
Isoform-Directed Control of c-Myc Functions: Understanding the Balance from Proliferation to Growth Arrest


Abstract
:1. Introduction
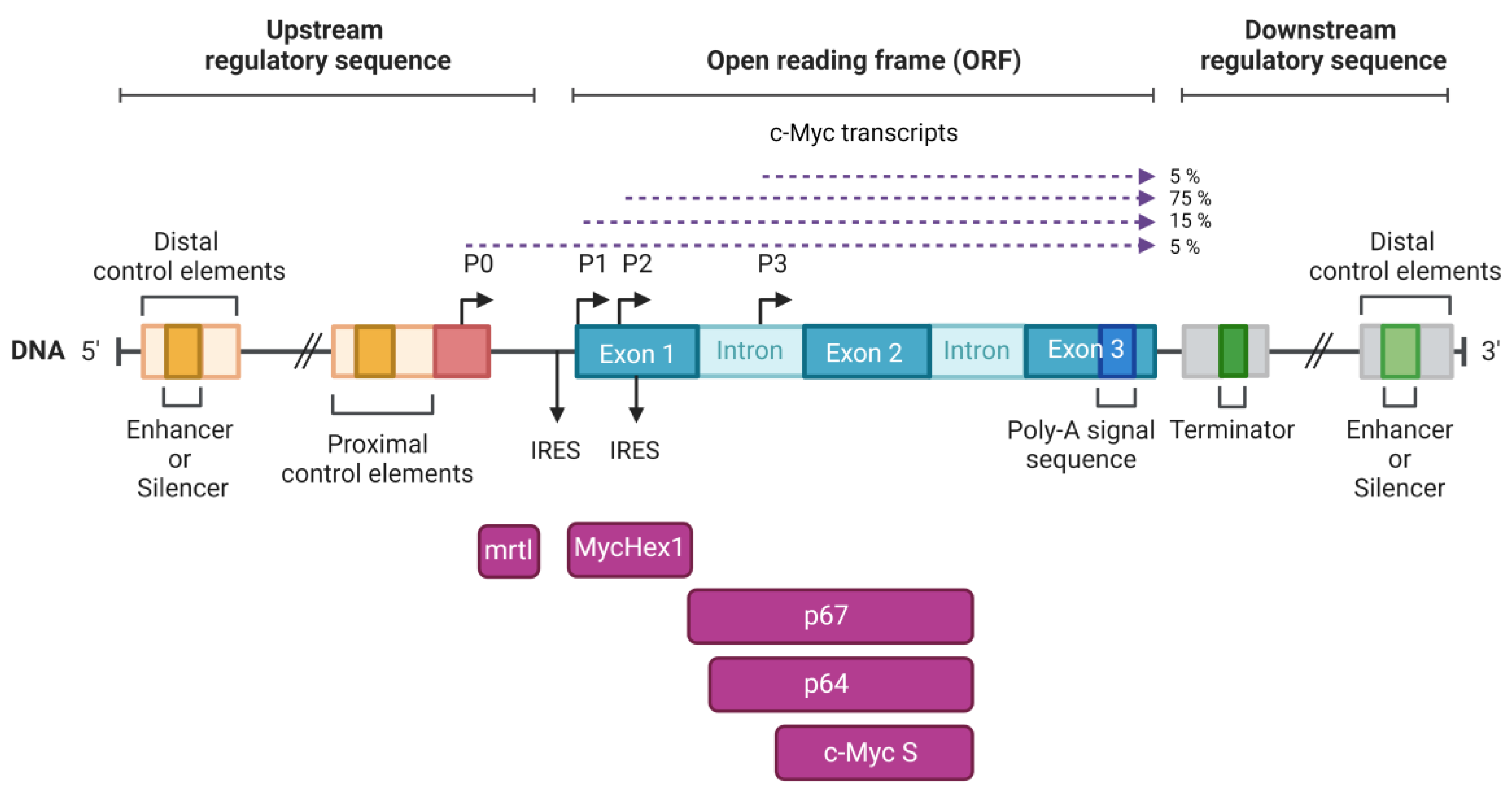
2. Structure of the c-Myc Locus
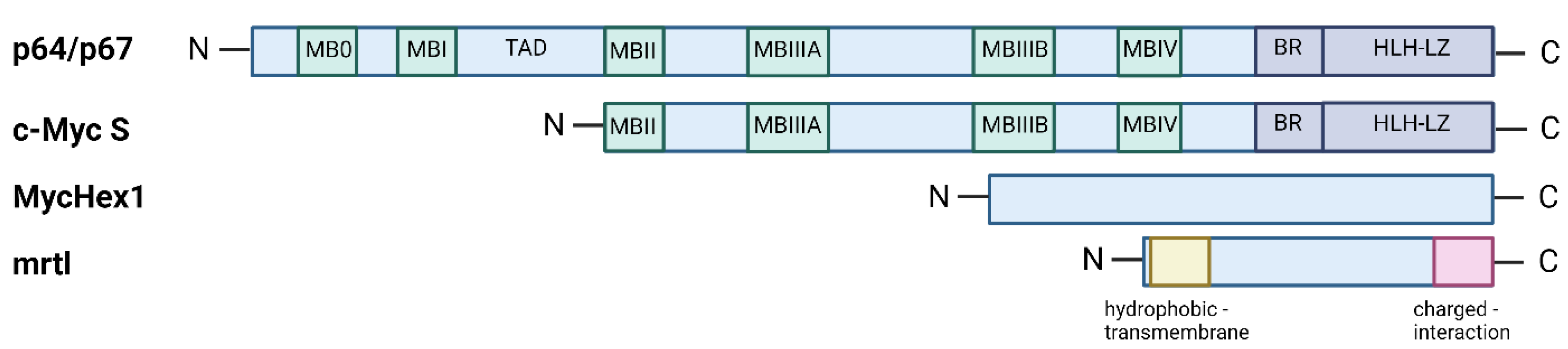
3. Two Main c-Myc Isoforms: p64 and p67
4. The Third Isoform c-Myc S
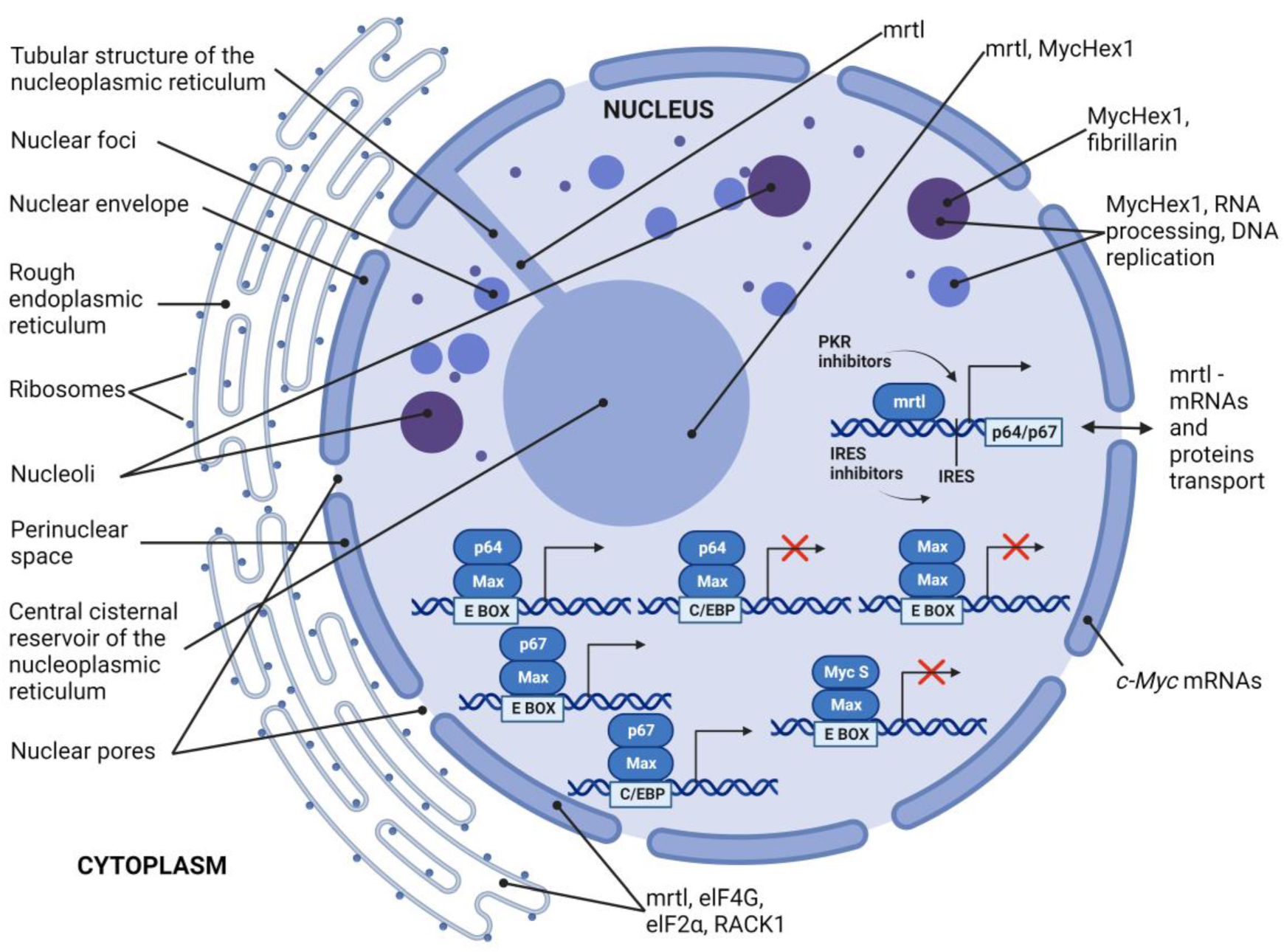
5. MycHex1 and mrtl
6. Targeting c-Myc in Cancer
7. Discussion and Summary
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amati, B.; Frank, S.R.; Donjerkovic, D.; Taubert, S. Function of the C-Myc Oncoprotein in Chromatin Remodeling and Transcription. Biochim. Biophys. Acta 2001, 1471, M135–M145. [Google Scholar] [CrossRef]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The C-Myc Target Gene Network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Shen, J.; Wu, M.; Arsura, M.; FitzGerald, M.; Suldan, Z.; Kim, D.W.; Hofmann, C.S.; Pianetti, S.; Romieu-Mourez, R.; et al. Repression of Transcription of the P27(Kip1) Cyclin-Dependent Kinase Inhibitor Gene by c-Myc. Oncogene 2001, 20, 1688–1702. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Wang, Y.; Liang, W.; Liu, L.; Pan, N.; Deng, H.; Li, L.; Zou, C.; Chan, F.L.; Zhou, Y. LRH-1 Drives Hepatocellular Carcinoma Partially through Induction of c-Myc and Cyclin E1, and Suppression of P21. Cancer Manag. Res. 2018, 10, 2389–2400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, N.; Zang, D.; Yu, J.; Li, J.; Di, W.; Guo, R.; Zhao, W.; Wang, H. C-Myc Promotes Tumor Proliferation and Anti-apoptosis by Repressing P21 in Rhabdomyosarcomas. Mol. Med. Rep. 2017, 16, 4089–4094. [Google Scholar] [CrossRef] [PubMed]
- Gruszka, R.; Zakrzewski, K.; Liberski, P.P.; Zakrzewska, M. mRNA and miRNA Expression Analyses of the MYC/E2F/miR-17-92 Network in the Most Common Pediatric Brain Tumors. Int. J. Mol. Sci. 2021, 22, 543. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; León, J. Myc and Cell Cycle Control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; Groudine, M. Control of C-Myc Regulation in Normal and Neoplastic Cells. Adv. Cancer Res. 1991, 56, 1–48. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC Oncogene—The Grand Orchestrator of Cancer Growth and Immune Evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Gao, F.Y.; Li, X.T.; Xu, K.; Wang, R.T.; Guan, X.X. C-MYC Mediates the Crosstalk between Breast Cancer Cells and Tumor Microenvironment. Cell Commun. Signal. 2023, 21, 28. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-Myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer Res. 2020, 40, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.; Jing, X.; Hu, K.; Wen, X.; Ye, L.; Zhang, X.; Qin, J.; Ye, J.; Lin, R.; Wang, Z.; et al. The C-MYC-WDR43 Signalling Axis Promotes Chemoresistance and Tumour Growth in Colorectal Cancer by Inhibiting P53 Activity. Drug Resist. Updat. 2023, 66, 100909. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of Apoptosis in Fibroblasts by C-Myc Protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B. MYC and the Control of Apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [PubMed]
- Kotulova, J.; Lonova, K.; Kubickova, A.; Vrbkova, J.; Kourilova, P.; Hajduch, M.; Dzubak, P. 2-Cl-IB-MECA Regulates the Proliferative and Drug Resistance Pathways, and Facilitates Chemosensitivity in Pancreatic and Liver Cancer Cell Lines. Int. J. Mol. Med. 2022, 49, 31. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Emerging Roles of Myc in Stem Cell Biology and Novel Tumor Therapies. J. Exp. Clin. Cancer Res. 2018, 37, 173. [Google Scholar] [CrossRef] [PubMed]
