
Mitochondrial DNA Variants at Low-Level Heteroplasmy and Decreased Copy Numbers in Chronic Kidney Disease (CKD) Tissues with Kidney Cancer
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
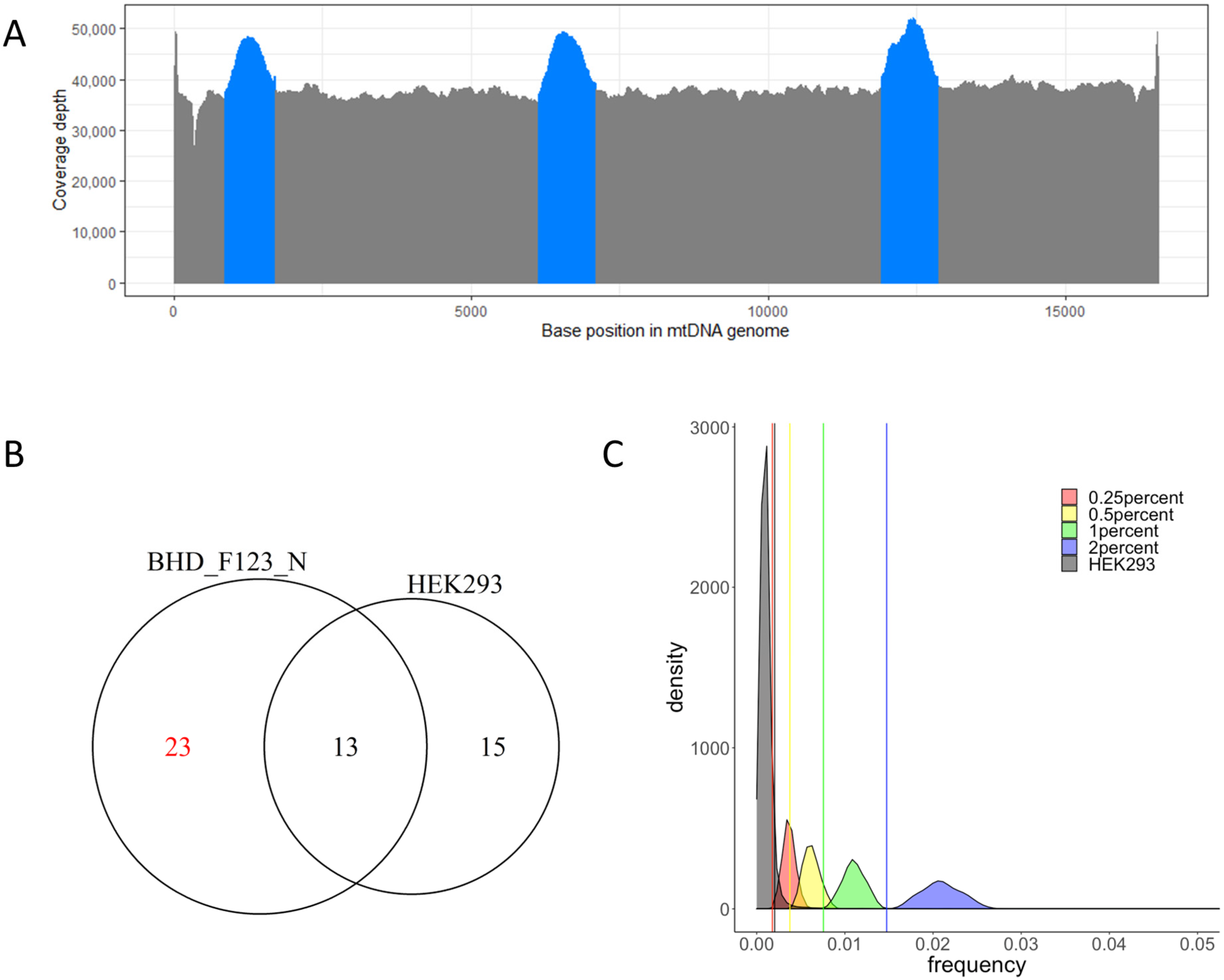
2.1. mtDNA Sequencing
2.2. Cutoff Determination and Variants Call
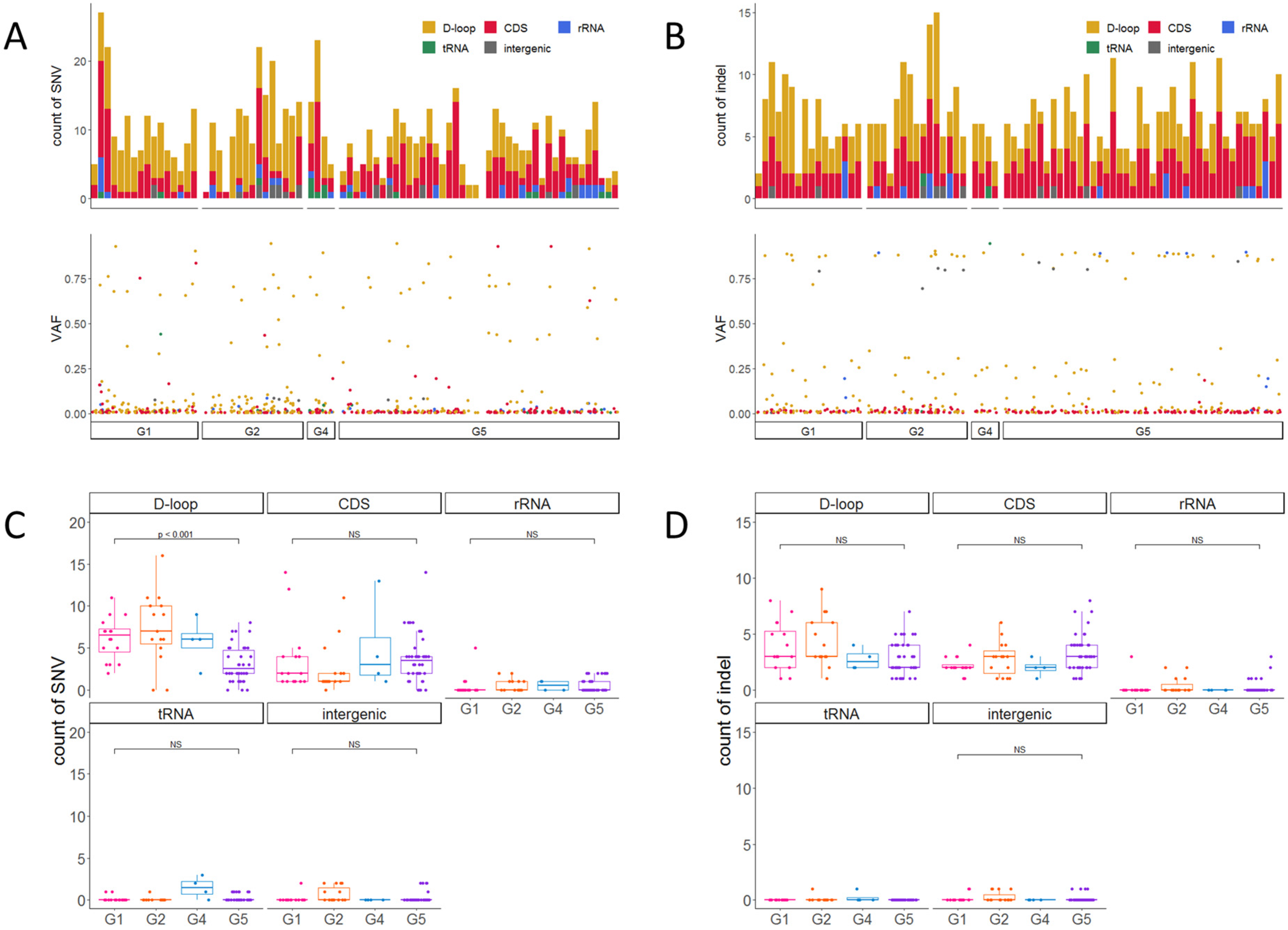
2.3. Accumulation of Heteroplasmic SNVs and Indels
2.4. Mutational Signatures of mtDNA
2.5. Homoplasmic mtDNA Variants
2.6. Deleterious mtDNA Variants
2.7. Decreased mtDNA-CN in Kidney Tissue with CKD Progression
3. Discussion
4. Materials and Methods
4.1. Clinical Samples
4.2. HEK293 Cell Culture
4.3. DNA Extraction
4.4. PCR-Based mtDNA Enrichment and Sequencing
4.5. Variant Calling
4.6. In Silico Admixture
4.7. Mutational Signature Analysis
4.8. Variant Annotation
4.9. Evaluation Using Cancer and Blood
4.10. mtDNA Copy Number (mtDNA-CN)
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Aryaman, J.; Johnston, I.G.; Jones, N.S. Mitochondrial Heterogeneity. Front. Genet. 2019, 9, 718. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef]
- Gao, R.; Ma, S.L. Is Mitochondria DNA Variation a Biomarker for AD? Genes 2022, 13, 1789. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghamdi, B.A.; Al-Shamrani, J.M.; El-Shehawi, A.M.; Al-Johani, I.; Al-Otaibi, B.G. Role of mitochondrial DNA in diabetes Mellitus Type I and Type II. Saudi J. Biol. Sci. 2022, 29, 103434. [Google Scholar] [CrossRef] [PubMed]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Yin, X.-M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 299–318. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Müller, M.J. Specific metabolic rates of major organs and tissues across adulthood: Evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 2010, 92, 1369–1377. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lin, P.H. Mitochondria Damage and Kidney Disease. Adv. Exp. Med. Biol. 2017, 982, 529–551. [Google Scholar]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Marquez-Expósito, L.; Rodrigues-Diez, R.; Sanz, A.B.; Guiteras, R.; Doladé, N.; Rubio-Soto, I.; Manonelles, A.; Codina, S.; Ortiz, A.; et al. Molecular Mechanisms of Kidney Injury and Repair. Int. J. Mol. Sci. 2022, 23, 1542. