Two alternative approaches, our SP2 protocol and ethyl acetate extraction (EE), were systematically benchmarked for the off-line clean-up of plant samples with regard to SDS. Additionally, the SP2 clean-up protocol was applied for the removal of PEG. The experiments were designed for peptide input amounts of 250 ng and 10 µg. Samples of Arabidopsis thaliana tryptic peptides, generated by FASP, were subsequently contaminated, as follows: (1) SDS to three levels of a final concentration of 0.1%, 1%, and 5%, or (2) PEG to three levels of a final concentration of 0.01%, 0.1%, and 1% (SP2 clean-up only). As an additional variable for the EE workflow, the optimal number of iterations for a given SDS concentration was also assessed for the clean-up of a 250-ng sample. The differences between the two clean-up methods were evaluated based on the LC-MS/MS data processed by the DIA-NN program using the following characteristics: (1) the number of quantified peptides, (2) the peptides’ signal characteristics (intensity, area, peak width, and retention time), and (3) the physical-chemical properties (hydrophobicity and molecular weight). The results were compared to the control peptide digest after FASP without any artificial contamination or clean-up. In the case of the 10-µg peptide input, the intensities were normalized according to the injection volume. Only the peptides quantified in at least two replicates of at least one sample category of the given comparison (control, clean-up of uncontaminated sample, or clean-up of samples with different levels of contamination) were used for the comparisons. The peptide intensity ratios were calculated to the control sample intensities, with the missing values being pairwise imputed by the minimum value observed for the given processing type to assess the qualitative changes.
The SP2 protocol was also tested for the clean-up of the 250-ng and 10-µg samples contaminated simultaneously by 5% SDS and 1% PEG. Finally, the SP2 protocol with no adjustments was applied for the clean-up of the 10-ng sample.
2.1. SDS Removal by Ethyl Acetate Extraction
In our common practice, three EE iterations are usually sufficient to clean up the samples from the residual SDS, e.g., after FASP, which might occur especially after processing higher amounts of protein loads. However, our aim was to evaluate the capabilities and limits of ethyl acetate extraction on different amounts of peptide samples; therefore, we tested this method with excessive SDS contaminations of up to 5% in combination with the increased number of iterations of up to 12.
