

Nicotinamide-Expanded Allogeneic Natural Killer Cells with CD38 Deletion, Expressing an Enhanced CD38 Chimeric Antigen Receptor, Target Multiple Myeloma Cells

 ,
,  ,
,
Abstract
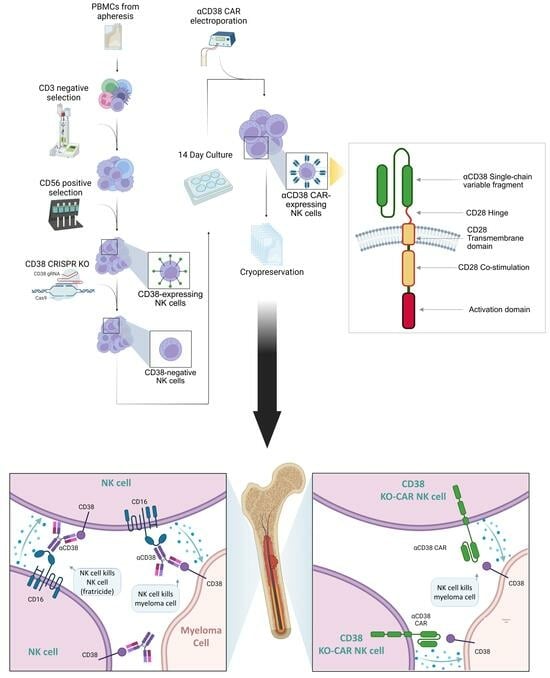
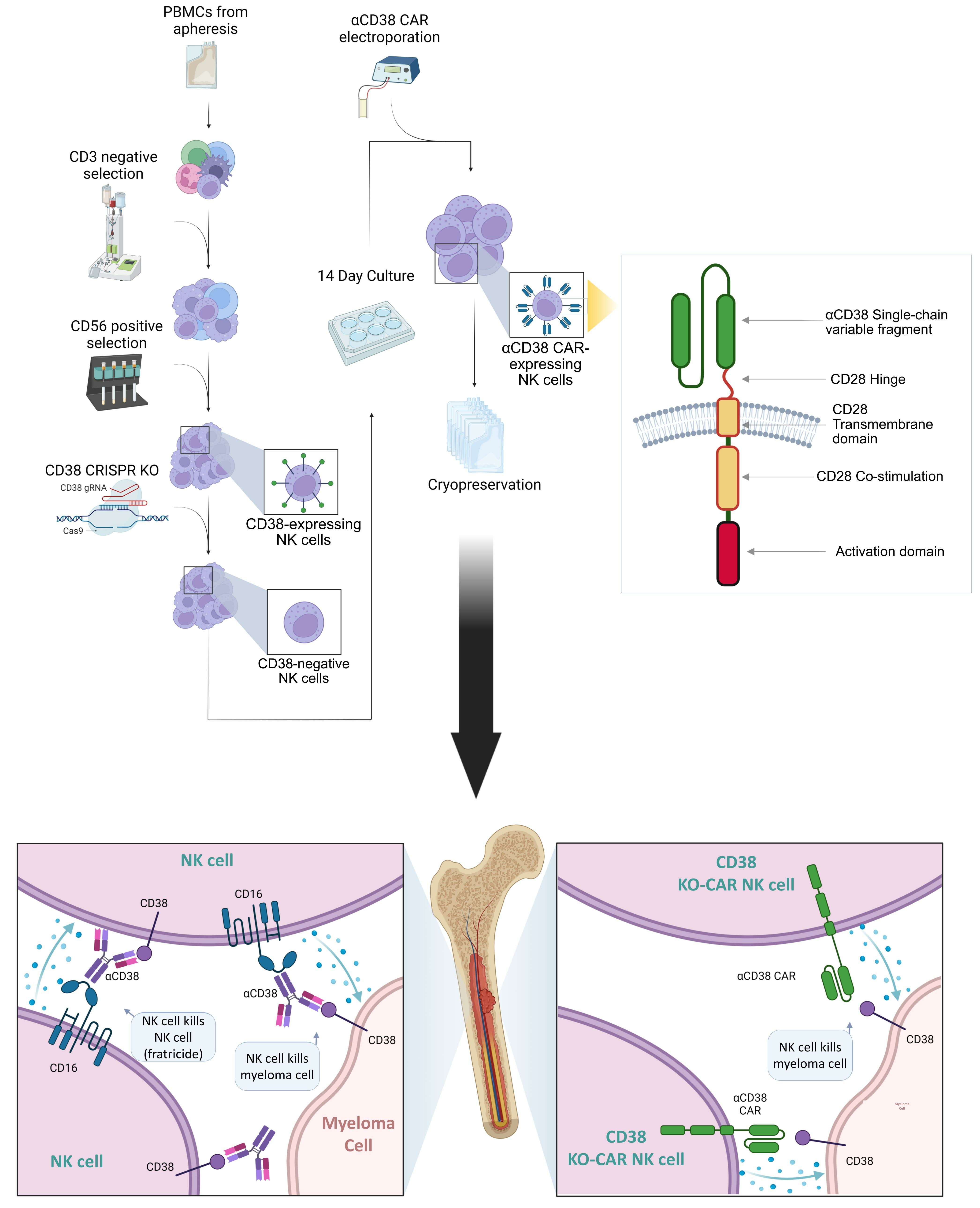
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
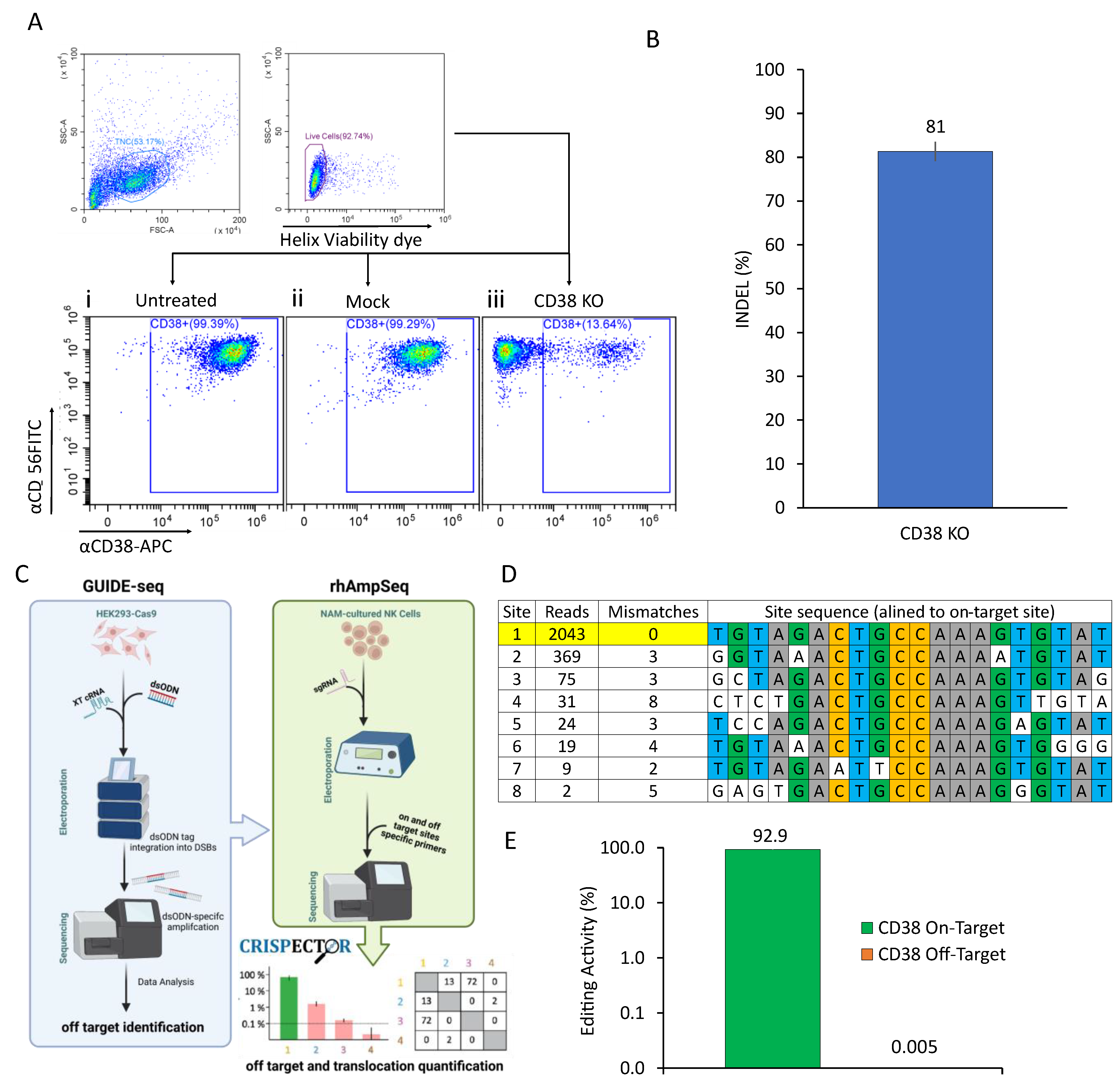
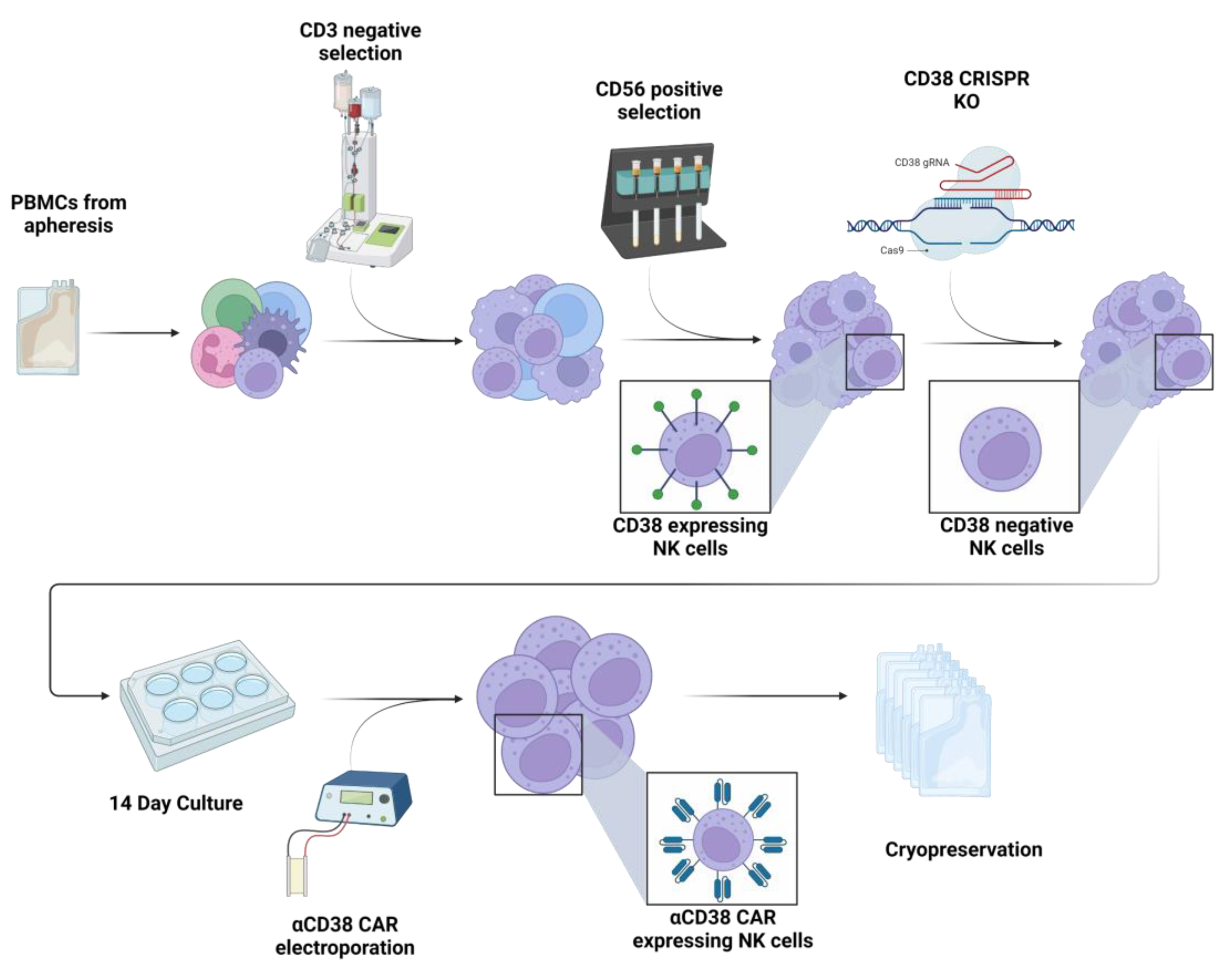
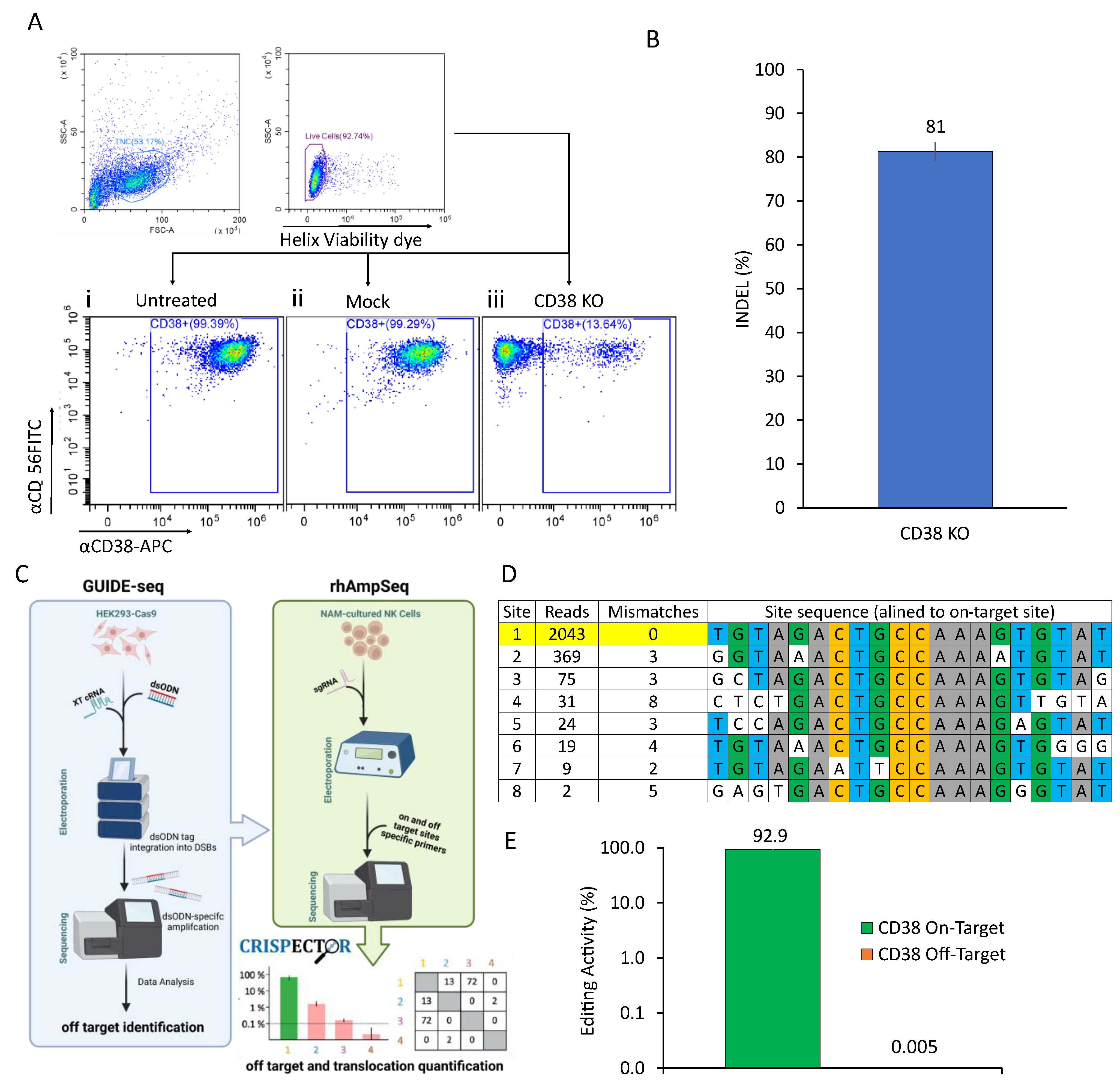
2.1. CRISPR-Based CD38 Knock-Out in Primary NK Cells Is Precise and Efficient
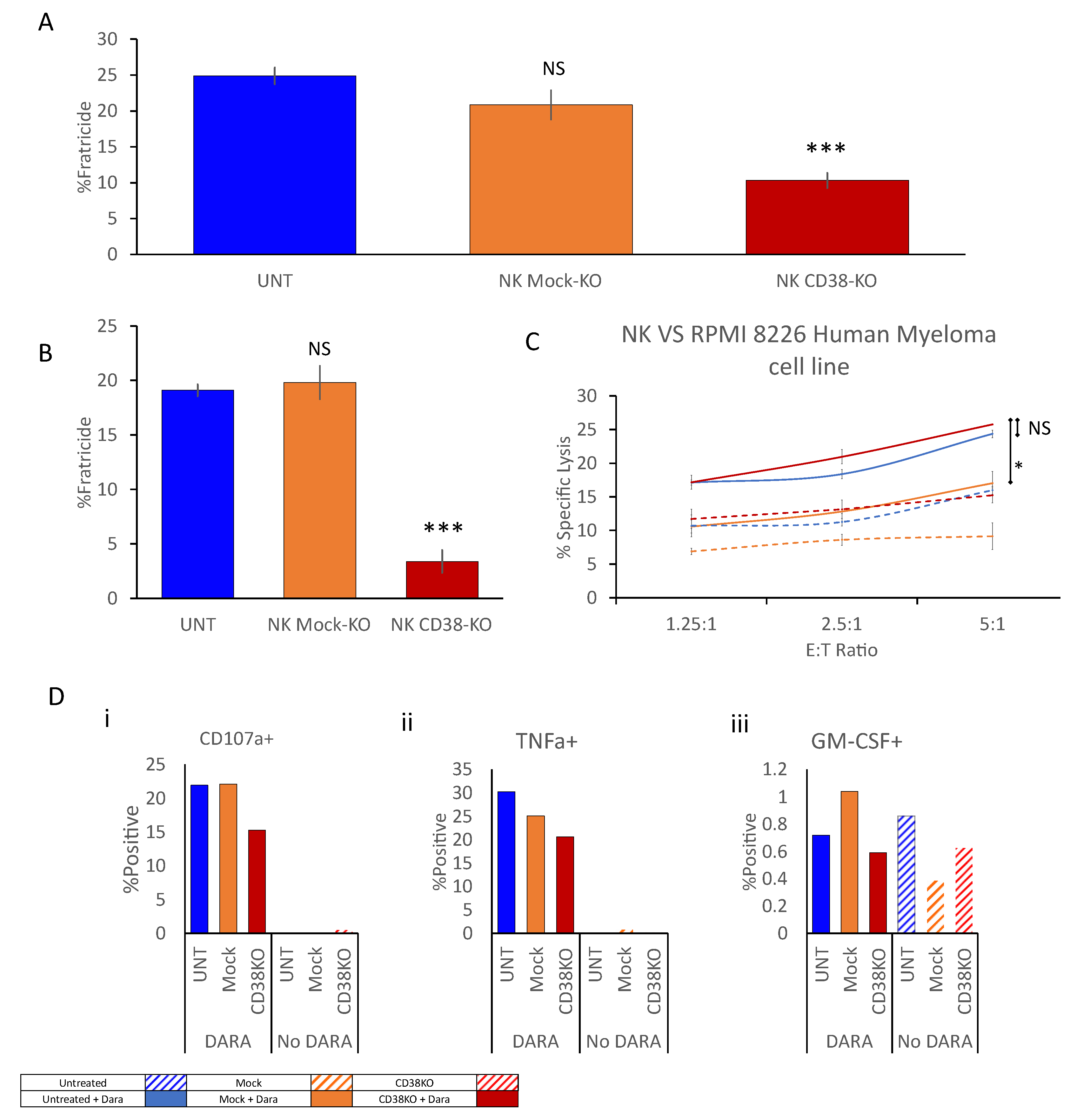
2.2. CD38 Knock-Out Eliminates αCD38-Mediated Fratricide
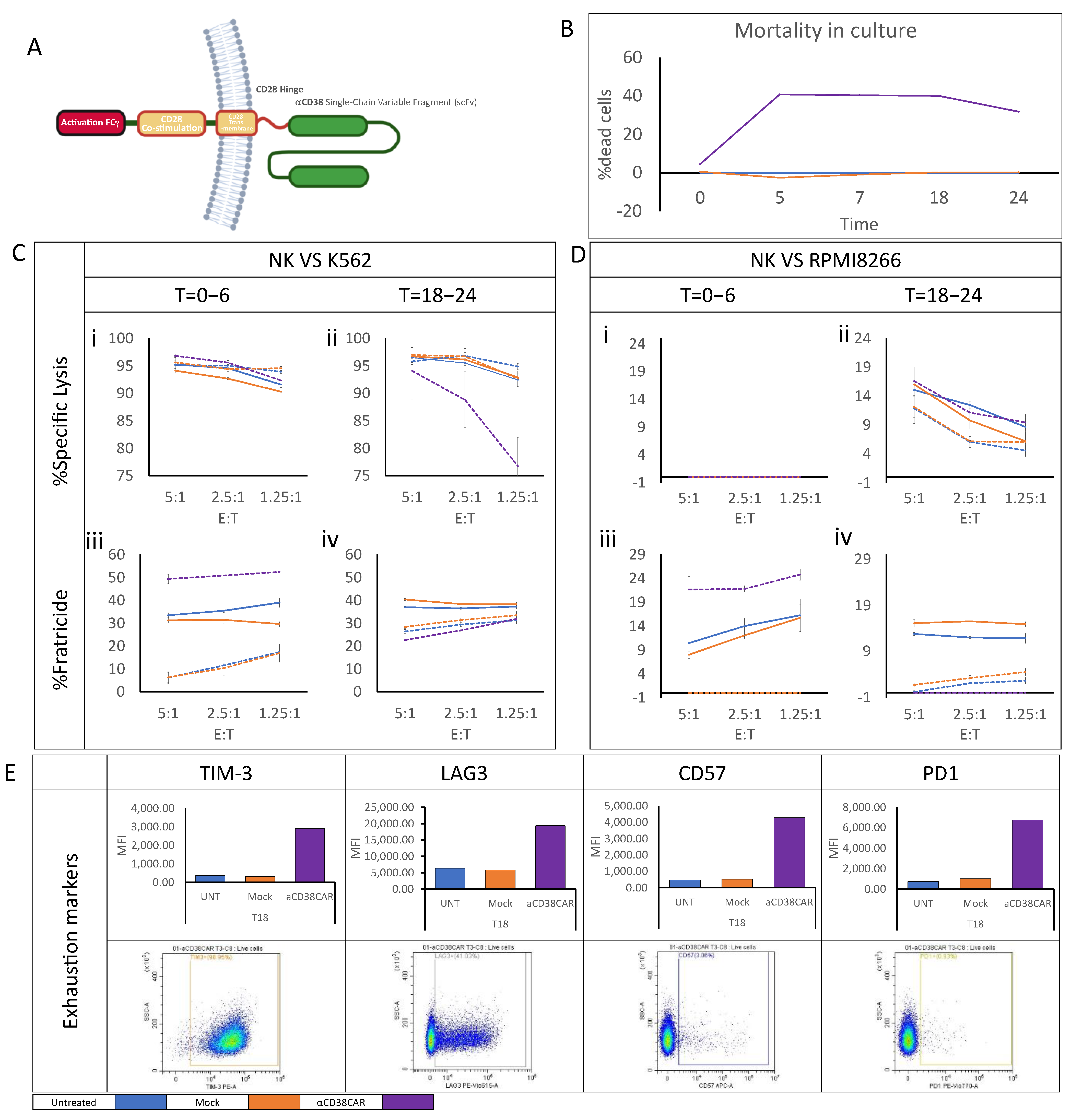
2.3. Anti-CD38 CAR Expression Results in Extensive Fratricide and Effector Cell Exhaustion
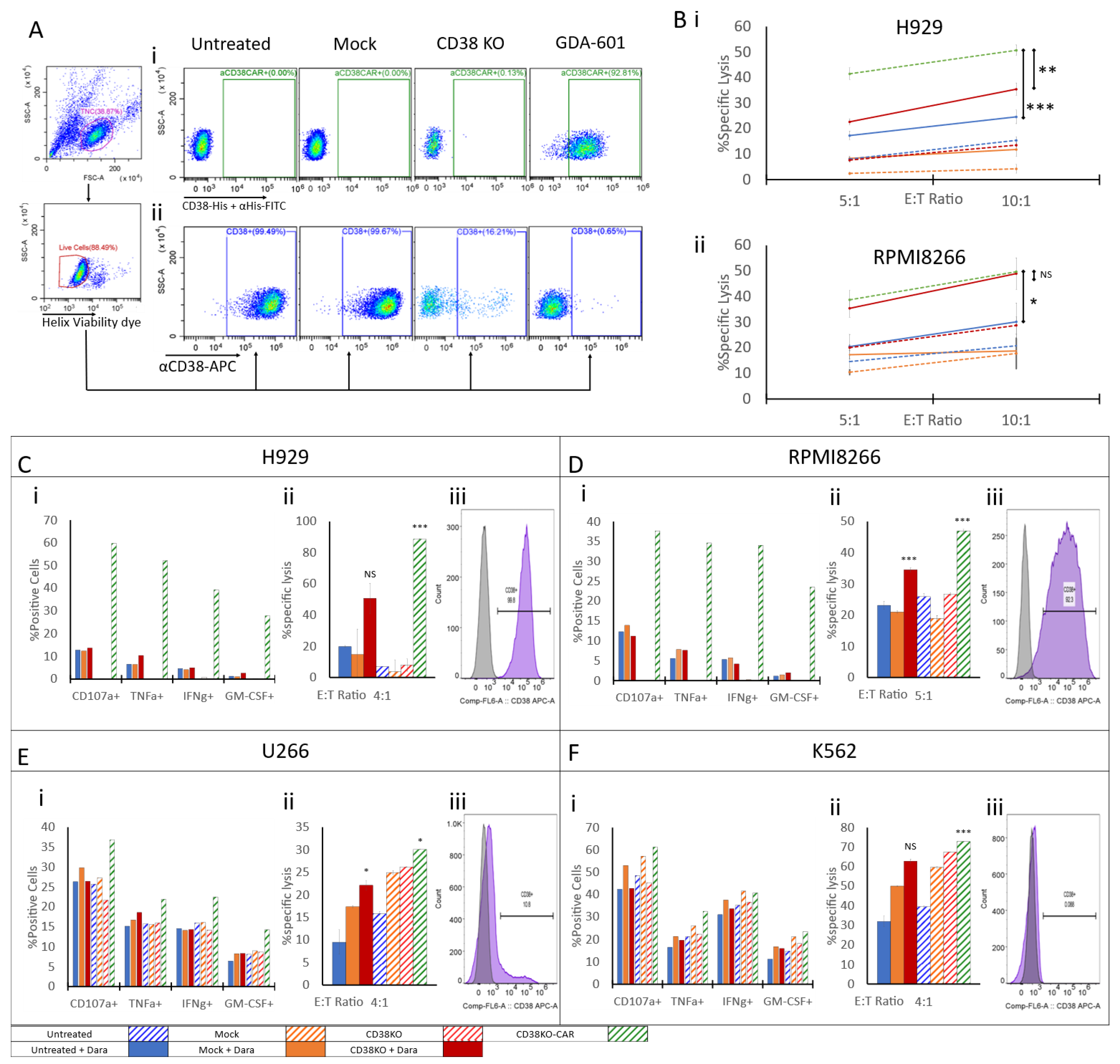
2.4. CD38 Knock-Out NK Cells Expressing Anti-CD38 CAR Show Enhanced Anti-Tumor Activity against MM Cell Lines
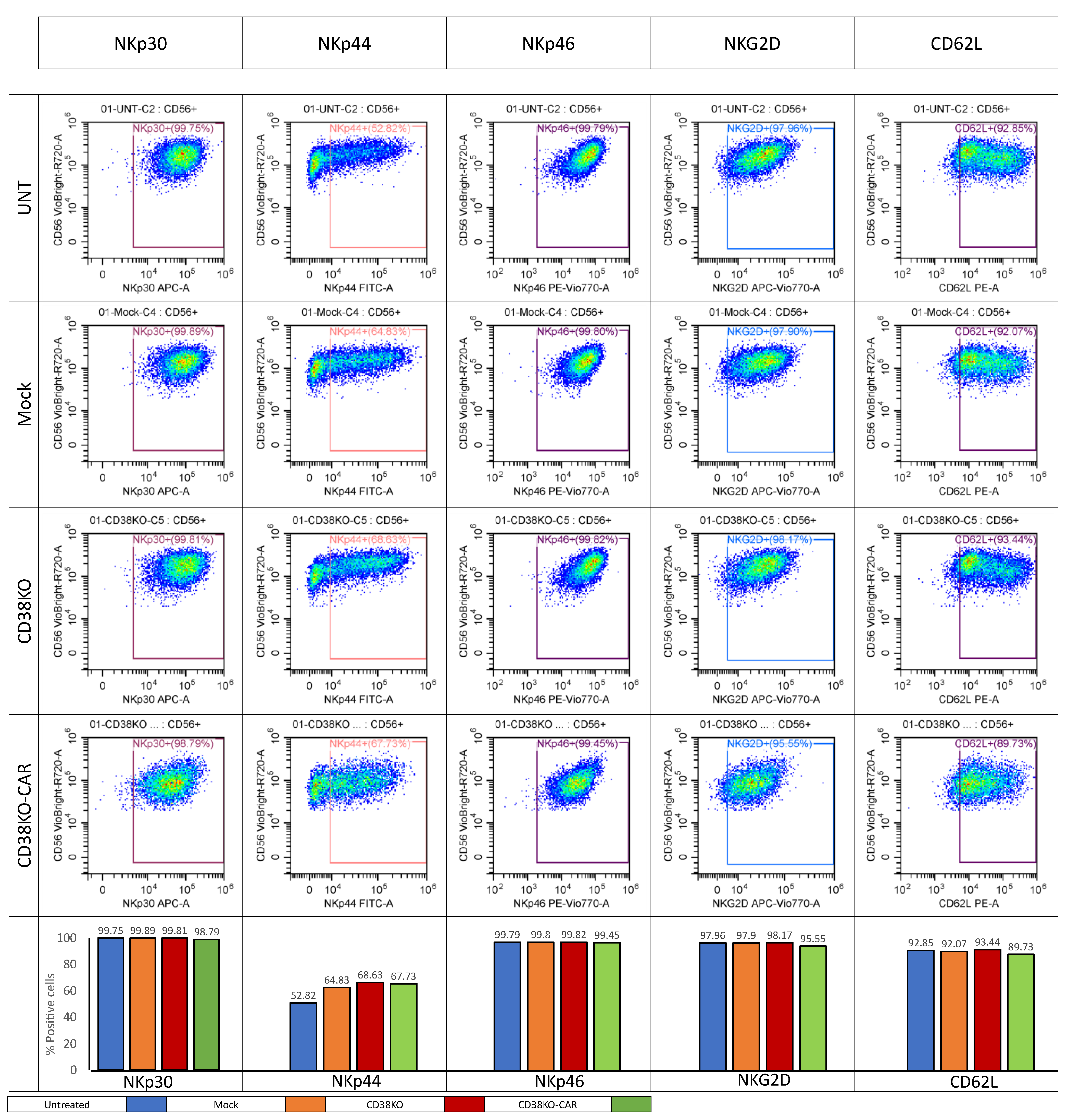
2.5. aCD38 CAR Expression Allows Specific CD38-Targeted Activity While Maintaining Native NK-Expressed Receptor Function
3. Discussion
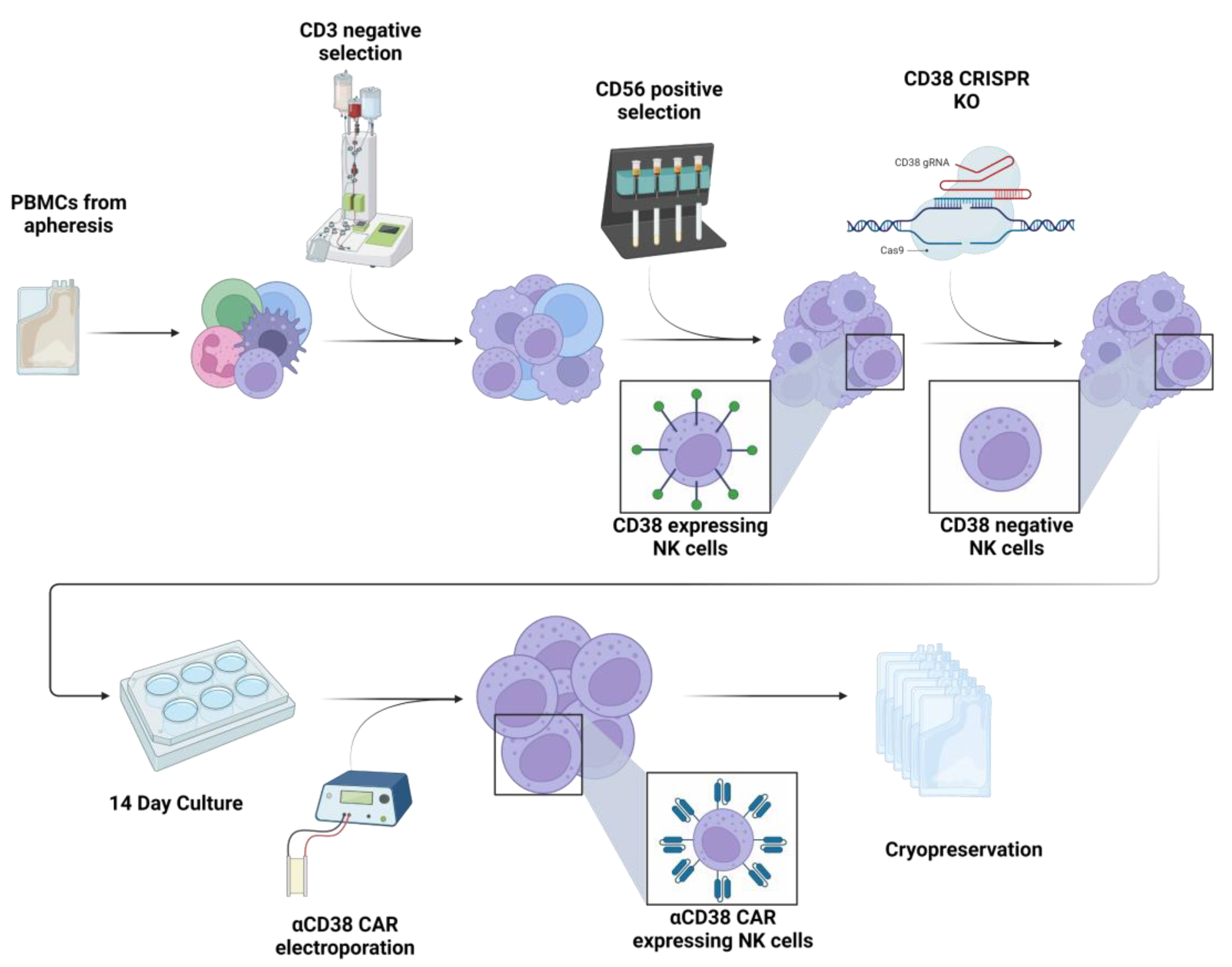
4. Materials and Methods
4.1. Cell Culture
4.2. Knock-Out
4.3. Off-Target Analysis
4.4. FACS Analysis
4.5. Killing and Fratricide Assays
4.6. Calcein-AM Release Assay
4.7. Potency
4.8. Single Axis Assay
4.9. Cryopreservation Protocol
4.10. Thawing Protocol
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rajkumar, S.V. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020, 109, 94. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Green, D.J.; Kwok, M.; Lee, S.; Coffey, D.G.; Holmberg, L.A.; Tuazon, S.; Gopal, A.K.; Libby, E.N. Diagnosis and Management of Multiple Myeloma: A Review. JAMA 2022, 327, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Ingegnere, T.; Escure, G.; Prodhomme, C.; Nudel, M.; Mitra, S.; Facon, T. Current state and next-generation CAR-T cells in multiple myeloma. Blood Rev. 2022, 54, 100929. [Google Scholar] [CrossRef] [PubMed]