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Johnson, T.A.; Maekawa, S.; Fujita, M.; An, J.; Ju, Y.-S.; Maejima, K.; Kanazashi, Y.; Jikuya, R.; Okawa, Y.; Sasagawa, S.; et al. Genomic features of renal cell carcinoma developed during end-stage renal disease and dialysis. Hum. Mol. Genet. 2023, 32, 290–303. [Google Scholar] [CrossRef]
- Guo, J.-H.; Shi, J.-M.; Shi, G.-P.; Wang, Y.; Chu, X.-F.; Wang, Z.-D.; Yao, S.; Sun, X.-H.; Wang, X.-F.; Zhu, Y.-S.; et al. Association Study of Mitochondrial DNA Haplogroup D and C5178A Polymorphisms with Chronic Kidney Disease. Genet. Test. Mol. Biomarkers 2021, 25, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Inagi, R. The subtle long-lasting burden of mitochondrial DNA variants. Nat. Rev. Nephrol. 2022, 18, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Guo, Z.; Xu, J.; Zhang, J.; Cui, L.; Zhang, H.; Zhang, S.; Ai, X. Association of sequence polymorphism in the mitochondrial D-loop with chronic kidney disease. Ren. Fail. 2014, 36, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Guo, Z.; Bai, Y.; Zhang, J.; Cui, L.; Zhang, H.; Zhang, S.; Ai, X. Single nucleotide polymorphisms in the D-loop region of mitochondrial DNA is associated with the kidney survival time in chronic kidney disease patients. Ren. Fail. 2015, 37, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-B.; Yang, Y.-H.; Lee, W.-C.; Liou, C.-W.; Lin, T.-K.; Chung, Y.-H.; Chuang, L.-Y.; Yang, C.-H.; Chang, H.-W. Sequence-Based Polymorphisms in the Mitochondrial D-Loop and Potential SNP Predictors for Chronic Dialysis. PLoS ONE 2012, 7, e41125. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, S.; Yin, C.; Guo, X.; Liu, M.; Yuan, Z.; Zhao, Z.; Jia, Y.; Xing, J. Optimized PCR-Based Enrichment Improves Coverage Uniformity and Mutation Detection in Mitochondrial DNA Next-Generation Sequencing. J. Mol. Diagn. 2020, 22, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Leuthner, T.C.; Meyer, J.N. Mitochondrial DNA Mutagenesis: Feature of and Biomarker for Environmental Exposures and Aging. Curr. Environ. Health Rep. 2021, 8, 294–308. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Song, M.; Tao, Z.; Wu, T.; He, Z.; Zhao, X.; Wu, K.; Liu, X.