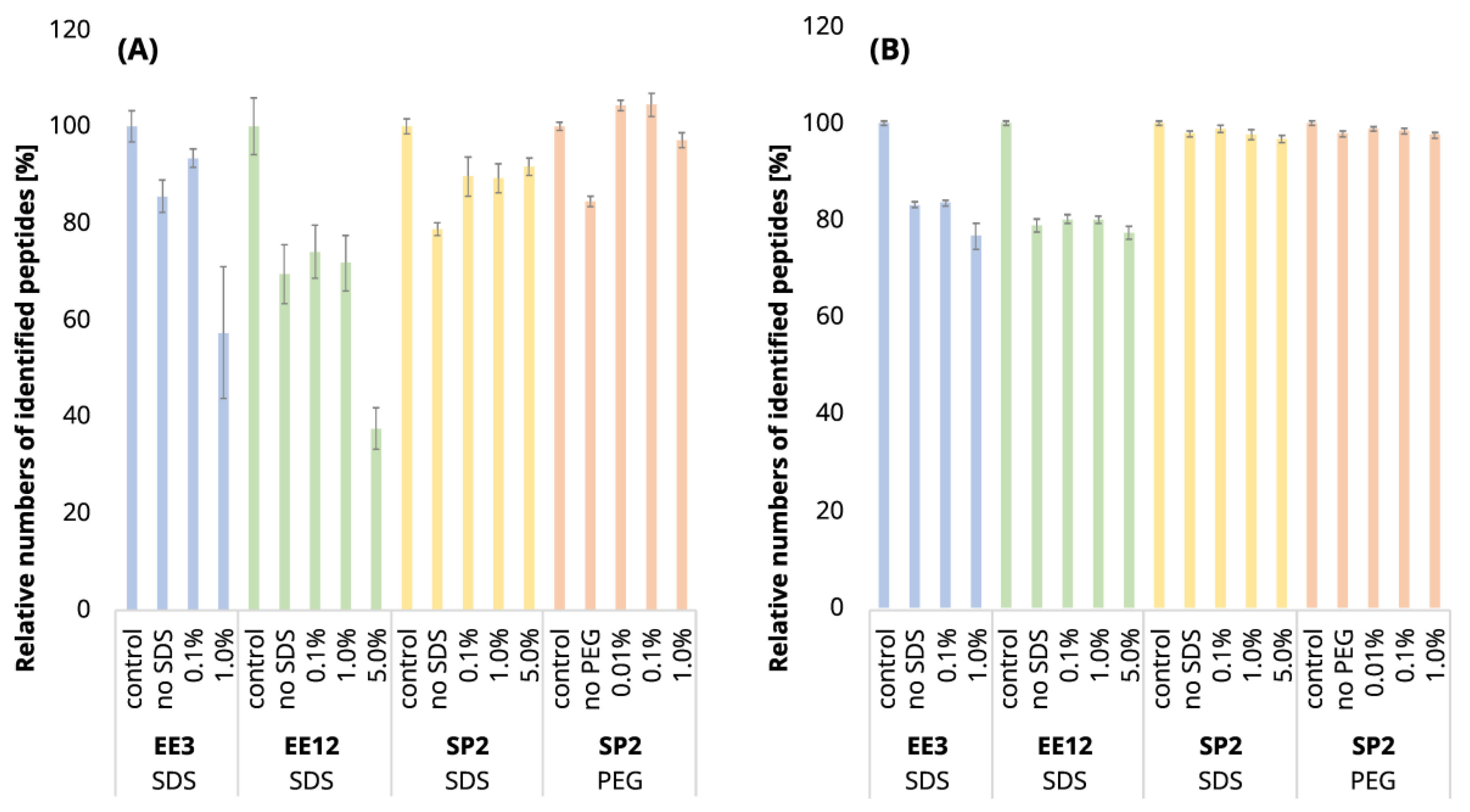
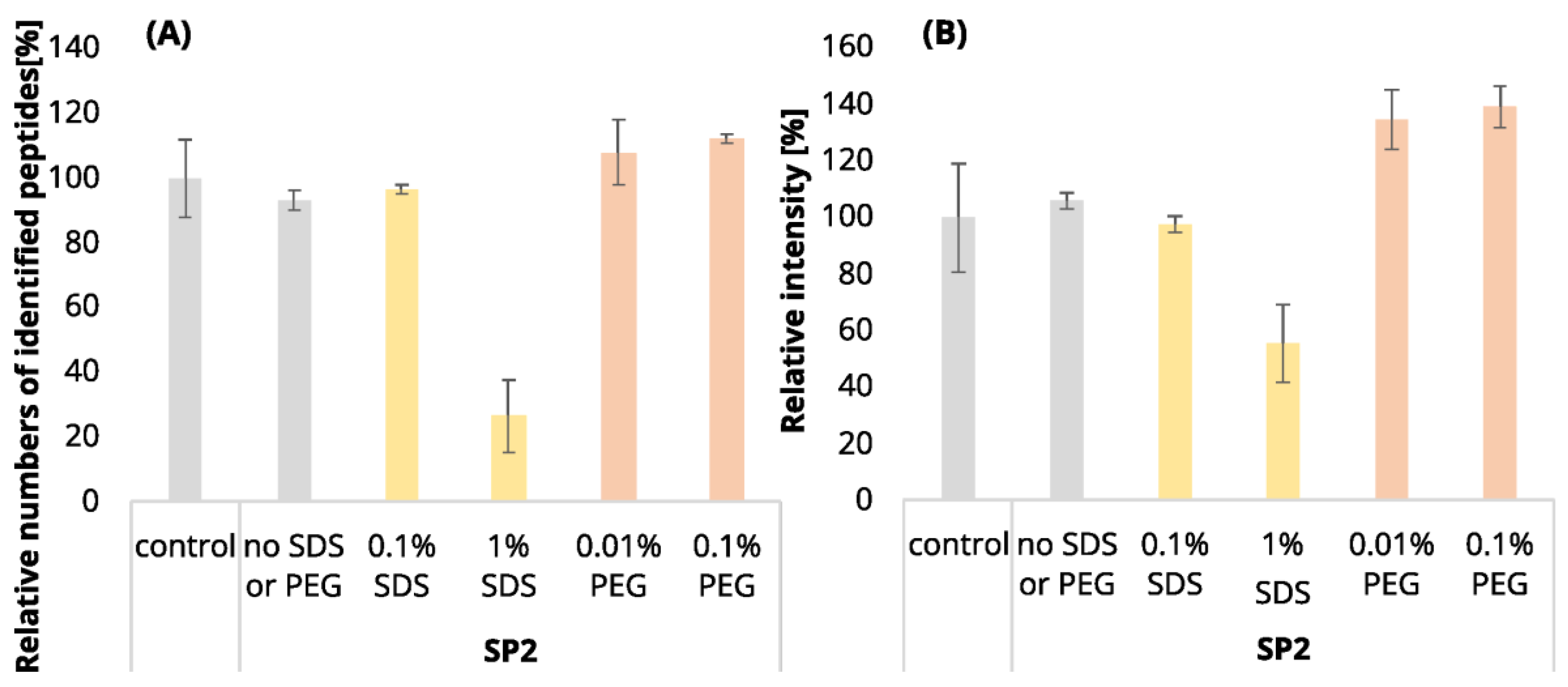
It is expectable that the sample losses will increase with the higher number of EE iterations. Indeed, we observed a loss of 30.6% of the identified peptides after EE with 12 iterations of an uncontaminated 250-ng peptide input, while in the case of 3 EE iterations, it was only 14.5% (compared to the control sample,
Figure 1A, blue and green bars;
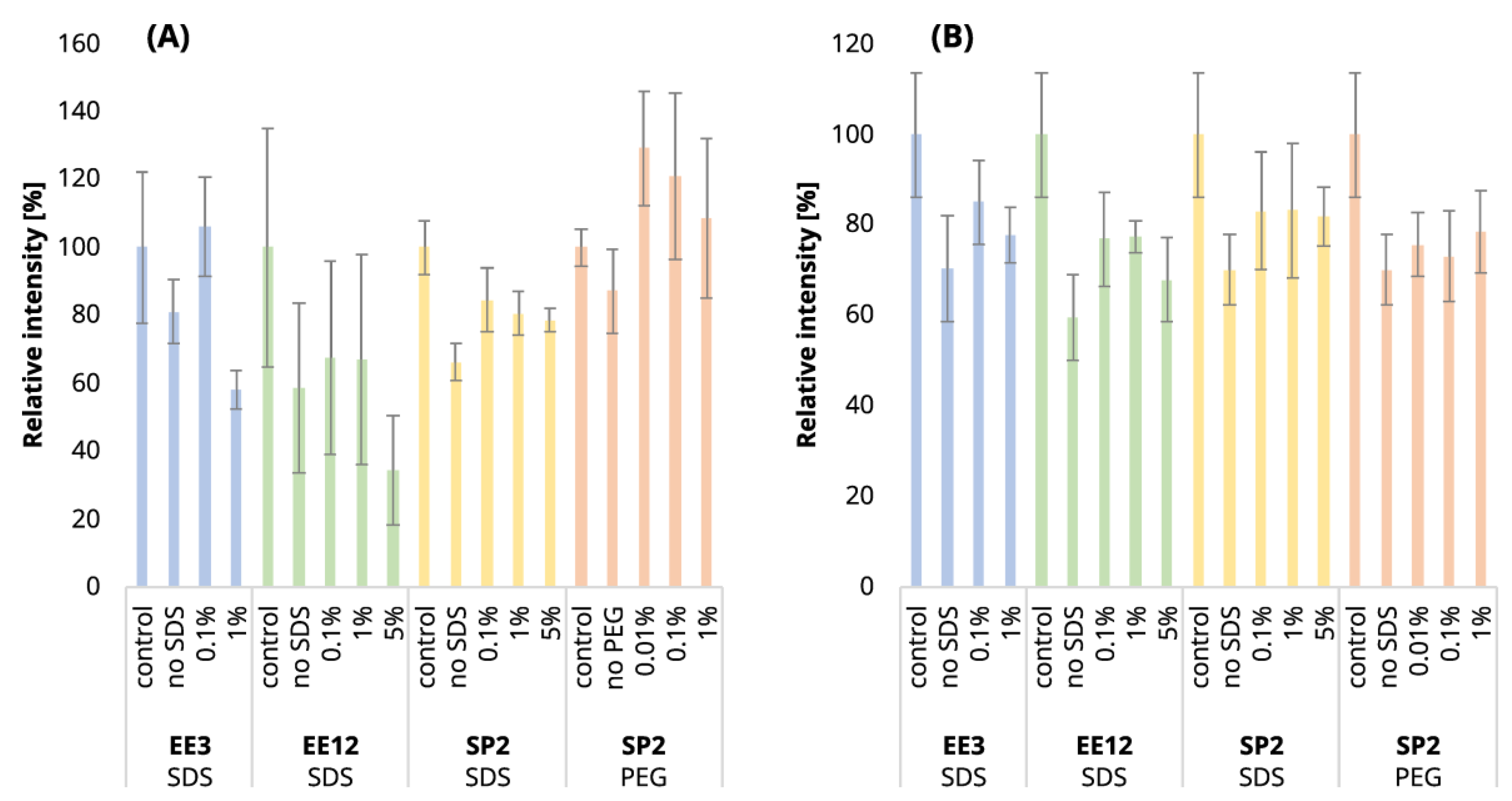
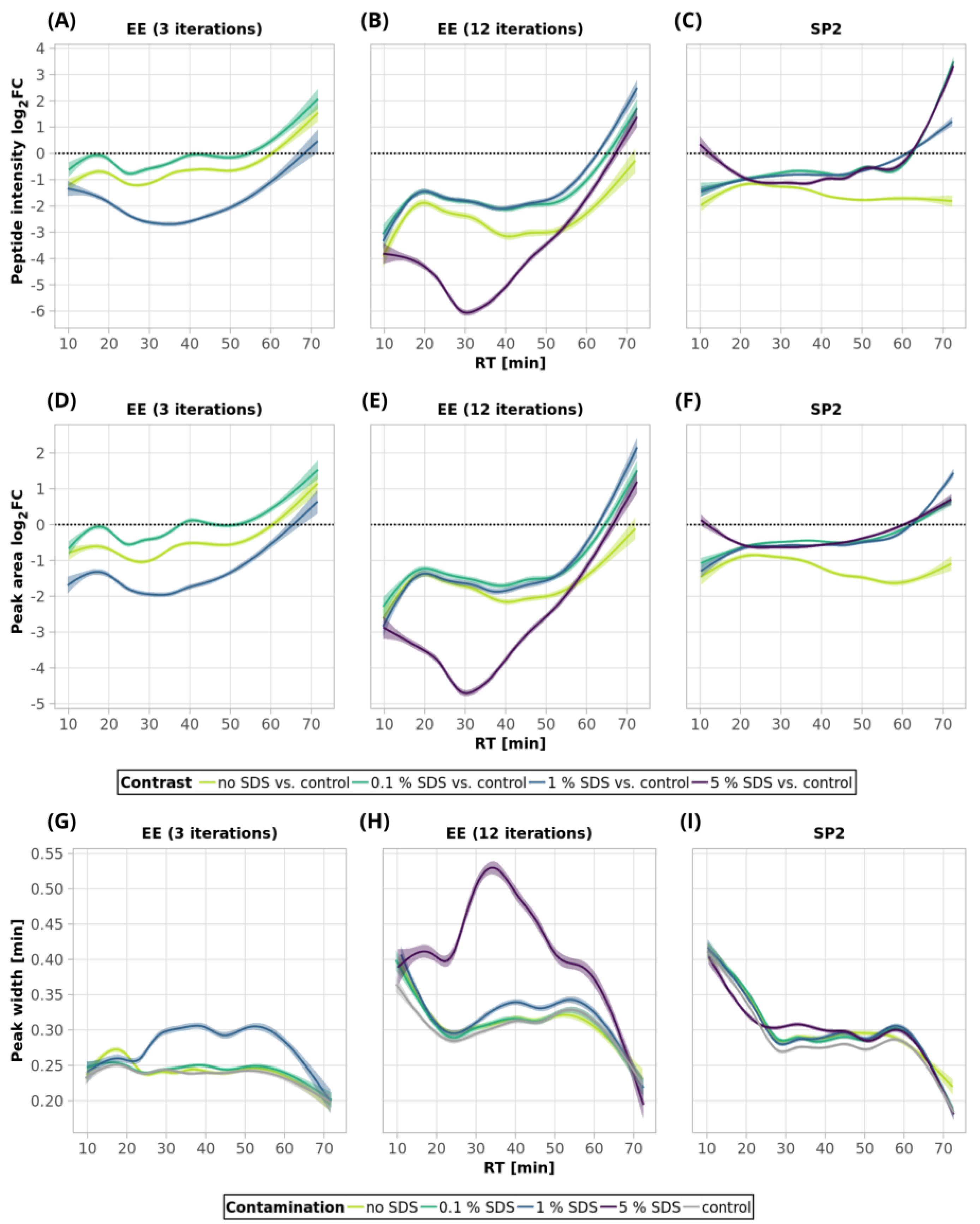
Table S1). The increased number of iterations was also reflected in the decrease in peptide intensities (
Figure 2A, blue and green bars). After 3 iterations, the mean intensities sum decreased by 19%, but 12 EE iterations led to a reduction of 41%, on average.
Using three EE iterations for the removal of 0.1% SDS contamination from a 250-ng peptide input sample was sufficient, and resulted in even more identified peptides (
Figure 1A,
Table S1) and lower quantitative losses compared to the processing of the uncontaminated sample (
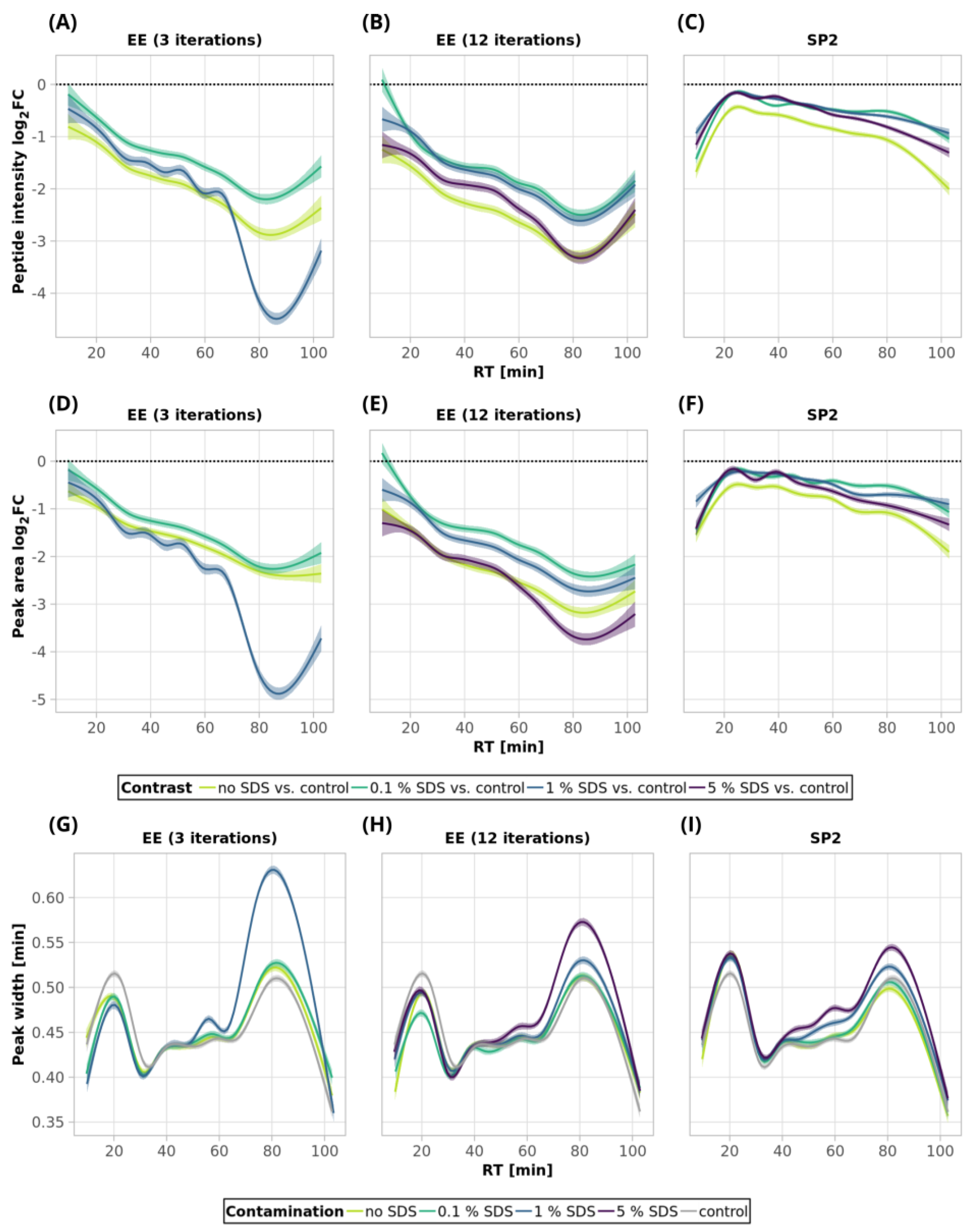
