
Disrupting Poly(ADP-ribosyl)ating Pathway Creates Premalignant Conditions in Mammalian Liver



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
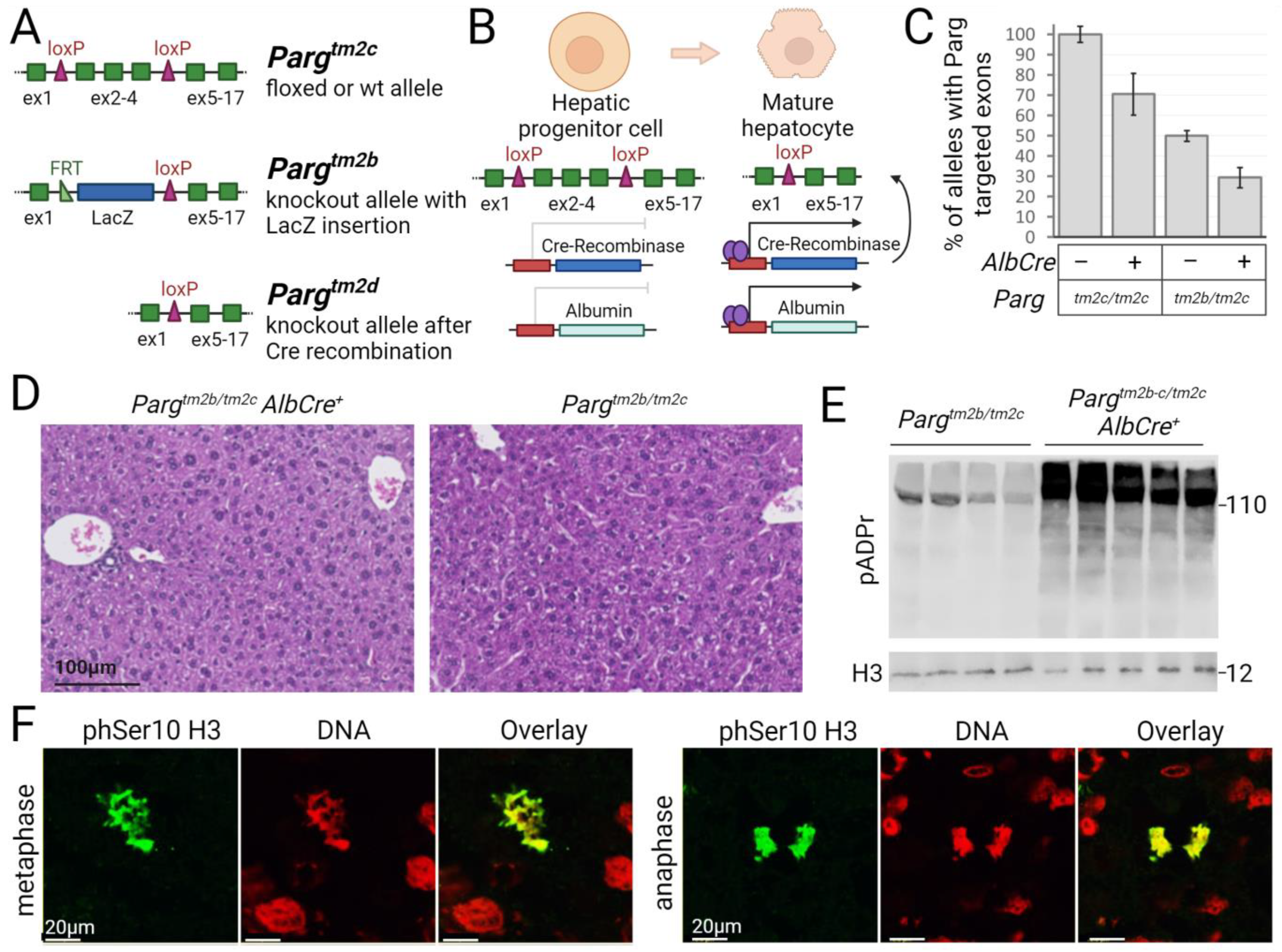
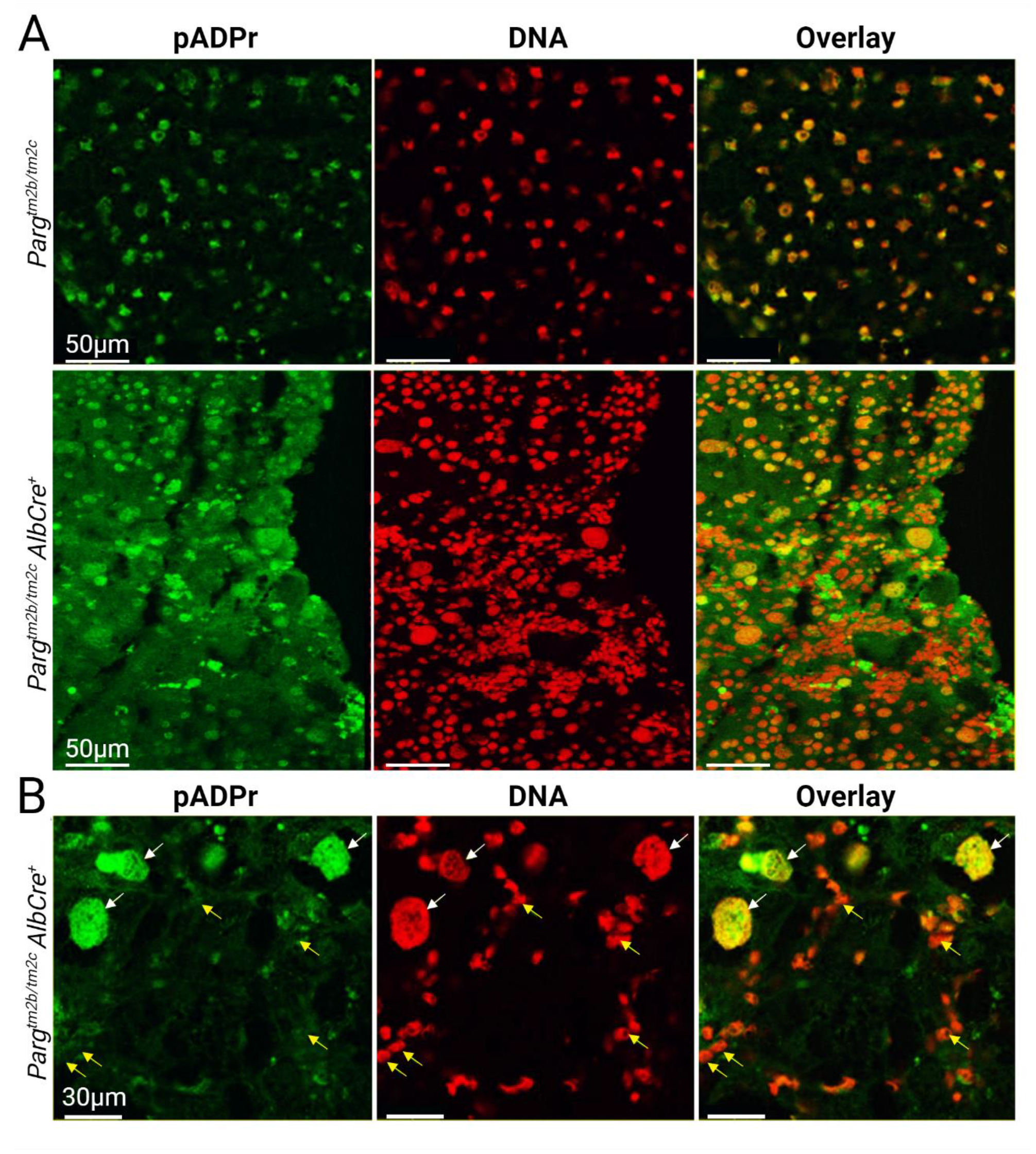
2.1. The loxP-Flanked Parg Gene Region Is Deleted in Hepatocytes of Mice Expressing the Albumin Promoter-Driven Cre Recombinase
2.2. The Regeneration Capacity of Parg Hepatocyte-Knockout Livers Is Not Compromised
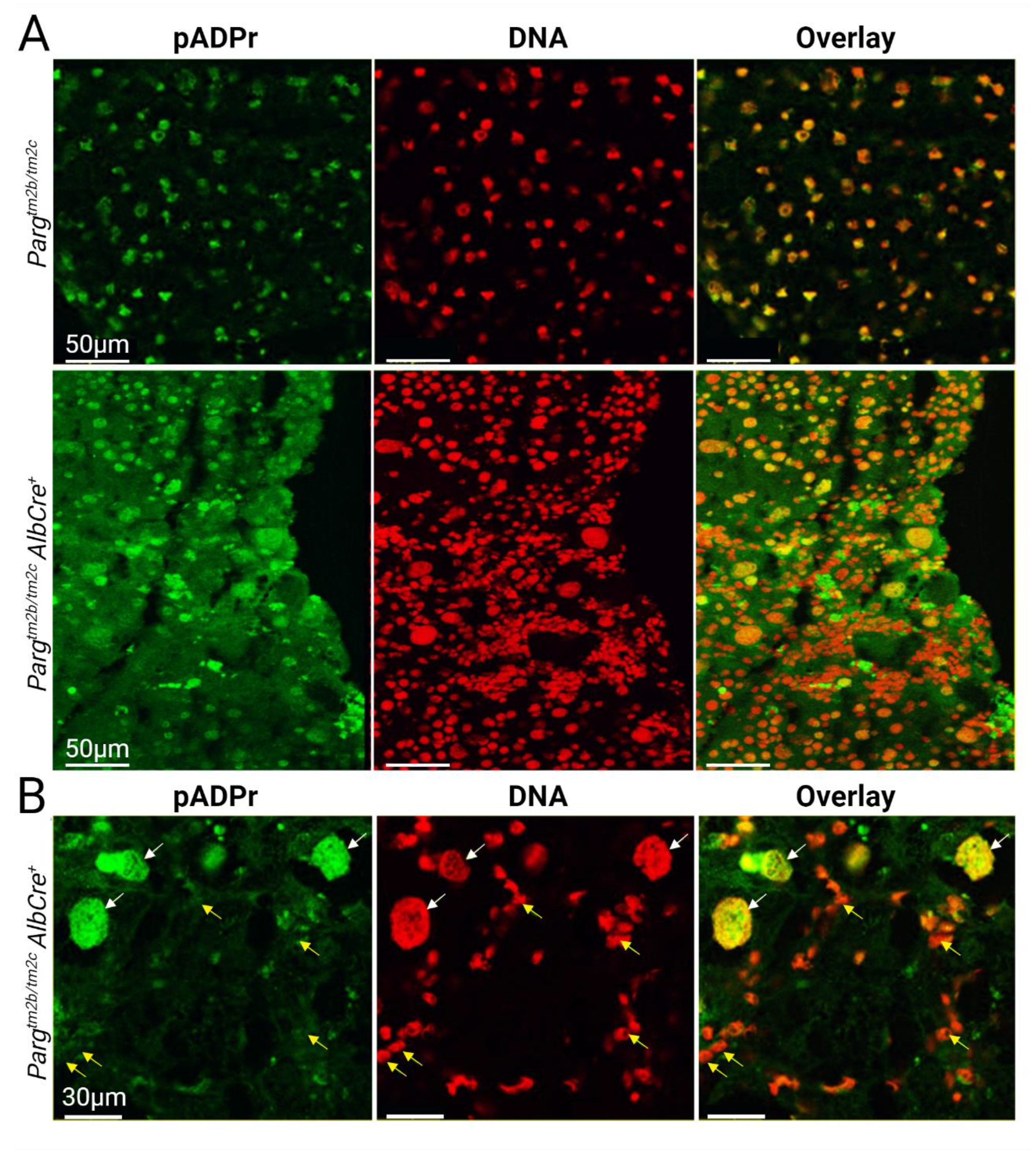
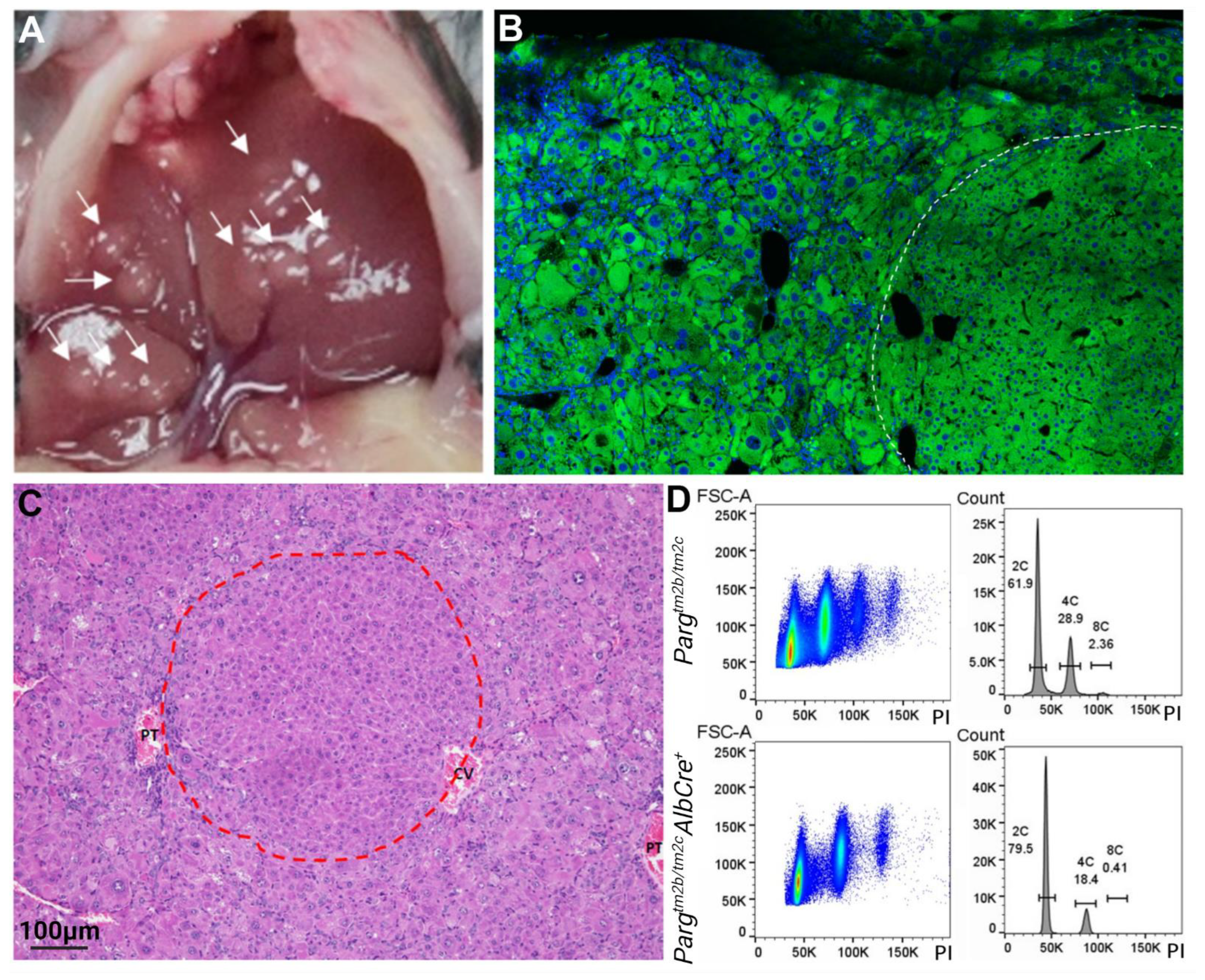
2.3. Liver Development and Functioning in the Absence of PARG in Hepatocytes Is Affected
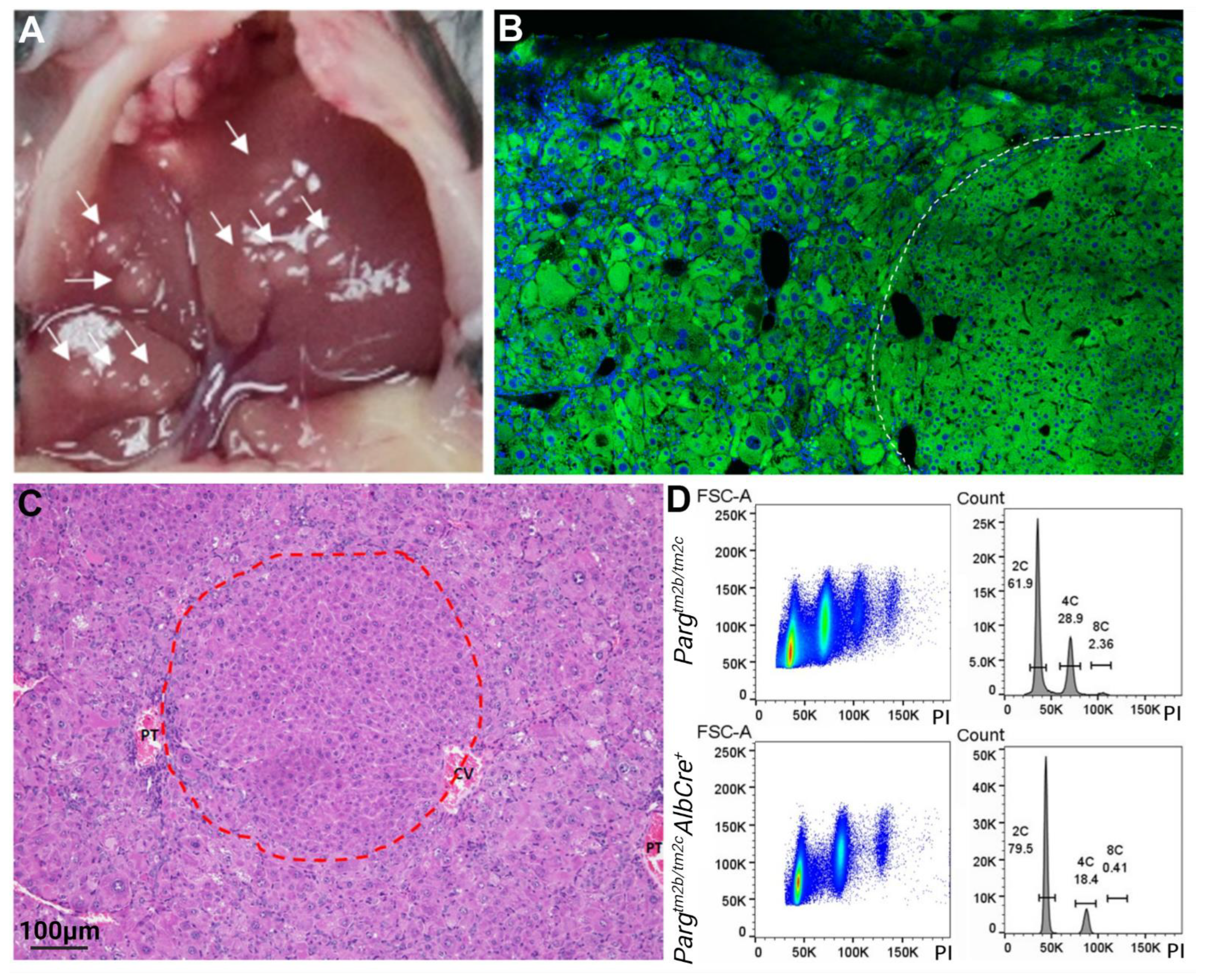
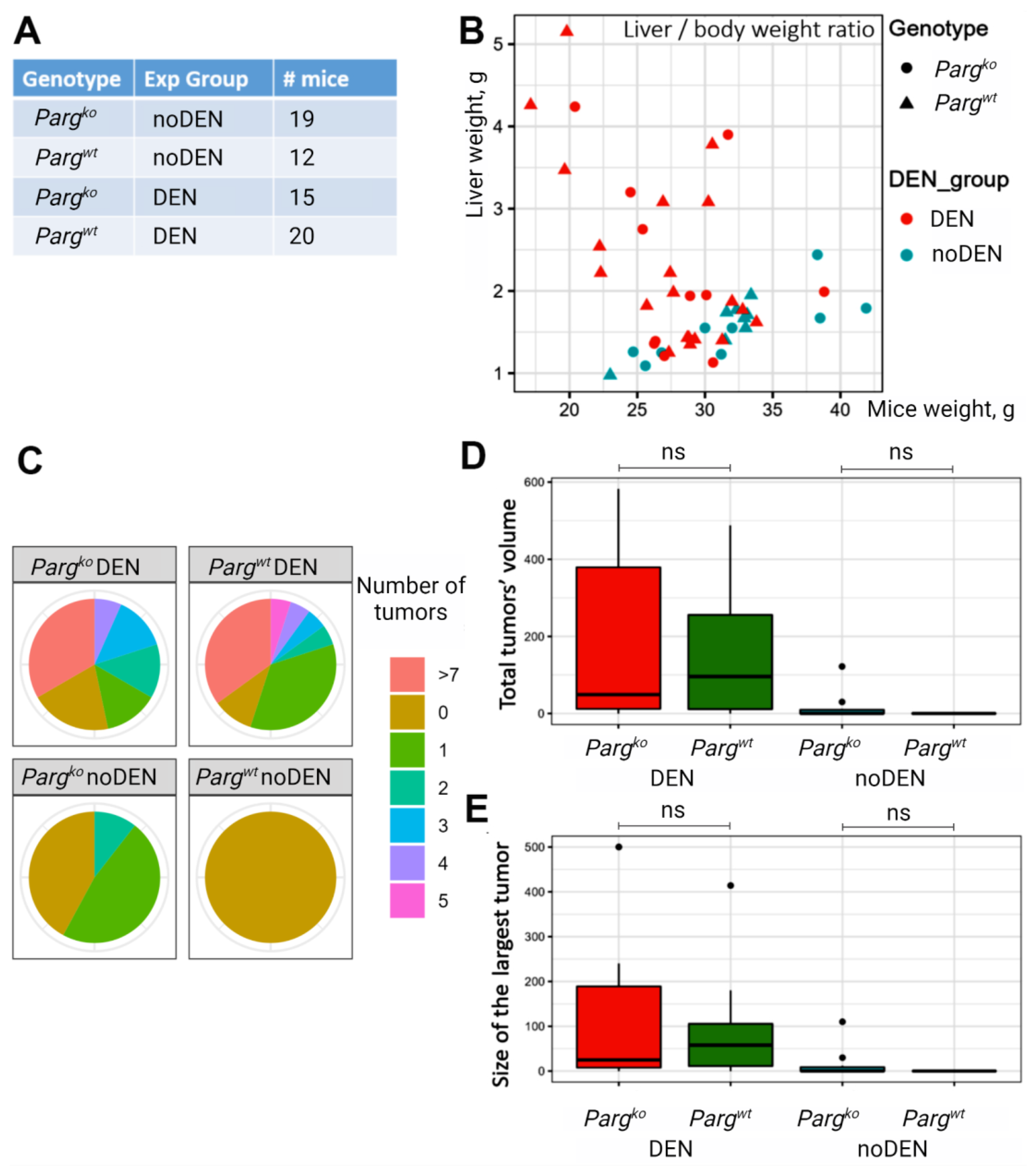
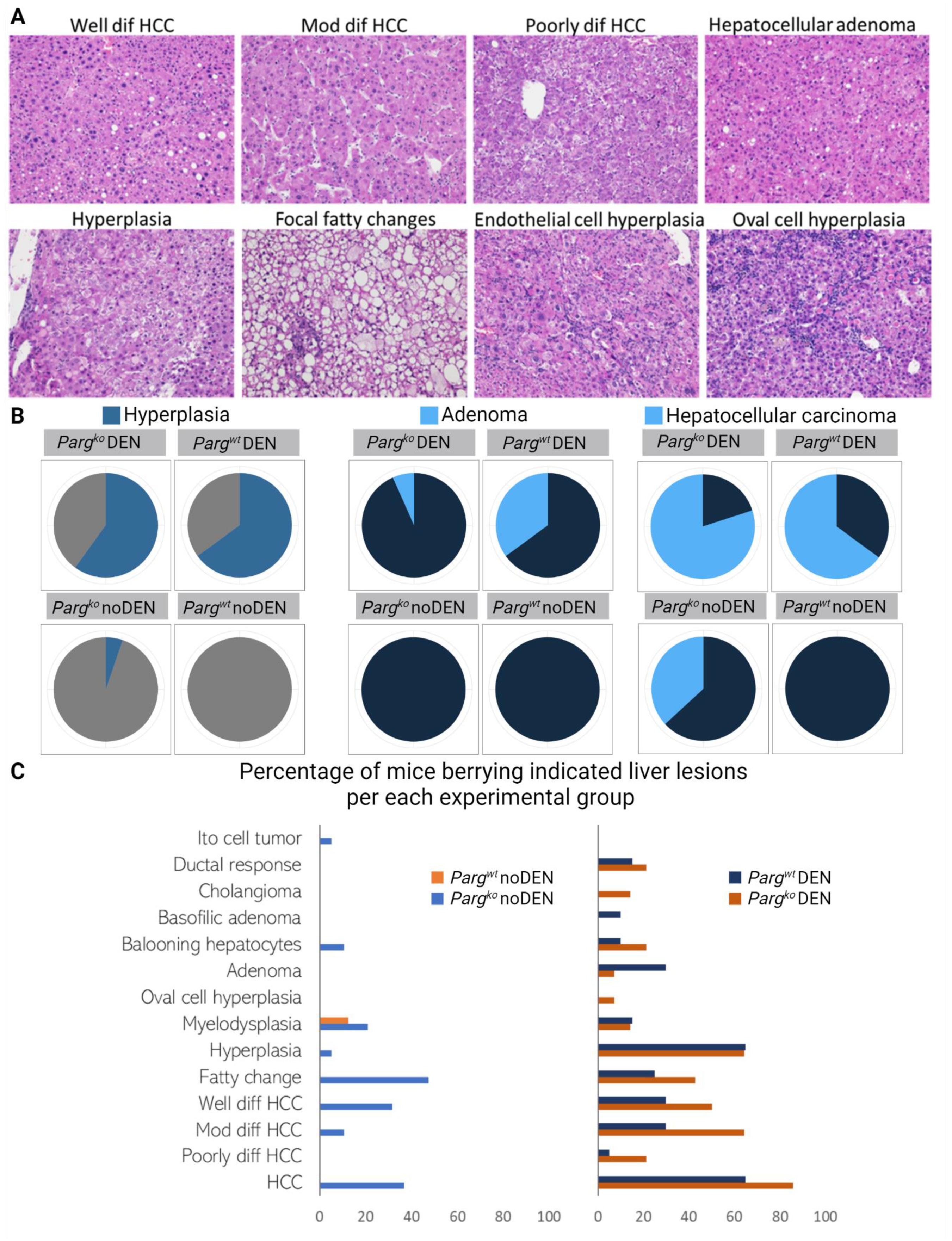
2.4. The Lack of PARG in Hepatocytes Predisposes Liver to Tumors
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Western Blotting
4.3. Chemically Induced Liver Tumor Generation
4.4. Tissue Histology and Immunofluorescence Staining
4.5. Nuclei Area Calculation in Liver Sections
4.6. Nuclei Isolation from Liver and Flow Cytometry
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Su, G.L.; Altayar, O.; O’Shea, R.; Shah, R.; Estfan, B.; Wenzell, C.; Sultan, S.; Falck-Ytter, Y. AGA Clinical Practice Guideline on Systemic Therapy for Hepatocellular Carcinoma. Gastroenterology 2022, 162, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Kennedy, E.B.; Abou-Alfa, G.K.; Beg, M.S.; Brower, S.T.; Gade, T.P.; Goff, L.; Gupta, S.; Guy, J.; Harris, W.P.; et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 4317–4345. [Google Scholar] [CrossRef]