-S. Copy number signature analysis tool and its application in prostate cancer reveals distinct mutational processes and clinical outcomes. PLoS Genet. 2021, 17, e1009557. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. eBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J. Leber’s hereditary optic neuropathy. New genetic considerations. Arch. Neurol. 1993, 50, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.C.; Davis, R.L.; Ravishankar, S.; Copty, J.; Kummerfeld, S.; Sue, C.M. Low disease risk and penetrance in Leber hereditary optic neuropathy. Am. J. Hum. Genet. 2023, 110, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.C.; Ruiz-Pesini, E.; Mishmar, D.; Procaccio, V.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Patil, U.; Baldi, P.; Wallace, D.C. MITOMASTER: A bioinformatics tool for the analysis of mitochondrial DNA sequences. Hum. Mutat. 2009, 30, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ratnaike, T.E.; Greene, D.; Wei, W.; Sanchis-Juan, A.; Schon, K.; Ameele, J.V.D.; Raymond, L.; Horvath, R.; Turro, E.; Chinnery, P. MitoPhen database: A human phenotype ontology-based approach to identify mitochondrial DNA diseases. Nucleic Acids Res. 2021, 49, 9686–9695. [Google Scholar] [CrossRef] [PubMed]
- Sonney, S.; Leipzig, J.; Lott, M.T.; Zhang, S.; Procaccio, V.; Wallace, D.C.; Sondheimer, N. Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput. Biol. 2017, 13, e1005867. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, Z.; Liang, W.; Wei, Z.; Ding, G. Roles of Mitochondrial DNA Damage in Kidney Diseases: A New Biomarker. Int. J. Mol. Sci. 2022, 23, 15166. [Google Scholar] [CrossRef]
- He, W.J.; Li, C.; Huang, Z.; Geng, S.; Rao, V.S.; Kelly, T.N.; Hamm, L.L.; Grams, M.E.; Arking, D.E.; Appel, L.J.; et al. Association of Mitochondrial DNA Copy Number with Risk of Progression of Kidney Disease. Clin. J. Am. Soc. Nephrol. 2022, 17, 966–975. [Google Scholar] [CrossRef]
- Jin, L.; Yu, B.; Armando, I.; Han, F. Mitochondrial DNA-Mediated Inflammation in Acute Kidney Injury and Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2021, 2021, 9985603. [Google Scholar] [CrossRef]
