ijms242417572