- Macečková, Z.; Kubíčková, A.; Sanctis, J.B.D.; Hajdúch, M. Effect of Glucocorticosteroids in Diamond-Blackfan Anaemia: Maybe Not as Elusive as It Seems. Int. J. Mol. Sci. 2022, 23, 1886. [Google Scholar] [CrossRef] [PubMed]
- Kubickova, A.; Maceckova, Z.; Vojta, P.; Ondra, M.; Volejnikova, J.; Koralkova, P.; Jungova, A.; Jahoda, O.; Mojzikova, R.; Hadacova, I.; et al. Missense Mutation in RPS7 Causes Diamond-Blackfan Anemia via Alteration of Erythrocyte Metabolism, Protein Translation and Induction of Ribosomal Stress. Blood Cells Mol. Dis. 2022, 97, 102690. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of Germline-Competent Induced Pluripotent Stem Cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Llombart, V.; Mansour, M.R. Therapeutic Targeting of “Undruggable” MYC. EBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef]
- Cole, M.D. The Myc Oncogene: Its Role in Transformation and Differentiation. Annu. Rev. Genet. 1986, 20, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, M.; Lüscher, B. Proteins of the Myc Network: Essential Regulators of Cell Growth and Differentiation. Adv. Cancer Res. 1996, 68, 109–182. [Google Scholar] [CrossRef]
- Lüscher, B.; Eisenman, R.N. New Light on Myc and Myb. Part I. Myc. Genes Dev. 1990, 4, 2025–2035. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; Birnie, G.D. Myc Oncogenes: The Enigmatic Family. Biochem. J. 1996, 314 Pt 3, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Hann, S.R.; Eisenman, R.N. Proteins Encoded by the Human C-Myc Oncogene: Differential Expression in Neoplastic Cells. Mol. Cell. Biol. 1984, 4, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, G.; Evan, G.I.; Bishop, J.M. The Protein Encoded by the Human Proto-Oncogene c-Myc. Proc. Natl. Acad. Sci. USA 1984, 81, 7742–7746. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A Helix-Loop-Helix Zipper Protein That Forms a Sequence-Specific DNA-Binding Complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Kretzner, L.; Blackwood, E.M.; Eisenman, R.N.; Weintraub, H. Sequence-Specific DNA Binding by the c-Myc Protein. Science 1990, 250, 1149–1151. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Ziff, E.B. Methylation-Sensitive Sequence-Specific DNA Binding by the c-Myc Basic Region. Science 1991, 251, 186–189. [Google Scholar] [CrossRef]
- Amati, B.; Brooks, M.W.; Levy, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic Activity of the C-Myc Protein Requires Dimerization with Max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Amin, C.; Wagner, A.J.; Hay, N. Sequence-Specific Transcriptional Activation by Myc and Repression by Max. Mol. Cell. Biol. 1993, 13, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Kretzner, L.; Blackwood, E.M.; Eisenman, R.N. Myc and Max Proteins Possess Distinct Transcriptional Activities. Nature 1992, 359, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Barrett, J.; Villa-Garcia, M.; Dang, C.V. An Amino-Terminal c-Myc Domain Required for Neoplastic Transformation Activates Transcription. Mol. Cell. Biol. 1990, 10, 5914–5920. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Bhatia, K.; Magrath, I.T.; Dang, C.V.; Dalla-Favera, R. Binding and Suppression of the Myc Transcriptional Activation Domain by P107. Science 1994, 264, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Maheswaran, S.; Lee, H.; Sonenshein, G.E. Intracellular Association of the Protein Product of the C-Myc Oncogene with the TATA-Binding Protein. Mol. Cell. Biol. 1994, 14, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Mac, J.; Siëbelt, F.; Ayer, D.E.; Eisenman, R.N. Myc-Max Heterodimers Activate a DEAD Box Gene and Interact with Multiple E Box-Related Sites in Vivo. EMBO J. 1996, 15, 4344–4357. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.E.; Farnham, P.J. Myc versus USF: Discrimination at the Cad Gene Is Determined by Core Promoter Elements. Mol. Cell. Biol. 1997, 17, 2529–2537. [Google Scholar] [CrossRef]