- Rao, M.; Jaber, B.L.; Balakrishnan, V.S. Chronic kidney disease and acquired mitochondrial myopathy. Curr. Opin. Nephrol. Hypertens. 2018, 27, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Gamboa, J.L.; Billings, F.T., 4th; Bojanowski, M.T.; Gilliam, L.A.; Yu, C.; Roshanravan, B.; Roberts, L.J., 2nd; Himmelfarb, J.; Ikizler, T.A.; Brown, N.J. Mitochondrial disfunction and oxidative stress in patients with chronic kidney disease. Physiol. Rep. 2016, 4, e12780. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Koch, R.; Westra, W.H.; Califano, J.A.; Ha, P.K.; Sidransky, D.; Koch, W.M. Mitochondrial DNA Mutation in Normal Margins and Tumors of Recurrent Head and Neck Squamous Cell Carcinoma Patients. Cancer Prev. Res. 2010, 3, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Nishimura, M.; Murayama, K.; Kuranobu, N.; Tojo, S.; Beppu, M.; Ishige, T.; Itoga, S.; Tsuchida, S.; Mori, M.; et al. A simple method for sequencing the whole human mitochondrial genome directly from samples and its application to genetic testing. Sci. Rep. 2019, 9, 17411. [Google Scholar] [CrossRef]
- Falk, M.J.; Pierce, E.A.; Consugar, M.; Xie, M.H.; Guadalupe, M.; Hardy, O.; Rappaport, E.F.; Wallace, D.C.; LeProust, E.; Gai, X. Mitochondrial disease genetic diagnostics: Optimized whole-exome analysis for all MitoCarta nuclear genes and the mitochondrial genome. Discov. Med. 2012, 14, 389–399. [Google Scholar] [PubMed]
- Naini, A.; Gilkerson, R.; Shanske, S.; Pang, J. Detection of mitochondrial DNA (mtDNA) mutations. Methods Cell Biol. 2020, 155, 383–400. [Google Scholar] [PubMed]
- Kaneva, K.; Merkurjev, D.; Ostrow, D.; Ryutov, A.; Triska, P.; Stachelek, K.; Cobrinik, D.; Biegel, J.A.; Gai, X. Detection of mitochondrial DNA variants at low level heteroplasmy in pediatric CNS and extra-CNS solid tumors with three different enrichment methods. Mitochondrion 2020, 51, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef]
- Fazzini, F.; Fendt, L.; Schönherr, S.; Forer, L.; Schöpf, B.; Streiter, G.; Losso, J.L.; Kloss-Brandstätter, A.; Kronenberg, F.; Weissensteiner, H. Analyzing Low-Level mtDNA Heteroplasmy—Pitfalls and Challenges from Bench to Benchmarking. Int. J. Mol. Sci. 2021, 22, 935. [Google Scholar] [CrossRef]