Figure 3A, green and light-green lines; detailed scatterplots for individual GAM curves can be seen in
Figure S1). We lost only 6.6% of the identified peptides and observed comparable mean sum intensity compared to the control sample (
Figure 1A and
Figure 2A,
Table S1), and the peak width profiles were not distorted (
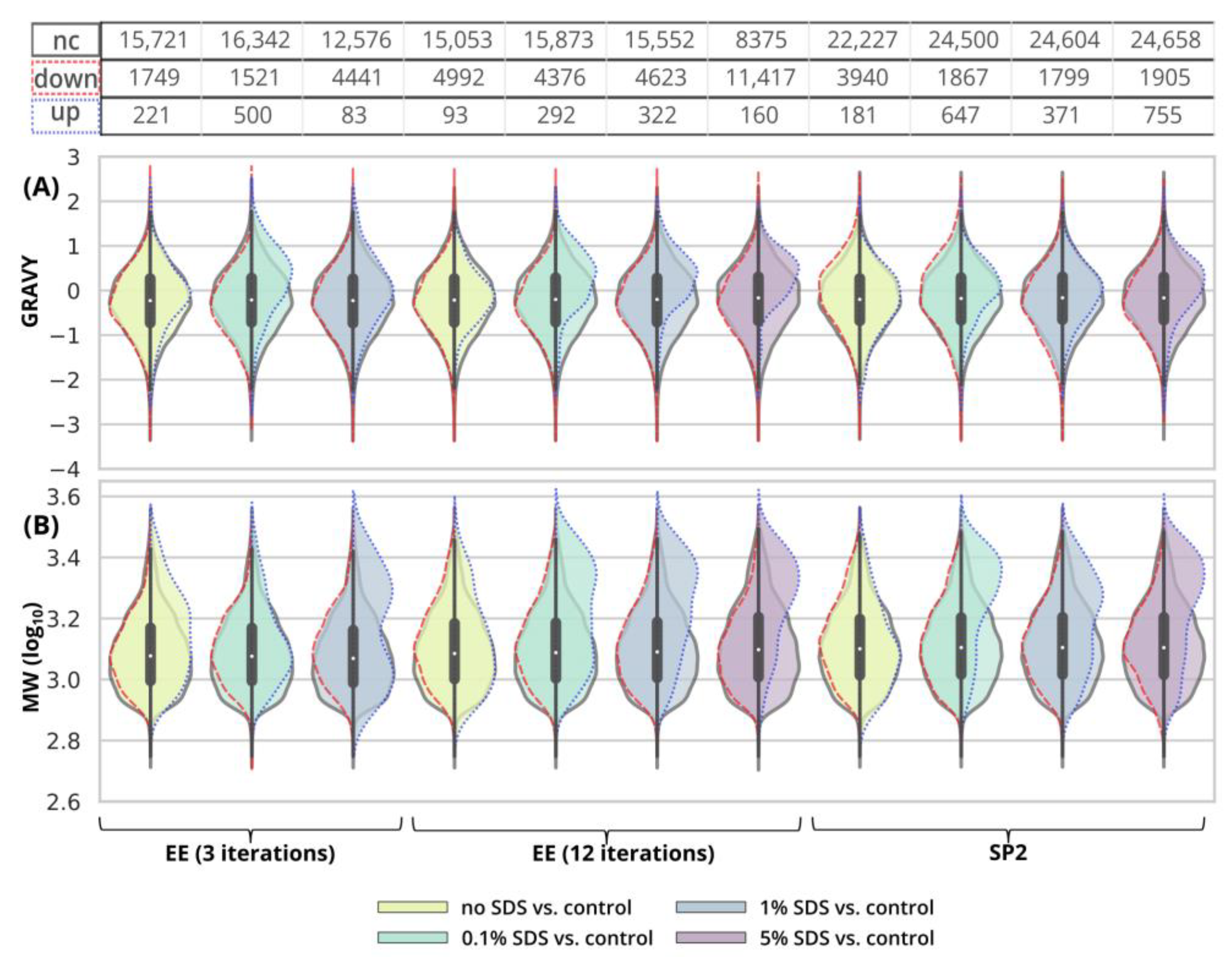
Figure 3G, green and light-green GAM curves). We observed that the peptides with 10× higher intensity compared to the control have a higher GRAVY index (
Figure 4A and
Figure S2), but the distribution of the molecular weight level practically did not change (
Figure 4B and
Figure S3). However, three EE iterations proved to be insufficient for the removal of 1% SDS contamination. We lost about half of the peptides (42.7%,
Figure 1A,
Table S1), and, furthermore, most of the identified peptides had decreased intensities compared to the control (
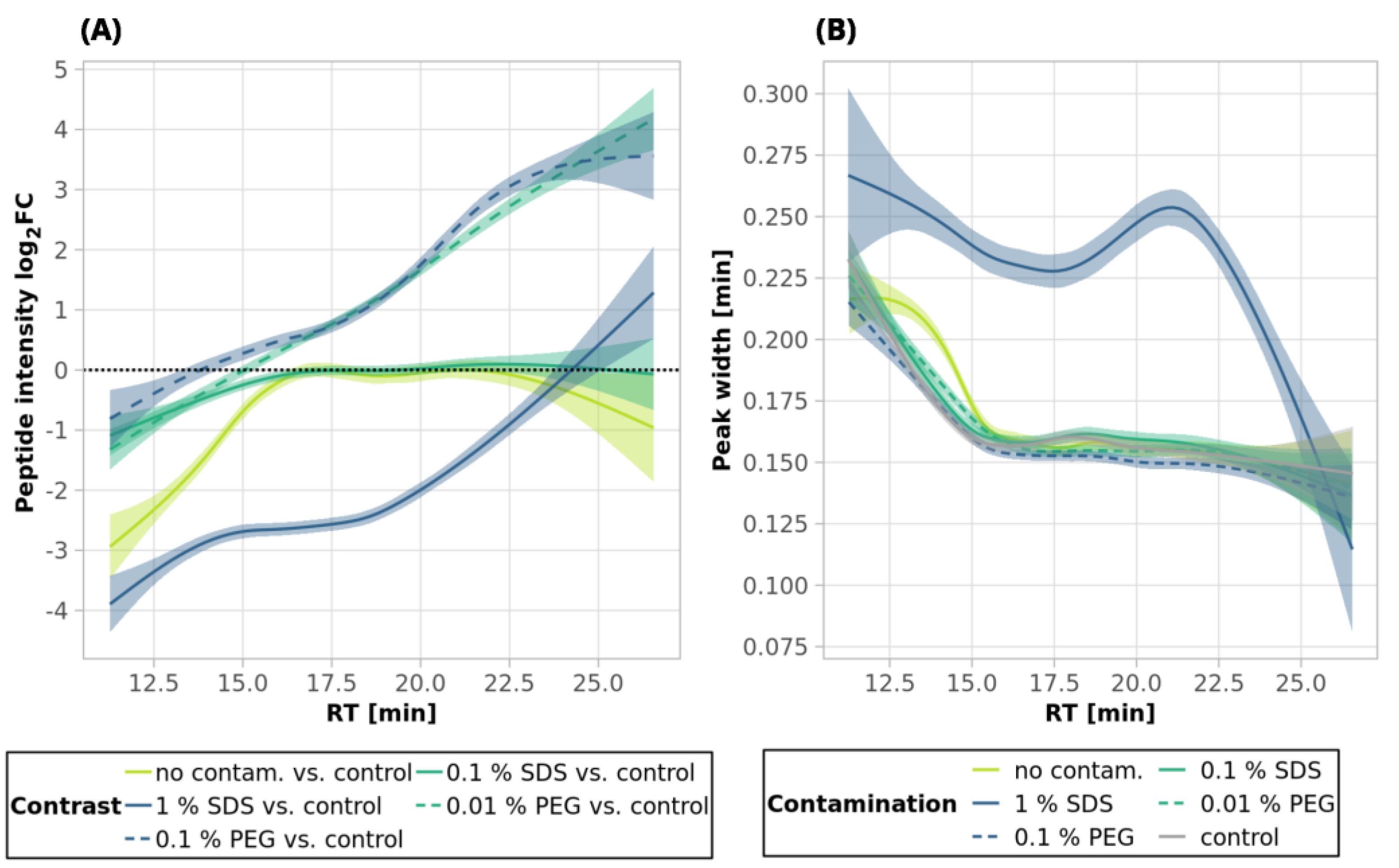
Figure 3A, blue line,
Figure S1), and the mean sum intensity decreased by 42% (
Figure 2A). The imperfect removal of 1% SDS is also documented by widening the peak widths (
Figure 3G; compare blue—1% SDS and green—0.1% SDS lines), indicating disturbed chromatographic separation.