- Cronin, K.A.; Scott, S.; Firth, A.U.; Sung, H.; Henley, S.J.; Sherman, R.L.; Siegel, R.L.; Anderson, R.N.; Kohler, B.A.; Benard, V.B.; et al. Annual Report to the Nation on the Status of Cancer, Part 1: National Cancer Statistics. Cancer 2022, 128, 4251–4284. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wen, Z. Survival Improvement and Prognosis for Hepatocellular Carcinoma: Analysis of the SEER Database. BMC Cancer 2021, 21, 1157. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Österreicher, C.H.; Takahashi, H.; Karin, M. Dietary and Genetic Obesity Promote Liver Inflammation and Tumorigenesis by Enhancing IL-6 and TNF Expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Splan, M.F.; Weiss, N.S.; McDonald, G.B.; Beretta, L.; Lee, S.P. Incidence and Predictors of Hepatocellular Carcinoma in Patients with Cirrhosis. Clin. Gastroenterol. Hepatol. 2007, 5, 938–945.e4. [Google Scholar] [CrossRef]
- Heidelbaugh, J.J.; Bruderly, M. Cirrhosis and Chronic Liver Failure: Part I. Diagnosis and Evaluation. Am. Fam. Physician 2006, 74, 756–762. [Google Scholar]
- White, D.L.; Kanwal, F.; El–Serag, H.B. Association Between Nonalcoholic Fatty Liver Disease and Risk for Hepatocellular Cancer, Based on Systematic Review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef] [PubMed]
- Alter, M.J. Epidemiology of Hepatitis C Virus Infection. World J. Gastroenterol. 2007, 13, 2436–2441. [Google Scholar] [CrossRef] [PubMed]