- O’Connell, B.C.; Cheung, A.F.; Simkevich, C.P.; Tam, W.; Ren, X.; Mateyak, M.K.; Sedivy, J.M. A Large Scale Genetic Analysis of C-Myc-Regulated Gene Expression Patterns. J. Biol. Chem. 2003, 278, 12563–12573. [Google Scholar] [CrossRef]
- Hölzel, M.; Kohlhuber, F.; Schlosser, I.; Hölzel, D.; Lüscher, B.; Eick, D. Myc/Max/Mad Regulate the Frequency but Not the Duration of Productive Cell Cycles. EMBO Rep. 2001, 2, 1125–1132. [Google Scholar] [CrossRef]
- Choi, H.; Jackson, N.L.; Shaw, D.R.; Emanuel, P.D.; Liu, Y.L.; Tousson, A.; Meng, Z.; Blume, S.W. Mrtl-A Translation/Localization Regulatory Protein Encoded within the Human c-Myc Locus and Distributed throughout the Endoplasmic and Nucleoplasmic Reticular Network. J. Cell Biochem. 2008, 105, 1092–1108. [Google Scholar] [CrossRef]
- Arabi, A.; Wu, S.; Ridderstråle, K.; Bierhoff, H.; Shiue, C.; Fatyol, K.; Fahlén, S.; Hydbring, P.; Söderberg, O.; Grummt, I.; et al. C-Myc Associates with Ribosomal DNA and Activates RNA Polymerase I Transcription. Nat. Cell Biol. 2005, 7, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. C-Myc Binds to Human Ribosomal DNA and Stimulates Transcription of rRNA Genes by RNA Polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roman, N.; Grandori, C.; Eisenman, R.N.; White, R.J. Direct Activation of RNA Polymerase III Transcription by C-Myc. Nature 2003, 421, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Gardner, C.; Li, E.R.; Arnold, I.; Watt, F.M. Evidence That Myc Activation Depletes the Epidermal Stem Cell Compartment by Modulating Adhesive Interactions with the Local Microenvironment. Development 2003, 130, 2793–2808. [Google Scholar] [CrossRef] [PubMed]
- Shiio, Y.; Donohoe, S.; Yi, E.C.; Goodlett, D.R.; Aebersold, R.; Eisenman, R.N. Quantitative Proteomic Analysis of Myc Oncoprotein Function. EMBO J. 2002, 21, 5088–5096. [Google Scholar] [CrossRef] [PubMed]
- Mateyak, M.K.; Obaya, A.J.; Adachi, S.; Sedivy, J.M. Phenotypes of C-Myc-Deficient Rat Fibroblasts Isolated by Targeted Homologous Recombination. Cell Growth Differ. 1997, 8, 1039–1048. [Google Scholar] [PubMed]
- Sakamuro, D.; Eviner, V.; Elliott, K.J.; Showe, L.; White, E.; Prendergast, G.C. C-Myc Induces Apoptosis in Epithelial Cells by Both P53-Dependent and P53-Independent Mechanisms. Oncogene 1995, 11, 2411–2418. [Google Scholar]
- Blume, S.W.; Miller, D.M.; Guarcello, V.; Shrestha, K.; Meng, Z.; Snyder, R.C.; Grizzle, W.E.; Ruppert, J.M.; Gartland, G.L.; Stockard, C.R.; et al. Inhibition of Tumorigenicity by the 5′-Untranslated RNA of the Human c-Myc P0 Transcript. Exp. Cell Res. 2003, 288, 131–142. [Google Scholar] [CrossRef]
- Marin, M.C.; Hsu, B.; Stephens, L.C.; Brisbay, S.; McDonnell, T.J. The Functional Basis of C-Myc and Bcl-2 Complementation during Multistep Lymphomagenesis in Vivo. Exp. Cell Res. 1995, 217, 240–247. [Google Scholar] [CrossRef]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc Signaling via the ARF Tumor Suppressor Regulates P53-Dependent Apoptosis and Immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef]
- Klein, G. Dysregulation of Lymphocyte Proliferation by Chromosomal Translocations and Sequential Genetic Changes. Bioessays 2000, 22, 414–422. [Google Scholar] [CrossRef]