- Reina-Ortiz, C.; Giraldos, D.; Azaceta, G.; Palomera, L.; Marzo, I.; Naval, J.; Villalba, M.; Anel, A. Harnessing the Potential of NK Cell-Based Immunotherapies against Multiple Myeloma. Cells 2022, 11, 392. [Google Scholar] [CrossRef] [PubMed]
- Bottino, C.; Castriconi, R.; Moretta, L.; Moretta, A. Cellular ligands of activating NK receptors. Trends Immunol. 2005, 26, 221–226. [Google Scholar] [CrossRef]
- Shemesh, A.; Brusilovsky, M.; Kundu, K.; Ottolenghi, A.; Campbell, K.S.; Porgador, A. Splice variants of human natural cytotoxicity receptors: Novel innate immune checkpoints. Cancer Immunol. Immunother. 2018, 67, 1871–1883. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Elahi, R.; Heidary, A.H.; Hadiloo, K.; Esmaeilzadeh, A. Chimeric Antigen Receptor-Engineered Natural Killer (CAR NK) Cells in Cancer Treatment; Recent Advances and Future Prospects. Stem Cell Rev. Rep. 2021, 17, 2081–2106. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shmuel, A.; Biber, G.; Barda-Saad, M. Unleashing Natural Killer Cells in the Tumor Microenvironment–The Next Generation of Immunotherapy? Front Immunol. 2020, 11, 275. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K. Adoptive cell therapy using engineered natural killer cells. Bone Marrow Transplant. 2019, 54, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2020, 18, 85–100. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B Clin. Cytom. 2013, 84, 207–217. [Google Scholar] [CrossRef]
- Zonder, J.A.; Mohrbacher, A.F.; Singhal, S.; Van Rhee, F.; Bensinger, W.I.; Ding, H.; Fry, J.; Afar, D.E.; Singhal, A.K. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012, 120, 552–559. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]
- De Weers, M.; Tai, Y.T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Plesner, T.; Krejcik, J. Daratumumab for the Treatment of Multiple Myeloma. Front. Immunol. 2018, 9, 1228. [Google Scholar] [CrossRef]