In an attempt to remove higher SDS contaminations, we applied 12 EE iterations, which we considered as an acceptable processing maximum, to 1% and 5% SDS concentrations. The removal of 1% SDS contamination from the 250-ng peptide input sample was satisfactory with respect to the chromatographic behavior (
Figure 3H, blue line), but we observed qualitative and quantitative losses comparable to those obtained for the processing of the uncontaminated input sample
Figure 1A and
Figure 2A,
Table S1). In the case of the removal of 5% SDS contamination, even 12 iterations of EE were not sufficient, as evident from the shifted elution profile of iRT peptides (not shown), as well as the increased peak widths in general (
Figure 3H, magenta line). As a result, the number of detected peptides decreased by 62.6% and 46.1% compared to the control and processed uncontaminated samples under the given conditions, respectively (
Figure 1A,
Table S1). We observed a decrease in the mean intensities sum by 65.5% (
Figure 2A) and corresponding high quantitative losses of individual peptides (
Figure 3B, magenta line,
Figure S1) compared to the control sample.
As SDS widens the peak widths, in addition, we evaluated the quantitative changes based on the peak areas and observed similar trends to those seen for the intensities (
Figure 3D–F).
Since even 12 EE iterations cannot remove the 5% SDS contamination, and the procedure was accompanied by substantial losses in the number of identified peptides (
Figure 1A,
Table S1), we continued in search for the optimal number of EE iterations only for the removal of 1% SDS contamination. The results obtained from the initial experiment showed that three EE iterations are not sufficient for the removal of 1% SDS from 250-ng peptide input samples, so we have tested a higher number of iterations (six and nine iterations) in order to select the optimal conditions for the removal of 1% SDS from 250 ng of peptides. The chromatographic peaks narrowed, as the result of the successful SDS removal, already after the application of six EE iterations, with comparable losses as those seen for three EE iterations (
Figure S4B). Nine EE iterations further slightly improved the peak widths (
Figure S4B) but led to higher losses (
Figure S4A).
In conclusion, three EE iterations were sufficient to remove contamination up to 0.1% SDS from 250-ng
A. thaliana peptide samples (
Figure 1A,
Figure 2A and
Figure 3,
Table S1), and six EE iterations are recommended for the removal of contaminations up to 1% SDS. We have not found sufficient EE conditions for the removal of 5% SDS contamination.
In the case of 10 µg of uncontaminated peptide input, the number of identified peptides decreased by 16.8% after EE with 3 iterations and by 21.1% after 12 iterations (
Figure 1B,
Table S1). The sum intensity decreased by 29.5 and 40.3% after 3 and 12 EE iterations, respectively (
Figure 2B, blue and green bars). Three EE iterations satisfactorily removed 0.1% SDS, the number of identified peptides decreased by 16.4%, and the mean sum of intensities was lower by 15% compared to the control. The peak widths are comparable to those of the control (
Figure 5G, green line).
The 1% SDS was the highest SDS concentration that we attempted to clean-up by three EE iterations in the case of the 10-µg input. The number of identified peptides decreased by 23.3% compared to the control, and the mean sum of peptide intensities decreased by 22.1% (
Figure 1B and
Figure 2B and
Table S1). The peptides with the most decreased intensities were observed in the second half of the gradient, which correlates with increased peak widths (
Figure 5A,G, blue line, and
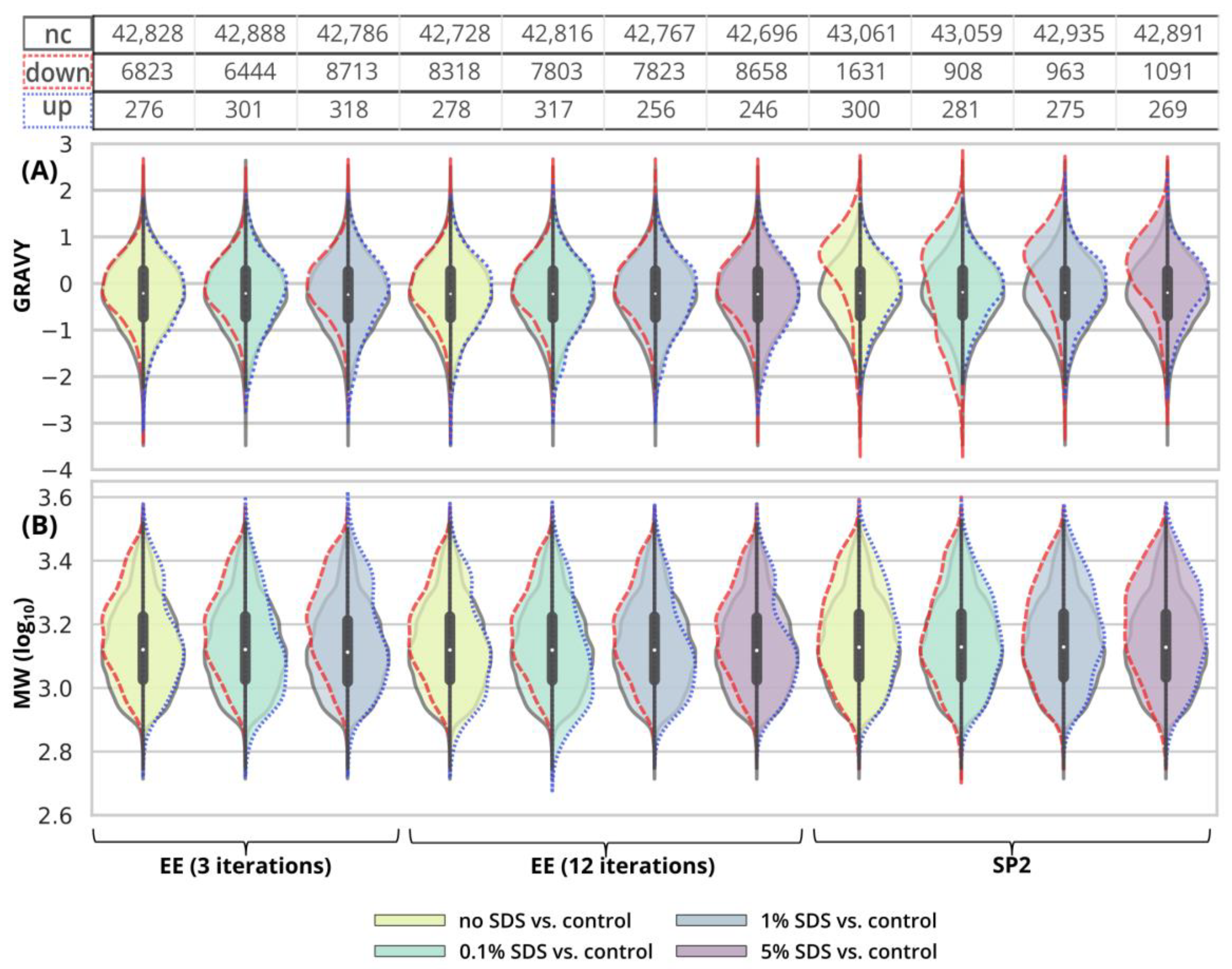
Figure S5). No distinct trends in hydrophobicity and molecular weight of peptides with reduced intensity were observed (
Figure 6,
Figures S6 and S7).