- Ge, K.; Duhadaway, J.; Sakamuro, D.; Wechsler-Reya, R.; Reynolds, C.; Prendergast, G.C. Losses of the Tumor Suppressor BIN1 in Breast Carcinoma Are Frequent and Reflect Deficits in Programmed Cell Death Capacity. Int. J. Cancer 2000, 85, 376–383. [Google Scholar] [PubMed]
- Martin, S.J.; Green, D.R. Protease Activation during Apoptosis: Death by a Thousand Cuts? Cell 1995, 82, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, H.O.; McCurrach, M.E.; O’Neill, J.; Zhang, K.; Lowe, S.W.; Lazebnik, Y.A. Oncogene-Dependent Apoptosis in Extracts from Drug-Resistant Cells. Genes Dev. 1997, 11, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Isaacs, J.T.; Ottaviano, Y.L.; Davidson, N.E. Programmed Cell Death in an Estrogen-Independent Human Breast Cancer Cell Line, MDA-MB-468. Cancer Res. 1992, 52, 3418–3424. [Google Scholar] [PubMed]
- Fukasawa, K.; Wiener, F.; Vande Woude, G.F.; Mai, S. Genomic Instability and Apoptosis Are Frequent in P53 Deficient Young Mice. Oncogene 1997, 15, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative Reading Frames of the INK4a Tumor Suppressor Gene Encode Two Unrelated Proteins Capable of Inducing Cell Cycle Arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Tumor Surveillance via the ARF-P53 Pathway. Genes Dev. 1998, 12, 2984–2991. [Google Scholar] [CrossRef]
- Watt, R.; Nishikura, K.; Sorrentino, J.; ar-Rushdi, A.; Croce, C.M.; Rovera, G. The Structure and Nucleotide Sequence of the 5′ End of the Human c-Myc Oncogene. Proc. Natl. Acad. Sci. USA 1983, 80, 6307–6311. [Google Scholar] [CrossRef]
- Bentley, D.L.; Groudine, M. Novel Promoter Upstream of the Human C-Myc Gene and Regulation of c-Myc Expression in B-Cell Lymphomas. Mol. Cell. Biol. 1986, 6, 3481–3489. [Google Scholar] [CrossRef]
- Ray, D.; Robert-Lézénès, J. Coexistence of a C-Myc mRNA Initiated in Intron 1 with the Normal c-Myc mRNA and Similar Regulation of Both Transcripts in Mammalian Cells. Oncogene Res. 1989, 5, 73–78. [Google Scholar] [PubMed]
- Nanbru, C.; Prats, A.C.; Droogmans, L.; Defrance, P.; Huez, G.; Kruys, V. Translation of the Human C-Myc P0 Tricistronic mRNA Involves Two Independent Internal Ribosome Entry Sites. Oncogene 2001, 20, 4270–4280. [Google Scholar] [CrossRef] [PubMed]
- Hann, S.R.; King, M.W.; Bentley, D.L.; Anderson, C.W.; Eisenman, R.N. A Non-AUG Translational Initiation in c-Myc Exon 1 Generates an N-Terminally Distinct Protein Whose Synthesis Is Disrupted in Burkitt’s Lymphomas. Cell 1988, 52, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Marcu, K.B.; Bossone, S.A.; Patel, A.J. Myc Function and Regulation. Annu. Rev. Biochem. 1992, 61, 809–860. [Google Scholar] [CrossRef] [PubMed]
- Hann, S.R.; Dixit, M.; Sears, R.C.; Sealy, L. The Alternatively Initiated C-Myc Proteins Differentially Regulate Transcription through a Noncanonical DNA-Binding Site. Genes Dev. 1994, 8, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Cory, S. Activation of Cellular Oncogenes in Hemopoietic Cells by Chromosome Translocation. Adv. Cancer Res. 1986, 47, 189–234. [Google Scholar] [CrossRef] [PubMed]
- Battey, J.; Moulding, C.; Taub, R.; Murphy, W.; Stewart, T.; Potter, H.; Lenoir, G.; Leder, P. The Human C-Myc Oncogene: Structural Consequences of Translocation into the IgH Locus in Burkitt Lymphoma. Cell 1983, 34, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Bernard, O.; Cory, S.; Gerondakis, S.; Webb, E.; Adams, J.M. Sequence of the Murine and Human Cellular Myc Oncogenes and Two Modes of Myc Transcription Resulting from Chromosome Translocation in B Lymphoid Tumours. EMBO J. 1983, 2, 2375–2383. [Google Scholar] [CrossRef]