- Laubach, J.; Prada, C.P.; Richardson, P.; Longo, D. Daratumumab, Elotuzumab, and the Development of Therapeutic Monoclonal Antibodies in Multiple Myeloma. Clin. Pharmacol. Ther. 2017, 101, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Daratumumab: A Review in Relapsed and/or Refractory Multiple Myeloma. Drugs 2017, 77, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.J.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Casneuf, T.; Xu, X.S.; Adams, H.C., 3rd; Axel, A.E.; Chiu, C.; Khan, I.; Ahmadi, T.; Yan, X.; Lonial, S.; Plesner, T.; et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 2017, 1, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Maakaron, J.; McKenna, D.H.; Cao, Q.; DeFor, T.E.; He, F.; Janakiram, M.; Wangen, R.; Cayci, Z.; Grzywacz, B.; et al. Results of a Phase 1 Trial of Gda-201, Nicotinamide-Expanded Allogeneic Natural Killer (NK) Cells in Patients with Refractory Non-Hodgkin Lymphoma (NHL) and Multiple Myeloma. Blood 2020, 136 (Suppl. 1), 6. [Google Scholar] [CrossRef]
- BBachanova, V.; McKenna, D.H.; Luo, X.; E Defor, T.; Cooley, S.; Warlick, E.; Weisdorf, D.J.; Brachya, G.; Peled, T.; Miller, J.S. First-in-Human Phase I Study of Nicotinamide-Expanded Related Donor Natural Killer Cells for the Treatment of Relapsed/Refractory Non-Hodgkin Lymphoma and Multiple Myeloma. Biol. Blood Marrow Transpl. 2019, 25, S175–S176. [Google Scholar] [CrossRef]
- Bachanova, V.; McKenna, D.H.; Luo, X.; DeFor, T.E.; He, F.; Janakiram, M.; Warlick, E.D.; Wangen, R.; Cayci, Z.; Grzywacz, B.; et al. Results of a Phase 1 Trial of Gda-201, Nicotinamide-Expanded Allogeneic Natural Killer Cells (NAM-NK) in Patients with Refractory Non-Hodgkin Lymphoma (NHL) and Multiple Myeloma (MM). Blood 2019, 134 (Suppl. 1), 777. [Google Scholar] [CrossRef]
- Pittari, G.; Vago, L.; Festuccia, M.; Bonini, C.; Mudawi, D.; Giaccone, L.; Bruno, B. Restoring Natural Killer Cell Immunity against Multiple Myeloma in the Era of New Drugs. Front. Immunol. 2017, 8, 1444. [Google Scholar] [CrossRef] [PubMed]
- Venglar, O.; Bago, J.R.; Motais, B.; Hajek, R.; Jelinek, T. Natural Killer Cells in the Malignant Niche of Multiple Myeloma. Front. Immunol. 2022, 12, 5710. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, F.; Jiang, Y.; Li, S.; Liu, X.; Xu, Y.; Li, B.; Feng, X.; Zheng, C. Tim-3 Blockade Elicits Potent Anti-Multiple Myeloma Immunity of Natural Killer Cells. Front. Oncol. 2022, 12, 739976. [Google Scholar] [CrossRef] [PubMed]