- Weledji, E.P. Familial Hepatocellular Carcinoma: ‘A Model for Studying Preventive and Therapeutic Measures’. Ann. Med. Surg. 2018, 35, 129–132. [Google Scholar] [CrossRef]
- Dragani, T.A. Risk of HCC: Genetic Heterogeneity and Complex Genetics. J. Hepatol. 2010, 52, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Taddei, T.; Mistry, P.; Schilsky, M.L. Inherited Metabolic Disease of the Liver. Curr. Opin. Gastroenterol. 2008, 24, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, Y.; Patel, P.; Chu, J.; Yu, X.; Hernaez, R.; El-Serag, H.; Kanwal, F. Risk of Hepatocellular Carcinoma in Patients with Various HFE Genotypes. Dig. Dis. Sci. 2023, 68, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Marek, G.W.; Hlady, R.A.; Wagner, R.T.; Zhao, X.; Clark, V.C.; Fan, A.X.; Liu, C.; Brantly, M.; Robertson, K.D. Alpha-1 Antitrypsin Deficiency Liver Disease, Mutational Homogeneity Modulated by Epigenetic Heterogeneity with Links to Obesity. Hepatology 2019, 70, 51–66. [Google Scholar] [CrossRef]
- Gunjan, D.; Shalimar; Nadda, N.; Kedia, S.; Nayak, B.; Paul, S.B.; Gamanagatti, S.R.; Acharya, S.K. Hepatocellular Carcinoma: An Unusual Complication of Longstanding Wilson Disease. J. Clin. Exp. Hepatol. 2017, 7, 152–154. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Hagström, H.; Sun, J.; Bergman, D.; Shang, Y.; Yang, W.; Roelstraete, B.; Ludvigsson, J.F. Familial Coaggregation of MASLD with Hepatocellular Carcinoma and Adverse Liver Outcomes: Nationwide Multigeneration Cohort Study. J. Hepatol. 2023, 79, 1374–1384. [Google Scholar] [CrossRef]