- Bentley, D.L.; Groudine, M. A Block to Elongation Is Largely Responsible for Decreased Transcription of C-Myc in Differentiated HL60 Cells. Nature 1986, 321, 702–706. [Google Scholar] [CrossRef]
- Nepveu, A.; Marcu, K.B. Intragenic Pausing and Anti-Sense Transcription within the Murine c-Myc Locus. EMBO J. 1986, 5, 2859–2865. [Google Scholar] [CrossRef]
- Yang, J.Q.; Remmers, E.F.; Marcu, K.B. The First Exon of the C-Myc Proto-Oncogene Contains a Novel Positive Control Element. EMBO J. 1986, 5, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Nerlov, C. The C/EBP Family of Transcription Factors: A Paradigm for Interaction between Gene Expression and Proliferation Control. Trends Cell Biol. 2007, 17, 318–324. [Google Scholar] [CrossRef]
- Baluapuri, A.; Wolf, E.; Eilers, M. Target Gene-Independent Functions of MYC Oncoproteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Philipp, A.; Schneider, A.; Väsrik, I.; Finke, K.; Xiong, Y.; Beach, D.; Alitalo, K.; Eilers, M. Repression of Cyclin D1: A Novel Function of MYC. Mol. Cell. Biol. 1994, 14, 4032–4043. [Google Scholar] [CrossRef] [PubMed]
- Freytag, S.O.; Geddes, T.J. Reciprocal Regulation of Adipogenesis by Myc and C/EBP Alpha. Science 1992, 256, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Hann, S.R.; Sloan-Brown, K.; Spotts, G.D. Translational Activation of the Non-AUG-Initiated c-Myc 1 Protein at High Cell Densities Due to Methionine Deprivation. Genes Dev. 1992, 6, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Blalock, W.L.; Piazzi, M.; Bavelloni, A.; Raffini, M.; Faenza, I.; D’Angelo, A.; Cocco, L. Identification of the PKR Nuclear Interactome Reveals Roles in Ribosome Biogenesis, mRNA Processing and Cell Division. J. Cell Physiol. 2014, 229, 1047–1060. [Google Scholar] [CrossRef]
- Umek, R.M.; Friedman, A.D.; McKnight, S.L. CCAAT-Enhancer Binding Protein: A Component of a Differentiation Switch. Science 1991, 251, 288–292. [Google Scholar] [CrossRef]
- Xiao, Q.; Claassen, G.; Shi, J.; Adachi, S.; Sedivy, J.; Hann, S.R. Transactivation-Defective c-MycS Retains the Ability to Regulate Proliferation and Apoptosis. Genes Dev. 1998, 12, 3803–3808. [Google Scholar] [CrossRef]
- Spotts, G.D.; Patel, S.V.; Xiao, Q.; Hann, S.R. Identification of Downstream-Initiated c-Myc Proteins Which Are Dominant-Negative Inhibitors of Transactivation by Full-Length c-Myc Proteins. Mol. Cell. Biol. 1997, 17, 1459–1468. [Google Scholar] [CrossRef]
- Cazalla, D.; Zhu, J.; Manche, L.; Huber, E.; Krainer, A.R.; Cáceres, J.F. Nuclear Export and Retention Signals in the RS Domain of SR Proteins. Mol. Cell. Biol. 2002, 22, 6871–6882. [Google Scholar] [CrossRef]
- Galmozzi, E.; Casalini, P.; Iorio, M.V.; Casati, B.; Olgiati, C.; Ménard, S. HER2 Signaling Enhances 5′UTR-Mediated Translation of c-Myc mRNA. J. Cell Physiol. 2004, 200, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Notari, M.; Neviani, P.; Santhanam, R.; Blaser, B.W.; Chang, J.-S.; Galietta, A.; Willis, A.E.; Roy, D.C.; Caligiuri, M.A.; Marcucci, G.; et al. A MAPK/HNRPK Pathway Controls BCR/ABL Oncogenic Potential by Regulating MYC mRNA Translation. Blood 2006, 107, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Stoneley, M.; Paulin, F.E.; Le Quesne, J.P.; Chappell, S.A.; Willis, A.E. C-Myc 5′ Untranslated Region Contains an Internal Ribosome Entry Segment. Oncogene 1998, 16, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Le Quesne, J.P.; Stoneley, M.; Fraser, G.A.; Willis, A.E. Derivation of a Structural Model for the C-Myc IRES. J. Mol. Biol. 2001, 310, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.H.; Kim, S.