- D’souza, C.; Keam, S.P.; Yeang, H.X.A.; Neeson, M.; Richardson, K.; Hsu, A.K.; Canfield, R.; Bezman, N.; Robbins, M.; Quach, H.; et al. Myeloma natural killer cells are exhausted and have impaired regulation of activation. Haematologica 2021, 106, 2522–2526. [Google Scholar] [CrossRef] [PubMed]
- Iraqi, M.; Edri, A.; Greenshpan, Y.; Goldstein, O.; Ofir, N.; Bolel, P.; Abu Ahmad, M.; Zektser, M.; Campbell, K.S.; Rouvio, O.; et al. Blocking the PCNA/NKp44 Checkpoint to Stimulate NK Cell Responses to Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 4717. [Google Scholar] [CrossRef] [PubMed]
- Leivas, A.; Valeri, A.; Córdoba, L.; García-Ortiz, A.; Ortiz, A.; Sánchez-Vega, L.; Graña-Castro, O.; Fernández, L.; Carreño-Tarragona, G.; Pérez, M.; et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Van De Donk, N.W.C.J.; Usmani, S.Z. CD38 antibodies in multiple myeloma: Mechanisms of action and modes of resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Mei, H.; Li, C.; Jiang, H.; Zhao, X.; Huang, Z.; Jin, D.; Guo, T.; Kou, H.; Liu, L.; Tang, L.; et al. A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2021, 14, 161. [Google Scholar] [CrossRef]
- Tang, Y.; Yin, H.; Zhao, X.; Jin, D.; Liang, Y.; Xiong, T.; Li, L.; Tang, W.; Zhang, J.; Liu, M.; et al. High efficacy and safety of CD38 and BCMA bispecific CAR-T in relapsed or refractory multiple myeloma. J. Exp. Clin. Cancer Res. 2022, 41, 2. [Google Scholar] [CrossRef]
- Cui, Q.; Qian, C.; Xu, N.; Kang, L.; Dai, H.; Cui, W.; Song, B.; Yin, J.; Li, Z.; Zhu, X.; et al. CD38-directed CAR-T cell therapy: A novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J. Hematol. Oncol. 2021, 14, 82. [Google Scholar] [CrossRef]