- Kogiso, T.; Ogasawara, Y.; Horiuchi, K.; Taniai, M.; Tokushige, K. Analysis of Genetic Factors Associated with Fatty Liver Disease-Related Hepatocellular Carcinoma. Cancer Med. 2023, 12, 17798–17807. [Google Scholar] [CrossRef]
- Wang, Q. Cancer Predisposition Genes: Molecular Mechanisms and Clinical Impact on Personalized Cancer Care: Examples of Lynch and HBOC Syndromes. Acta Pharmacol. Sin. 2016, 37, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-Induced Carcinogenic Liver Injury in Mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef]
- Li, N.; Chen, J. ADP-Ribosylation: Activation, Recognition, and Removal. Mol. Cells 2014, 37, 9–16. [Google Scholar] [CrossRef]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-Ribosyl)Ation Reactions in the Regulation of Nuclear Functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Tulin, A.; Spradling, A. Chromatin Loosening by Poly(ADP)-Ribose Polymerase (PARP) at Drosophila Puff Loci. Science 2003, 299, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, N.; Kossenkov, A.V.; Tulin, A.V. Bookmarking Promoters in Mitotic Chromatin: Poly(ADP-Ribose)Polymerase-1 as an Epigenetic Mark. Nucleic Acids Res. 2014, 42, 7028–7038. [Google Scholar] [CrossRef]
- Carrillo, A. Transcription Regulation of TNF- -Early Response Genes by Poly(ADP-Ribose) Polymerase-1 in Murine Heart Endothelial Cells. Nucleic Acids Res. 2004, 32, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Hottiger, M.O. PARP-1 as Novel Coactivator of NF-κB in Inflammatory Disorders. In Poly(ADP-Ribosyl)ation; Bürkle, A., Ed.; Springer US: New York, NY, USA, 2006; pp. 75–90. ISBN 978-0-387-36005-8. [Google Scholar]