-K.; Kim, C.-Y.; Phi, J.H.; Jun, H.J.; Blume, S.W.; Choi, H.S. Physiological Expression and Accumulation of the Products of Two Upstream Open Reading Frames Mrtl and MycHex1 Along With P64 and P67 Myc From the Human C-Myc Locus. J. Cell. Biochem. 2016, 117, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Thiry, M.; Lafontaine, D.L.J. Birth of a Nucleolus: The Evolution of Nucleolar Compartments. Trends Cell Biol. 2005, 15, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.S.; Zhang, B.; Spector, D.L. Biogenesis and Function of Nuclear Bodies. Trends Genet. 2011, 27, 295–306. [Google Scholar] [CrossRef]
- Gazin, C.; Rigolet, M.; Briand, J.P.; Van Regenmortel, M.H.; Galibert, F. Immunochemical Detection of Proteins Related to the Human C-Myc Exon 1. EMBO J. 1986, 5, 2241–2250. [Google Scholar] [CrossRef]
- Fricker, M.; Hollinshead, M.; White, N.; Vaux, D. Interphase Nuclei of Many Mammalian Cell Types Contain Deep, Dynamic, Tubular Membrane-Bound Invaginations of the Nuclear Envelope. J. Cell Biol. 1997, 136, 531–544. [Google Scholar] [CrossRef]
- Broers, J.L.; Machiels, B.M.; van Eys, G.J.; Kuijpers, H.J.; Manders, E.M.; van Driel, R.; Ramaekers, F.C. Dynamics of the Nuclear Lamina as Monitored by GFP-Tagged A-Type Lamins. J. Cell Sci. 1999, 112 Pt 20, 3463–3475. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Krebs, M.; Boudreau, R.; Giorgi, G.; LeGros, M.; Larabell, C. Actin-Filled Nuclear Invaginations Indicate Degree of Cell de-Differentiation. Differentiation 2003, 71, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Lagace, T.A.; Ridgway, N.D. The Rate-Limiting Enzyme in Phosphatidylcholine Synthesis Regulates Proliferation of the Nucleoplasmic Reticulum. Mol. Biol. Cell 2005, 16, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Echevarría, W.; Leite, M.F.; Guerra, M.T.; Zipfel, W.R.; Nathanson, M.H. Regulation of Calcium Signals in the Nucleus by a Nucleoplasmic Reticulum. Nat. Cell Biol. 2003, 5, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Mickleburgh, I.; Burtle, B.; Hollås, H.; Campbell, G.; Chrzanowska-Lightowlers, Z.; Vedeler, A.; Hesketh, J. Annexin A2 Binds to the Localization Signal in the 3′ Untranslated Region of c-Myc mRNA. FEBS J. 2005, 272, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.K.; Young, D.W.; Javed, A.; Pratap, J.; Montecino, M.; van Wijnen, A.; Lian, J.B.; Stein, J.L.; Stein, G.S. Nuclear Microenvironments in Biological Control and Cancer. Nat. Rev. Cancer 2007, 7, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, T.; Marinkovic, D. Obscure Involvement of MYC in Neurodegenerative Diseases and Neuronal Repair. Mol. Neurobiol. 2021, 58, 4169–4177. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of Cancer: Toward Therapeutic Strategies to Directly Inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Ahmadi, S.E.; Rahimi, S.; Zarandi, B.; Chegeni, R.; Safa, M. MYC: A Multipurpose Oncogene with Prognostic and Therapeutic Implications in Blood Malignancies. J. Hematol. Oncol. 2021, 14, 121. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Soucek, L. The Long Journey to Bring a Myc Inhibitor to the Clinic. J. Cell Biol. 2021, 220, 202103090. [Google Scholar] [CrossRef]
- Vaklavas, C.; Meng, Z.; Choi, H.; Grizzle, W.E.; Zinn, K.R.; Blume, S.W. Small Molecule Inhibitors of IRES-Mediated Translation. Cancer Biol. Ther. 2015, 16, 1471–1485. [Google Scholar] [CrossRef] [PubMed]