- Hambach, J.; Riecken, K.; Cichutek, S.; Schütze, K.; Albrecht, B.; Petry, K.; Röckendorf, J.L.; Baum, N.; Kröger, N.; Hansen, T.; et al. Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs). Cells 2020, 9, 321. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; Van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Dobosy, J.R.; Rose, S.D.; Beltz, K.R.; Rupp, S.M.; Powers, K.M.; Behlke, M.A.; Walder, J.A. RNase H-dependent PCR (rhPCR): Improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Amit, I.; Iancu, O.; Levy-Jurgenson, A.; Kurgan, G.; McNeill, M.S.; Rettig, G.R.; Allen, D.; Breier, D.; Ben Haim, N.; Wang, Y.; et al. CRISPECTOR provides accurate estimation of genome editing translocation and off-target activity from comparative NGS data. Nat. Commun. 2021, 12, 3042. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Wang, Y.; Huang, Y.; Duan, Y.; Liang, Y.; Chen, J.; Jiang, J.; Shang, K.; Zhou, C.; Gu, Y.; et al. CD38-Specific CAR Integrated into CD38 Locus Driven by Different Promoters Causes Distinct Antitumor Activities of T and NK Cells. Adv. Sci. 2023, 10, e2207394. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.; Stikvoort, A.; Nolan, E.; Kirkham-McCarthy, L.; Khoruzhenko, S.; Shivakumar, R.; Zweegman, S.; Van de Donk, N.W.; Mutis, T.; Szegezdi, E.; et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica 2020, 107, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Fiegler, N.; Textor, S.; Arnold, A.; Rölle, A.; Oehme, I.; Breuhahn, K.; Moldenhauer, G.; Witzens-Harig, M.; Cerwenka, A. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood 2013, 122, 684–693. [Google Scholar] [CrossRef]
- Hirano, N.; Takahashi, T.; Ohtake, S.; Hirashima, K.; Leukemia, N.E. Expression of costimulatory molecules in human leukemias. Leukemia 1996, 10, 1168–1176. [Google Scholar]
- Mikkilineni, L.; Kochenderfer, J.N. CAR T cell therapies for patients with multiple myeloma. Nat. Rev. Clin. Oncol. 2020, 18, 71–84. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Lan, H.; Wu, J.; Xiao, Y. CAR-T cell therapy in multiple myeloma: Current limitations and potential strategies. Front. Immunol. 2023, 14, 1101495. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, B.; Hari, P.N.; Usmani, S.Z.; Hamadani, M. Chimeric antigen receptor T cell therapy in multiple myeloma: Promise and challenges. Bone Marrow Transplant. 2020, 56, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Abramson, H.N. Monoclonal Antibodies for the Treatment of Multiple Myeloma: An Update. Int. J. Mol. Sci. 2018, 19, 3924. [Google Scholar] [CrossRef] [PubMed]