- Tulin, A.; Stewart, D.; Spradling, A.C. The Drosophila Heterochromatic Gene Encoding Poly(ADP-Ribose) Polymerase (PARP) Is Required to Modulate Chromatin Structure during Development. Genes. Dev. 2002, 16, 2108–2119. [Google Scholar] [CrossRef]
- Ménissier de Murcia, J.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Amé, J.-C.; Dierich, A.; LeMeur, M.; et al. Functional Interaction between PARP-1 and PARP-2 in Chromosome Stability and Embryonic Development in Mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef]
- Hanai, S.; Kanai, M.; Ohashi, S.; Okamoto, K.; Yamada, M.; Takahashi, H.; Miwa, M. Loss of Poly(ADP-Ribose) Glycohydrolase Causes Progressive Neurodegeneration in Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 2004, 101, 82–86. [Google Scholar] [CrossRef]
- Koh, D.W.; Lawler, A.M.; Poitras, M.F.; Sasaki, M.; Wattler, S.; Nehls, M.C.; Stoger, T.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Failure to Degrade Poly(ADP-Ribose) Causes Increased Sensitivity to Cytotoxicity and Early Embryonic Lethality. Proc. Natl. Acad. Sci. USA 2004, 101, 17699–17704. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; Donson, A.M.; Kleinschmidt-DeMasters, B.K.; Gore, L.; Liu, A.K.; Foreman, N.K. PARP1 Expression in Pediatric Central Nervous System Tumors. Pediatr. Blood Cancer 2009, 53, 1227–1230. [Google Scholar] [CrossRef]