- Fujimoto, A.; Fujita, M.; Hasegawa, T.; Wong, J.H.; Maejima, K.; Oku-Sasaki, A.; Nakano, K.; Shiraishi, Y.; Miyano, S.; Yamamoto, G.; et al. Comprehensive analysis of indels in whole-genome microsatellite regions and microsatellite instability across 21 cancer types. Genome Res. 2020, 30, 334–346. [Google Scholar] [CrossRef]
- Lee, H.-C.; Chang, C.-M.; Chi, C.-W. Somatic mutations of mitochondrial DNA in aging and cancer progression. Ageing Res. Rev. 2010, 9 (Suppl. 1), S47–S58. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; Alexeyev, M. Mitochondrial DNA: Consensuses and Controversies. DNA 2022, 2, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genom. 2009, 36, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, L.V.; Tamirisa, A.; Kennedy, D.J.; Haller, S.T.; Budnyy, G.; Shapiro, J.I.; Malhotra, D. Mitochondrial impairment in the five-sixth nephrectomy model of chronic renal failure: Proteomic approach. BMC Nephrol. 2013, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Longley, M.J. Mitochondrial genome maintenance in health and disease. DNA Repair 2014, 19, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Sealfon, R.; Menon, R.; Eadon, M.T.; Lake, B.B.; Steck, B.; Anjani, K.; Parikh, S.; Sigdel, T.K.; Zhang, G.; et al. A reference tissue atlas for the human kidney. Sci. Adv. 2022, 8, eabn4965. [Google Scholar] [CrossRef]
- McEvoy, C.M.; Murphy, J.M.; Zhang, L.; Clotet-Freixas, S.; Mathews, J.A.; An, J.; Karimzadeh, M.; Pouyabahar, D.; Su, S.; Zaslaver, O.; et al. Single-cell profiling of healthy human kidney reveals features of sex-based transcriptional programs and tissue-specific immunity. Nat. Commun. 2022, 13, 7634. [Google Scholar] [CrossRef]
- Nadalutti, C.A.; Ayala-Peña, S.; Santos, J.H. Mitochondrial DNA damage as driver of cellular outcomes. Am. J. Physiol.-Cell Physiol. 2022, 322, C136–C150. [Google Scholar] [CrossRef]