- Gozzetti, A.; Ciofini, S.; Simoncelli, M.; Santoni, A.; Pacelli, P.; Raspadori, D.; Bocchia, M. Anti CD38 monoclonal antibodies for multiple myeloma treatment. Hum. Vaccines Immunother. 2022, 18, 2052658. [Google Scholar] [CrossRef] [PubMed]
- Lapietra, G.; Fazio, F.; Petrucci, M.T. Race for the Cure: From the Oldest to the Newest Monoclonal Antibodies for Multiple Myeloma Treatment. Biomolecules 2022, 12, 1146. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Allegra, A.; Di Chio, C.; Previti, S.; Zappalà, M.; Ettari, R. Monoclonal Antibodies: The Greatest Resource to Treat Multiple Myeloma. Int. J. Mol. Sci. 2023, 24, 3136. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Stege, C.A.M.; Zweegman, S.; van de Donk, N.W.C.J.; Nijhof, I.S. Resistance Mechanisms towards CD38−Directed Antibody Therapy in Multiple Myeloma. J. Clin. Med. 2020, 9, 1195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo–Expanded Autologous NK Cells. Clin. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Kararoudi, M.N.; Elmas, E.; Pereira, M.; Ali, S.A.; Imus, P.H.; Lee, D.A.; Ghiaur, G. CD38 Knockout Primary NK Cells to Prevent “Fratricide” and Boost Daratumumab Activity. Blood 2019, 134 (Suppl. 1), 870. [Google Scholar] [CrossRef]
- Merino, A.; Maakaron, J.; Bachanova, V. Advances in NK cell therapy for hematologic malignancies: NK source, persistence and tumor targeting. Blood Rev. 2023, 60, 101073. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Maus, M.V. Catch me if you can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput. Struct. Biotechnol. J. 2016, 14, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Epling-Burnette, P.K.; Bai, F.; Painter, J.S.; Rollison, D.E.; Salih, H.R.; Krusch, M.; Zou, J.; Ku, E.; Zhong, B.; Boulware, D.; et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood 2007, 109, 4816–4824. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-H.; Kim, J.-Y.; Kim, M.-J.; Chang, S.-H.; Park, Y.-S.; Son, C.-H.; Park, S.-J.; Chung, J.-S.; Lee, E.-Y.; Kim, S.-H.; et al. Quercetin Enhances Susceptibility to NK Cell-mediated Lysis of Tumor Cells Through Induction of NKG2D Ligands and Suppression of HSP70. J. Immunother. 2010, 33, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Tremblay-Mclean, A.; Coenraads, S.; Kiani, Z.; Dupuy, F.P.; Bernard, N.F. Expression of ligands for activating natural killer cell receptors on cell lines commonly used to assess natural killer cell function 11 Medical and Health Sciences 1107 Immunology. BMC Immunol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Garralda, E.; Del Conte, G.; Macchini, M.; Matos, I.; Klinghammer, K.F.; Saavedra Santa Gadea, O.; Fiedler, W.M.; Rolling, C.C.; Kebenko, M.; Raspagliesi, F.; et al. Activity results of the GATTO study, a phase Ib study combining the anti-TA-MUC1 antibody gatipotuzumab with the anti-EGFR tomuzotuximab or panitumumab in patients with refractory solid tumors. J. Clin. Oncol. 2021, 39 (Suppl. 15), 2522. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Setti, C.; Giordano, C.; Obino, V.; Greppi, M.; Pesce, S.; Marcenaro, E.; Rutigliani, M.; Provinciali, N.; Paleari, L.; et al. NK Cell-Based Immunotherapy in Colorectal Cancer. Vaccines 2022, 10, 1033. [Google Scholar] [CrossRef]
- Levy, E.; Reger, R.; Segerberg, F.; Lambert, M.; Leijonhufvud, C.; Baumer, Y.; Carlsten, M.; Childs, R. Enhanced Bone Marrow Homing of Natural Killer Cells Following mRNA Transfection With Gain-of-Function Variant CXCR4R334X. Front. Immunol. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Imamura, M.; Shook, D.; Kamiya, T.; Shimasaki, N.; Chai, S.M.H.; Coustan-Smith, E.; Imai, C.; Campana, D. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood 2014, 124, 1081–1088. [Google Scholar] [CrossRef]
- Wang, B.; Iriguchi, S.; Waseda, M.; Ueda, N.; Ueda, T.; Xu, H.; Minagawa, A.; Ishikawa, A.; Yano, H.; Ishi, T.; et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat. Biomed. Eng. 2021, 5, 429–440. [Google Scholar] [CrossRef]