- Cavallo, F.; Graziani, G.; Antinozzi, C.; Feldman, D.R.; Houldsworth, J.; Bosl, G.J.; Chaganti, R.S.K.; Moynahan, M.E.; Jasin, M.; Barchi, M. Reduced Proficiency in Homologous Recombination Underlies the High Sensitivity of Embryonal Carcinoma Testicular Germ Cell Tumors to Cisplatin and Poly (Adp-Ribose) Polymerase Inhibition. PLoS ONE 2012, 7, e51563. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Huebner, A.J.; Clement, K.; Walsh, R.M.; Savol, A.; Lin, K.; Gu, H.; Di Stefano, B.; Brumbaugh, J.; Kim, S.-Y.; et al. Prolonged Mek1/2 Suppression Impairs the Developmental Potential of Embryonic Stem Cells. Nature 2017, 548, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Donizy, P.; Wu, C.-L.; Mull, J.; Fujimoto, M.; Chłopik, A.; Peng, Y.; Shalin, S.C.; Selim, M.A.; Puig, S.; Fernandez-Figueras, M.-T.; et al. Up-Regulation of PARP1 Expression Significantly Correlated with Poor Survival in Mucosal Melanomas. Cells 2020, 9, 1135. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Zhao, Y.; Gao, D.; Xing, J.; Liu, H. High PARP-1 Expression Is Associated with Tumor Invasion and Poor Prognosis in Gastric Cancer. Oncol. Lett. 2016, 12, 3825–3835. [Google Scholar] [CrossRef]
- Karpova, Y.; Guo, D.; Makhov, P.; Haines, A.M.; Markov, D.A.; Kolenko, V.; Tulin, A.V. Poly(ADP)-Ribosylation Inhibition: A Promising Approach for Clear Cell Renal Cell Carcinoma Therapy. Cancers 2021, 13, 4973. [Google Scholar] [CrossRef]