EE extraction with twelve iterations was tested for the clean-up of the 10-µg peptide input from the whole range of SDS contamination levels (0.1–5%). After the clean-up of 0.1%, 1%, and 5% SDS, we identified 19.8, 20.0, and 22.6% fewer peptides compared to the control, and the mean sum of intensities decreased by 23.1, 23.0, and 32.1%, respectively (
Figure 1B and
Figure 2B and
Table S1). The same trends were observable not only in the intensities, but also in the peptides’ areas (
Figure 5E). In the case of 5% SDS, the peak widths were slightly increased, but they are comparable to those of the control for the lower SDS concentrations (
Figure 5H). We did not find any distinct trends in qualitative or quantitative behavior of the peptides concerning hydrophobicity or molecular weight (
Figure 6,
Figures S6 and S7).
Thus, EE extraction with 3 iterations can be applied for the clean-up of SDS contaminations up to 1% and with 12 iterations even up to 5%, with reasonable losses compared to those of the control for the 10-µg peptide input. We observed a positive effect of the presence of the SDS during EE on the suppression of losses caused by the clean-up step (compared to the uncontaminated sample), especially on the quantitative level (
Figure 5A,B).
2.2. SDS Removal by SP2
After the initial optimization of the SP2 protocol in terms of solution volume and bead amount (see the final protocol in the
Supplementary Material), we applied the SP2 procedure for the removal of SDS concentration up to 5% from both peptide inputs analogically, as for EE.
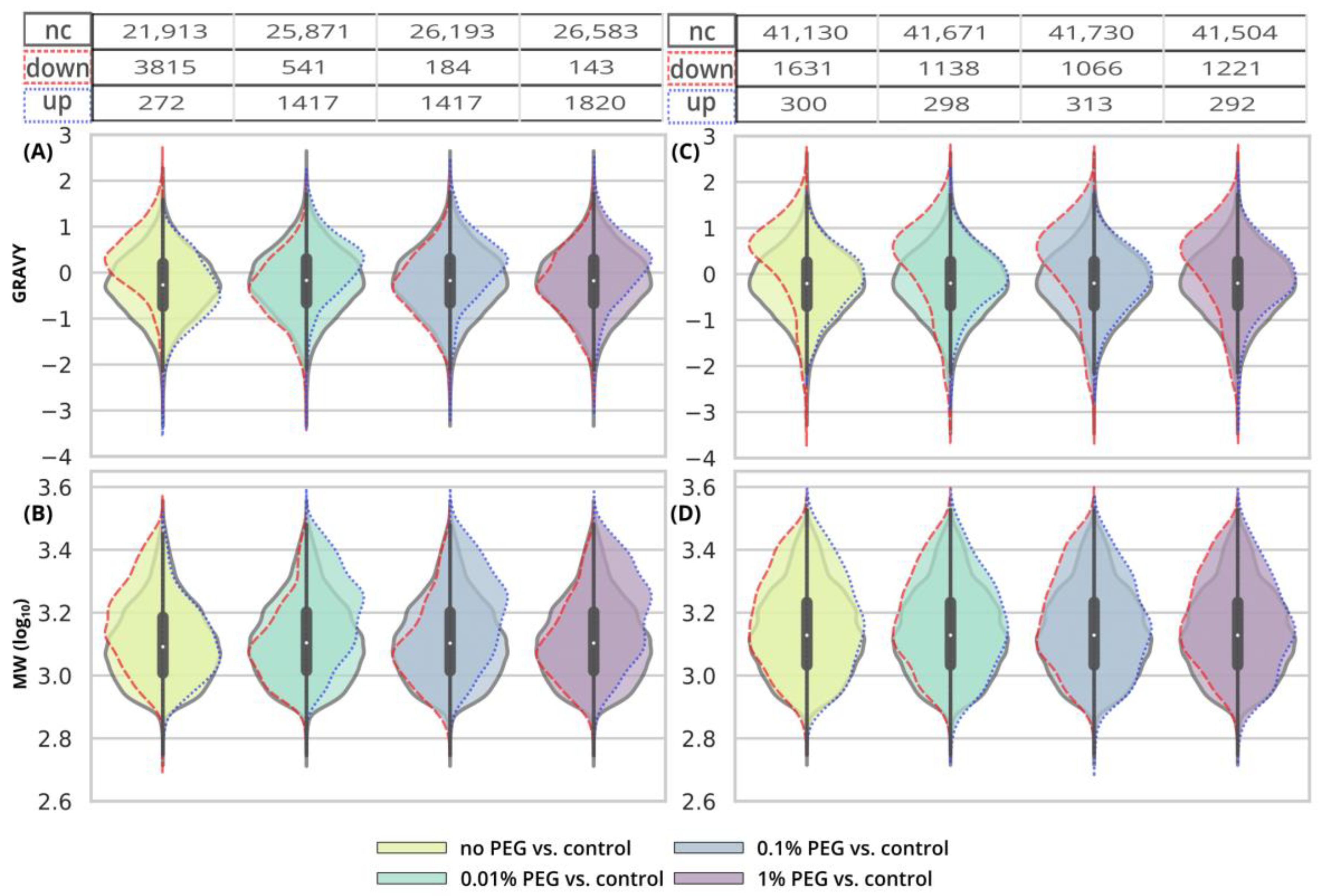
After the clean-up of the 250-ng peptide input without any contamination, we observed the loss of 21.3% of the identified peptides compared to the control peptide sample, and the average intensities decreased by 33.6% (
Figure 1A and
Figure 2A yellow bars,
Table S1). The results from the analyses of the peptide solutions contaminated with 0.1% SDS showed similar trends to the EE clean-up process. SDS contamination, again, resulted in the reduction in quantitative losses (
Figure 2A and
Figure 3C and
Figure S1) and more identified peptides compared to the processing of the uncontaminated sample (
Figure 1A,
Table S1). Specifically, we lost only 10.4% of the identified peptides, and the average intensity decreased only by 15.4% when the 250-ng peptide input sample was contaminated with 0.1% SDS. Moreover, hydrophobic peptides eluting at the end of the LC gradient displayed higher intensities compared to those in the analysis of the control sample (
Figure 3C). Similar trends as those seen for 0.1% SDS contamination were observed also for 1% and 5% SDS. Comparable peaks’ widths for all contamination levels indicated complete removal of SDS from all of the contaminated 250-ng peptide input samples up to 5% concentration (
Figure 3I).
Inspecting the data in terms of hydrophobicity (GRAVY index) and molecular weight revealed that, after SP2 treatment of the uncontaminated sample, we lost the peptides with a slightly decreased GRAVY index (
Figure 4A and
Figure S2), but the distribution of the molecular weight did not change (
Figure 4B and
Figure S3). As for the GRAVY indexes, we did not observe any distinct differences among the contamination levels; however, in the case of 5% SDS, there was a small population of peptides with increased intensities compared to the control group, with higher, more hydrophobic GRAVY values (
Figure 4A and
Figure S2). In the presence of SDS in the sample subjected to the SP2 protocol, the peptides with increased intensities compared to the control had a decreased molecular weight (
Figure 4B and
Figure S3).
The 10-µg input was processed in the same way via the SP2 protocol. We observed similar losses in terms of the number of identified peptides regardless of the presence or concentration levels of SDS (3.2% at most,
Figure 1B,
Table S1). However, the average peptide intensities sum decreased by 29.9% (
Figure 2B) in the absence of the SDS in the sample. If the sample was contaminated with SDS, the intensity did not decline as dramatically (by 18.1% on average with the 5% SDS contamination at most,
Figure 2B). The quantitative losses increased with the retention time (
Figure 5C), and the same trend was also visible in the quantitative changes based on the area (
Figure 5F). The minor chromatographic peaks widening compared to the control sample was observed in the samples contaminated with 5% and 1% SDS; however, the peak widths were narrower than the corresponding ones after the EE processing for either 3 or 12 iterations (
Figure 5G,I; magenta lines).