- Kang, H.J.; Bartholomae, C.C.; Paruzynski, A.; Arens, A.; Kim, S.; Yu, S.S.; Hong, Y.; Joo, C.-W.; Yoon, N.-K.; Rhim, J.-W.; et al. Retroviral Gene Therapy for X-linked Chronic Granulomatous Disease: Results From Phase I/II Trial. Mol. Ther. 2011, 19, 2092–2101. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edri, A.; Ben-Haim, N.; Hailu, A.; Brycman, N.; Berhani-Zipori, O.; Rifman, J.; Cohen, S.; Yackoubov, D.; Rosenberg, M.; Simantov, R.; et al. Nicotinamide-Expanded Allogeneic Natural Killer Cells with CD38 Deletion, Expressing an Enhanced CD38 Chimeric Antigen Receptor, Target Multiple Myeloma Cells. Int. J. Mol. Sci. 2023, 24, 17231. https://doi.org/10.3390/ijms242417231
Edri A, Ben-Haim N, Hailu A, Brycman N, Berhani-Zipori O, Rifman J, Cohen S, Yackoubov D, Rosenberg M, Simantov R, et al. Nicotinamide-Expanded Allogeneic Natural Killer Cells with CD38 Deletion, Expressing an Enhanced CD38 Chimeric Antigen Receptor, Target Multiple Myeloma Cells. International Journal of Molecular Sciences. 2023; 24(24):17231. https://doi.org/10.3390/ijms242417231
Chicago/Turabian StyleEdri, Avishay, Nimrod Ben-Haim, Astar Hailu, Nurit Brycman, Orit Berhani-Zipori, Julia Rifman, Sherri Cohen, Dima Yackoubov, Michael Rosenberg, Ronit Simantov, and et al. 2023. "Nicotinamide-Expanded Allogeneic Natural Killer Cells with CD38 Deletion, Expressing an Enhanced CD38 Chimeric Antigen Receptor, Target Multiple Myeloma Cells" International Journal of Molecular Sciences 24, no. 24: 17231. https://doi.org/10.3390/ijms242417231
APA StyleEdri, A., Ben-Haim, N., Hailu, A., Brycman, N., Berhani-Zipori, O., Rifman, J., Cohen, S., Yackoubov, D., Rosenberg, M., Simantov, R., Teru, H., Kurata, K., Anderson, K. C., Hendel, A., Pato, A., & Geffen, Y. (2023). Nicotinamide-Expanded Allogeneic Natural Killer Cells with CD38 Deletion, Expressing an Enhanced CD38 Chimeric Antigen Receptor, Target Multiple Myeloma Cells. International Journal of Molecular Sciences, 24(24), 17231. https://doi.org/10.3390/ijms242417231