- Karpova, Y.; Johnson, S.J.; Bordet, G.; Guo, D.; Ghatak, A.; Markov, D.A.; Tulin, A.V. Upregulation of PARG in Prostate Cancer Cells Suppresses Their Malignant Behavior and Downregulates Tumor-Promoting Genes. Biomed. Pharmacother. 2022, 153, 113504. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Karpova, Y.; Guo, D.; Ghatak, A.; Markov, D.A.; Tulin, A.V. PARG Suppresses Tumorigenesis and Downregulates Genes Controlling Angiogenesis, Inflammatory Response, and Immune Cell Recruitment. BMC Cancer 2022, 22, 557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, C.; Jiang, Y.; Huang, K.; Liu, F.; Du, M.; Luo, X.; Huang, D.; Huang, K. Yin and Yang Regulation of Liver X Receptor α Signaling Control of Cholesterol Metabolism by Poly(ADP-Ribose) Polymerase 1. Int. J. Biol. Sci. 2020, 16, 2868–2882. [Google Scholar] [CrossRef]
- Li, L.; Zhong, S.; Li, R.; Liang, N.; Zhang, L.; Xia, S.; Xu, X.; Chen, X.; Chen, S.; Tao, Y.; et al. Aldehyde Dehydrogenase 2 and PARP1 Interaction Modulates Hepatic HDL Biogenesis by LXRα-Mediated ABCA1 Expression. JCI Insight 2022, 7, e155869. [Google Scholar] [CrossRef]
- Ju, C.; Liu, C.; Yan, S.; Wang, Y.; Mao, X.; Liang, M.; Huang, K. Poly(ADP-Ribose) Polymerase-1 Is Required for Hepatocyte Proliferation and Liver Regeneration in Mice. Biochem. Biophys. Res. Commun. 2019, 511, 531–535. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Cao, Z.; Horváth, B.; Park, O.; Wang, H.; Erdelyi, K.; Holovac, E.; Wang, Y.; Liaudet, L.; et al. Poly (ADP-Ribose) Polymerase-1 Is a Key Mediator of Liver Inflammation and Fibrosis. Hepatology 2014, 59, 1998–2009. [Google Scholar] [CrossRef]