After the SP2 treatment of the 10-µg input, we observed hydrophobic peptides with quantitative losses regardless of the presence or concentration of SDS in the sample (
Figure 6A and
Figure S6). We did not observe any major changes in molecular weight distribution (
Figure 6B and
Figure S7).
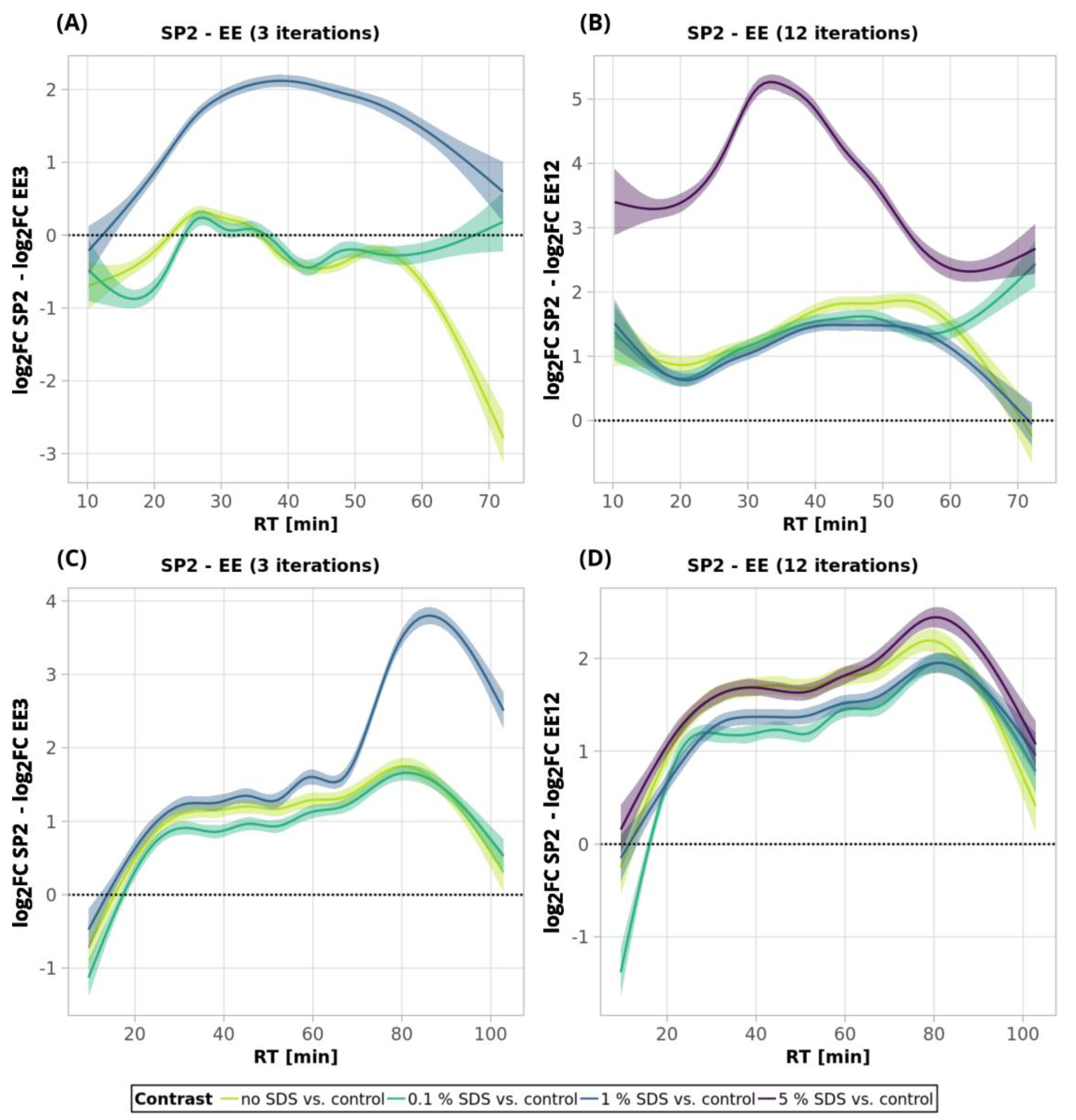
To summarize the comparison of the performance of ethyl acetate extraction and SP2 for SDS removal, we compared the changes in the intensities of individual peptides obtained using respective clean-up methods to the respective control (no contamination, no clean-up processing) experiment (
Figure 7). The SP2 outperformed EE in all of the tested scenarios, except for the 250-ng peptide amount with no SDS contamination and three EE iterations. This might be explained by the generally higher peptide losses of the SP2 compared to the EE (three iterations).
2.3. PEG Removal by SP2
After SDS contamination, we applied the SP2 protocol for the removal of another common contamination—polyethylene glycol (PEG)—which cannot be removed by ethyl acetate extraction.
Three different concentrations (0.01, 0.1, and 1%) of a wide range of PEG polymer length mixtures (from PEG 200 to PEG 8000) were tested.
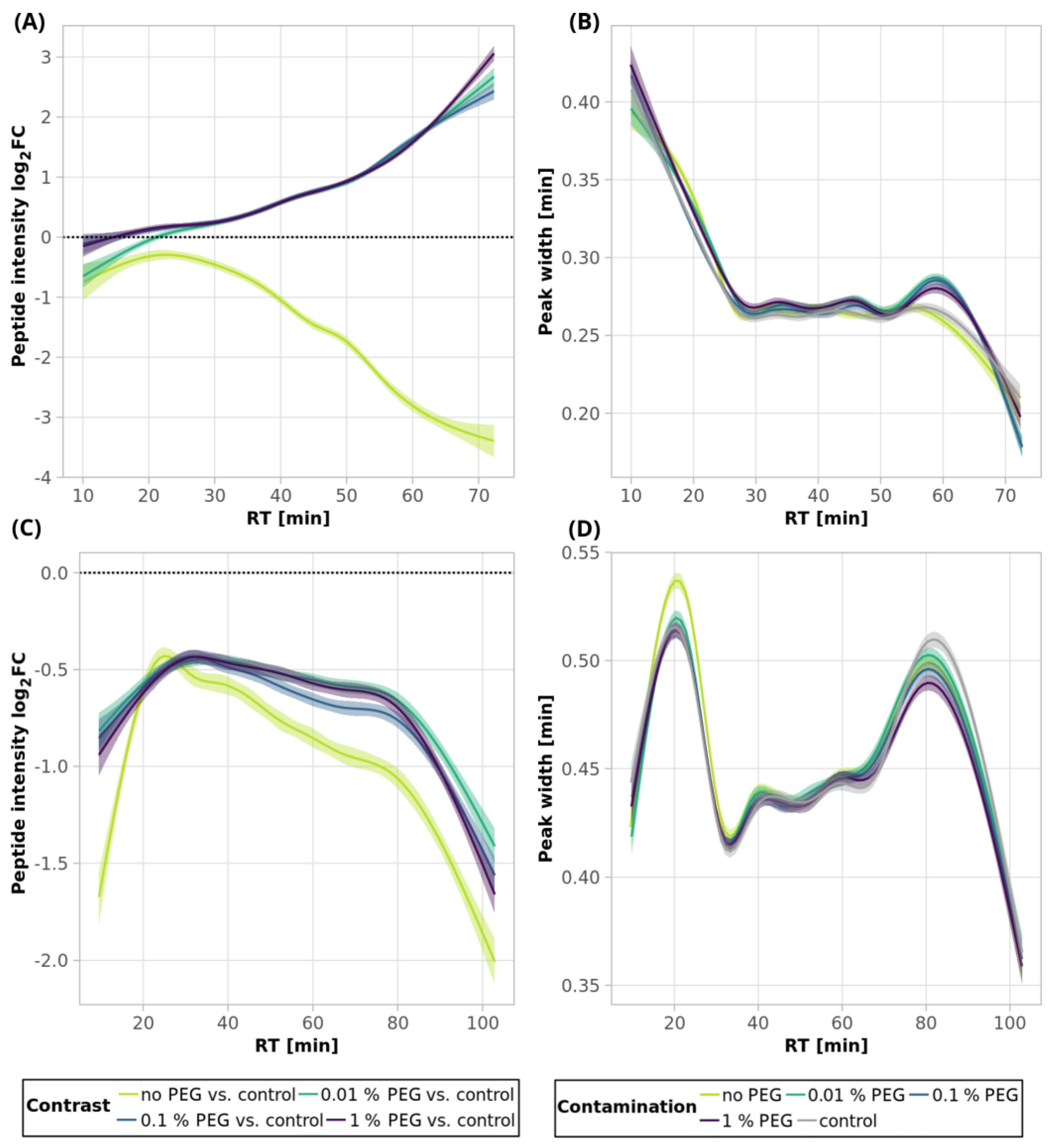
Similarly to the SDS contamination removal, we detected a decreased number of peptides (84.4%) after SP2 treatment of the uncontaminated sample with a lower mean sum intensity (87.1%) than that found for the control sample using 250-ng peptide input (
Figure 1A and
Figure 2A,
Table S1). The quantitative losses increased throughout the retention time (
Figure 8A, light-green line and
Figure S8), which, together with the GRAVY value distribution (
Figure 9A and
Figure S9A), points to the existence of more hydrophobic peptides losses during the SP2 process.