- Levin, A.; Stevens, P.E. Summary of KDIGO 2012 CKD Guideline: Behind the scenes, need for guidance, and a framework for moving forward. Kidney Int. 2014, 85, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Kechin, A.; Boyarskikh, U.; Kel, A.; Filipenko, M. cutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J. Comput. Biol. 2017, 24, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, H.; Forer, L.; Fuchsberger, C.; Schöpf, B.; Kloss-Brandstätter, A.; Specht, G.; Kronenberg, F.; Schönherr, S. mtDNA-Server: Next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 2016, 44, W64–W69. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Davis, C.F.; Ricketts, C.J.; Wang, M.; Yang, L.; Cherniack, A.D.; Shen, H.; Buhay, C.; Kang, H.; Kim, S.C.; Fahey, C.C.; et al. The Somatic Genomic Landscape of Chromophobe Renal Cell Carcinoma. Cancer Cell 2014, 26, 319–330. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanazashi, Y.; Maejima, K.; Johnson, T.A.; Sasagawa, S.; Jikuya, R.; Hasumi, H.; Matsumoto, N.; Maekawa, S.; Obara, W.; Nakagawa, H. Mitochondrial DNA Variants at Low-Level Heteroplasmy and Decreased Copy Numbers in Chronic Kidney Disease (CKD) Tissues with Kidney Cancer. Int. J. Mol. Sci. 2023, 24, 17212. https://doi.org/10.3390/ijms242417212
Kanazashi Y, Maejima K, Johnson TA, Sasagawa S, Jikuya R, Hasumi H, Matsumoto N, Maekawa S, Obara W, Nakagawa H. Mitochondrial DNA Variants at Low-Level Heteroplasmy and Decreased Copy Numbers in Chronic Kidney Disease (CKD) Tissues with Kidney Cancer. International Journal of Molecular Sciences. 2023; 24(24):17212. https://doi.org/10.3390/ijms242417212
Chicago/Turabian StyleKanazashi, Yuki, Kazuhiro Maejima, Todd A. Johnson, Shota Sasagawa, Ryosuke Jikuya, Hisashi Hasumi, Naomichi Matsumoto, Shigekatsu Maekawa, Wataru Obara, and Hidewaki Nakagawa. 2023. "Mitochondrial DNA Variants at Low-Level Heteroplasmy and Decreased Copy Numbers in Chronic Kidney Disease (CKD) Tissues with Kidney Cancer" International Journal of Molecular Sciences 24, no. 24: 17212. https://doi.org/10.3390/ijms242417212
APA StyleKanazashi, Y., Maejima, K., Johnson, T. A., Sasagawa, S., Jikuya, R., Hasumi, H., Matsumoto, N., Maekawa, S., Obara, W., & Nakagawa, H. (2023). Mitochondrial DNA Variants at Low-Level Heteroplasmy and Decreased Copy Numbers in Chronic Kidney Disease (CKD) Tissues with Kidney Cancer. International Journal of Molecular Sciences, 24(24), 17212. https://doi.org/10.3390/ijms242417212