- Vaklavas, C.; Grizzle, W.E.; Choi, H.; Meng, Z.; Zinn, K.R.; Shrestha, K.; Blume, S.W. IRES Inhibition Induces Terminal Differentiation and Synchronized Death in Triple-Negative Breast Cancer and Glioblastoma Cells. Tumour Biol. 2016, 37, 13247–13264. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sun, F.; Cheng, Y.; Holmes, B.; Dhakal, B.; Gera, J.F.; Janz, S.; Lichtenstein, A. Critical Role for Cap-Independent c-MYC Translation in Progression of Multiple Myeloma. Mol. Cancer Ther. 2022, 21, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.; Lee, J.; Landon, K.A.; Benavides-Serrato, A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Mechanistic Target of Rapamycin (mTOR) Inhibition Synergizes with Reduced Internal Ribosome Entry Site (IRES)-Mediated Translation of Cyclin D1 and c-MYC mRNAs to Treat Glioblastoma. J. Biol. Chem. 2016, 291, 14146–14159. [Google Scholar] [CrossRef] [PubMed]
- Piazzi, M.; Bavelloni, A.; Faenza, I.; Blalock, W. Glycogen Synthase Kinase (GSK)-3 and the Double-Strand RNA-Dependent Kinase, PKR: When Two Kinases for the Common Good Turn Bad. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118769. [Google Scholar] [CrossRef] [PubMed]
- Vaklavas, C.; Zinn, K.R.; Samuel, S.L.; Meng, Z.; Grizzle, W.E.; Choi, H.; Blume, S.W. Translational Control of the Undifferentiated Phenotype in ER-positive Breast Tumor Cells: Cytoplasmic Localization of ERα and Impact of IRES Inhibition. Oncol. Rep. 2018, 39, 2482–2498. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Indirect c-Myc inhibition | BET family inhibitors | JQ1, Birabresib (OTX015, MK-8628), Molibresib (GSK525762), RO6870810 (RG6146, TEN-0), FT-1101 (CC-95775), ZEN-3694, BMS-986158, AZD5153, BI894999, CPI-0610, GSK2820151, INCB057643, INCB054329 and GS-5829, TEN-010, ABBV-075, PROTACs ARV-771, and ARV-825 |
| BCR inhibition | Ibrutinib, ARQ531 | |
| eIF4A inhibition | Silvestrol | |
| Indirect c-Myc inhibition | PI3K inhibition | Idelalisib, TGR-1202, Fimepinostat (CUDC-907), BR101801 |
| CDK inhibition | Dinacyclib, TG02, KB-0742,THZ1 and THZ2, aminopyrimidines, triazane derivatives, carbamoyl sulfoximide, 4-(4-fluoro-2-methoxyphenyl)-N-1,3,5-triazin-2-amine | |
| PIM1 inhibition | AZD1208, SGI-1776, TP-3654 (SGI-9481), MEN1703, PIM447 | |
| PIN1 inhibition | KPT-6566, Retinoid ATRA, BJP-06-005-3, Sulfopin, PIM447, SEL24 (MEN1703) | |
| PP2A modulation | DT-061, FTY720, OP449, Perphenazine, LB-100 | |
| SKP2 inhibition | SZL-P1-41, FKA, Dioscin, SKPin C1 | |
| USP7 inhibition | P22077, XL177A, GNE-6640, GNE6776, FT671 | |
| JAK2/STAT3 inhibition | MTAP-26, and MTAP-27, WP1066, WP1130, and WP1129 | |
| NF-ĸB inhibition | Guggulsterone | |
| Src kinase inhibition | Saracatinib | |
| FBXW7 activation | Oridonin, HAO472 | |
| Aurora-A inhibition | Alisertib (MLN8054, MLN8237), CD532 | |
| Aurora-B inhibition | AZD1152 | |
| PLK-1 inhibition | BI6727 | |
| HUWE1 inhibition | BI8622 a BI8626 | |
| HDAC inhibition | Entinostat, Tucidinostat, CUDC-907 | |
| Direct c-Myc inhibition | G quadruplex stabilisation | CX-3543, APTO-253, IZCZ-3, cationic porphyrins (TMPyP4), quarfloxin, DM039, ruthenium complexes (Se2Py3, Se2SAP) |
| Antisense oligonucleotides | AVI- 4126, MYC-ASO, INX-3280, INX-6295 | |
| Miniproteins and protein domains | OmoMYCs (OMO-103, OMO-1, FPPa-OmoMYC), Bac- ELP-H1, PNDD1, ME47, Mad, alfa-helix peptide H1 | |
| Myc/Max interaction disruption | ME47, EN4, 3jc48-3, pyrazolo [1,5-a]-pyrimidines (MYCro1, MYCro2 a Mycro3), KJ-Pyr-9 (Kröhnke pyridine), MYCMI-6, MYCMI-7, MYCi975, MYCi361, KSI-3716, MYRA-A, MI1-PD, KI-MS2-008, quinolone derivatives (KSI-1449, KSI-2302, and KSI-3716), substituted pyrazole compounds (NUCC-0176242, and NUCC-0176248), IIA6B17, 10058-F4, 10074-G5, JY-3-094, JKY-2-169, SaJM589 | |