However, we observed a higher number of detected peptides and mean quantities compared to the control sample in the presence of 0.01 and 0.1% PEG contamination in the sample followed by SP2 clean-up (
Figure 1A and
Figure 2A, orange bars,
Table S1). The contamination of the sample by 1% PEG resulted in the reduction in detected peptides after the SP2 clean-up by 3.0%, compared to control (
Figure 1A,
Table S1). The beneficial effect of PEG on quantitative changes is reflected in
Figure 8A and
Figure S8, where it can be noticed that the peak intensities increase more for later eluting, thus showing more hydrophobic peptides. This is supported by the increased intensity dependence on GRAVY (
Figure 9A and
Figure S9A).
The beneficial effect of PEG contamination might be related to incomplete PEG removal, in agreement with our previous study on sample losses in LC-MS vials [
19], despite the different PEG used in these studies. The previous study used single PEG 20,000, while here we have used a mixture of PEGs in order to better mimic various PEG contamination sources. We have indeed noticed the presence of some residual PEG after SP2 clean-up of the 250-ng peptide input samples with initial 0.1 and 1% PEG concentrations during the quality control analyses (
Figure S10). Quality control analyses were run in a single-column setup (without the trap column), enabling better detectability of the PEG contamination in the on-line-connected mass spectrometer. Compared to the corresponding injection of PEG (0.01%), the residual PEG concentrations are estimated to be below 0.002%. As expected, based on our experience with PEG utilization to minimize sample losses [
19], it is evident that the residual PEG in the sample did not influence the peak width (
Figure 8B).
After the SP2 clean-up of the 10-µg peptides contaminated with all three concentrations of PEG, the number of identified peptides slightly decreased (less than 4%) compared to the unprocessed control sample (
Figure 1B, orange bars,
Table S1), as well as the mean sum of intensities (
Figure 2B, orange bars). The quantitative losses were more evident with an increasing retention time (
Figure 8C and
Figure S11). However, the numbers of identified peptides are comparable to those obtained by SP2 processing of the uncontaminated sample, and the mean sums of intensities are even higher for all of the tested PEG concentrations (
Figure 1B and
Figure 2B,
Table S1). The peak widths are comparable for all of the samples, regardless of clean-up processing (
Figure 8D). We observed a small population of hydrophobic peptides with decreased intensities but without any distinct change in the distribution of molecular weight (
Figure 9C,D and
Figure S12).
2.4. SP2 Clean-Up of SDS and PEG Combined Contamination
Next, we tested the SP2 protocol for removing the combination of 5% SDS and 1% PEG (maximum applied concentrations).
After cleaning the contaminated sample containing 250 ng of peptides, we lost only 2.9% of the identified peptides, and the mean sum of intensities increased by 7.6% in comparison with the control (
Table S1,
Figure S13A). We observed both scenarios, including peptides with decreased (approximately 1st half of the LC gradient) as well as increased (approximately 2nd half of the LC gradient) intensities after the SP2 process (
Figure S14A). The GRAVY indexes and molecular weight distributions are shown in
Figure S15A,B. The peak widths are comparable to the control and the uncontaminated sample after SP2 (
Figure S14B), but a bit wider, demonstrating close-to-complete SDS contamination removal.
In the case of the 10-µg peptide input, we identified 4.0% fewer peptides compared to the control, and the mean sum of intensities decreased by 16% (
Table S1 and
Figure S13B). We observed mainly peptides with decreased intensities, with the negative effect being more profound with an increasing retention time of the peptide (
Figure S14C). The peak widths were mostly slightly increased compared to the control and uncontaminated sample after SP2 (
Figure S14D), suggesting higher residual SDS contamination compared to the 250-ng peptide variant. The distribution of intensity log
2FC values based on the GRAVY indexes showed decreased intensities mainly for the more hydrophobic peptides, with no apparent trend in MW (
Figure S15C,D), a slightly different situation compared to the 10-µg input, where the same peptide categories showed increased intensities (
Figure S15A,B).
2.5. SP2 Clean-Up of 10-ng Peptide Input Sample
In order to evaluate the applicability of our SP2 protocol to lower the peptide input amounts used routinely on highly sensitive instrumentation, we tested this method on a 10-ng peptide input. We tried to clean up the sample individually from SDS (0.1 and 1%) and PEG (0.01 and 0.1%) contamination.
After the SP2 treatment of the uncontaminated sample, we lost 6.9% of the identified peptides (
Figure 10A,
Table S2), and the mean sum of intensity increased by 5.6% (
Figure 10B). The SP2 clean-up of the 10-ng peptide sample contaminated with 0.1% SDS resulted in the loss of 3.5% of the identified peptides, and the mean sum of intensities decreased by only 2.4% (
Figure 10A,B,
Table S2). The peak widths are comparable to the control and the uncontaminated samples after the clean-up (
Figure 11B, green and light-green continuous lines). However, in the case of 1% SDS, we encountered the limits of the protocol. We observed a loss of 73.6% of the identified peptides, and the mean sum of intensities decreased by 44.7% (
Figure 10A,B,
Table S2). There were quantitative losses throughout the gradient (
Figure 11A, blue continuous line), and the peak widths increased, which indicates presence of the SDS in the sample even after the SP2 clean-up (
Figure 11B, continuous blue line).
After the SP2 clean-up of the 10-ng peptide input contaminated with 0.01 and 0.1% PEG, we identified 7.9 and 12.4% more peptides compared to the control, respectively (
Figure 10A,
Table S2). The intensities increased by 34.8 and 39.1%, respectively (
Figure 10B). The increase in intensities is observable for the majority of peptides throughout the gradient, but increases with increasing retention time (
Figure 11A, dashed lines). The peak widths are comparable to the control and the cleaned-up uncontaminated samples, as expected (
Figure 11B, dashed lines).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}