| Max/Max homodimers sabilization | KI-MS2-008, NSC13728 |
| p64 Myc (c-Myc2) | p67 Myc (c-Myc1) | mrtl | MycHex1 | c-Myc S | |
|---|---|---|---|---|---|
| Structure | well known | contains additional 14 amino acids at its N terminus compared to p64 Myc | N-terminal region single transmembrane domain, C-terminal sequence interaction domain with homology to RNA-binding proteins | highly basic protein, capable of homo-oligomerization | c-Myc S lacks the N-terminal transactivation domain |
| Expression | predominant gene product of the c-Myc locus | lost in many tumours | unknown | IRES facilitates translation of the MycHex1 | higher levels of c-Myc S have been transiently observed during the rapid growth phase of several cell types |
| p64 Myc (c-Myc2) | p67 Myc (c-Myc1) | mrtl | MycHex1 | c-Myc S | |
| Function | oncogenic properties, p64 c-Myc isoform transactivates via the canonical EMS sequence and fails to transactivate the EFII enhancer element via the C/EBP binding site | growth inhibitory properties, p67 is a potent and specific transactivator of the enhancer element EFII via the C/EBP binding site and also transactivates via the canonical EMS sequence, mediates growth inhibitory response under nutrient depletion or contact inhibition | regulates c-Myc translation and localization to the nucleus, contributes to the role of the c-Myc locus in oncogenesis (IRES), might be part of a complex which regulates the translation, localization, or processing of mRNA | possibly involved in replication, RNA processing, and formation of nuclear bodies | c-Myc S protein lacks transactivation capacity, but it is able to inhibit p64 and p67, which suggests a dominant-negative inhibitory function |
| Onthology | conserved in chimpanzee, Rhesus monkey, dog, cow, mouse, rat, chicken, zebrafish, and frog | mrtl and MycHex1 are found only in primates | human, mouse, and avian cells | ||
| Subcellular localisation | mainly nucleus and cytoplasm | mainly nucleus and cytoplasm | nuclear envelope, ER, tubular and cisternal structures of the NR | colocalizes with fibrillarin | mainly nucleus and cytoplasm |
| Additional information | stoichiometric balance between p64 and p67 is important for cellular metabolism regulation and proliferation | colocalize in the central cisternal reservoir of the nucleoplasmic reticulum | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubickova, A.; De Sanctis, J.B.; Hajduch, M. Isoform-Directed Control of c-Myc Functions: Understanding the Balance from Proliferation to Growth Arrest. Int. J. Mol. Sci. 2023, 24, 17524. https://doi.org/10.3390/ijms242417524
Kubickova A, De Sanctis JB, Hajduch M. Isoform-Directed Control of c-Myc Functions: Understanding the Balance from Proliferation to Growth Arrest. International Journal of Molecular Sciences. 2023; 24(24):17524. https://doi.org/10.3390/ijms242417524
Chicago/Turabian StyleKubickova, Agata, Juan Bautista De Sanctis, and Marian Hajduch. 2023. "Isoform-Directed Control of c-Myc Functions: Understanding the Balance from Proliferation to Growth Arrest" International Journal of Molecular Sciences 24, no. 24: 17524. https://doi.org/10.3390/ijms242417524
APA StyleKubickova, A., De Sanctis, J. B., & Hajduch, M. (2023). Isoform-Directed Control of c-Myc Functions: Understanding the Balance from Proliferation to Growth Arrest. International Journal of Molecular Sciences, 24(24), 17524. https://doi.org/10.3390/ijms242417524