- Li, J.; Dou, D.; Li, P.; Luo, W.; Lv, W.; Zhang, C.; Song, X.; Yang, Y.; Zhang, Y.; Xu, Y.; et al. PARP-1 Serves as a Novel Molecular Marker for Hepatocellular Carcinoma in a Southern Chinese Zhuang Population. Tumour Biol. 2017, 39, 1010428317706914. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Du, M.; Tan, X.; Yang, L.; Li, X.; Jiang, Y.; Wang, C.; Zhang, F.; Zhu, F.; Cheng, M.; et al. PARP1-Mediated PPARα Poly(ADP-Ribosyl)Ation Suppresses Fatty Acid Oxidation in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2017, 66, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual Roles for Glucokinase in Glucose Homeostasis as Determined by Liver and Pancreatic Beta Cell-Specific Gene Knock-Outs Using Cre Recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Prigge, J.R.; Wiley, J.A.; Talago, E.A.; Young, E.M.; Johns, L.L.; Kundert, J.A.; Sonsteng, K.M.; Halford, W.P.; Capecchi, M.R.; Schmidt, E.E. Nuclear Double-Fluorescent Reporter for in Vivo and Ex Vivo Analyses of Biological Transitions in Mouse Nuclei. Mamm. Genome 2013, 24, 389–399. [Google Scholar] [CrossRef]
- Postic, C.; Magnuson, M.A. DNA Excision in Liver by an Albumin-Cre Transgene Occurs Progressively with Age. Genesis 2000, 26, 149–150. [Google Scholar] [CrossRef]
- Gu, X.; Huang, D.; Ci, L.; Shi, J.; Zhang, M.; Yang, H.; Wang, Z.; Sheng, Z.; Sun, R.; Fei, J. Fate Tracing of Hepatocytes in Mouse Liver. Sci. Rep. 2017, 7, 16108. [Google Scholar] [CrossRef]
- Weisend, C.M.; Kundert, J.A.; Suvorova, E.S.; Prigge, J.R.; Schmidt, E.E. Cre Activity in Fetal albCre Mouse Hepatocytes: Utility for Developmental Studies. Genesis 2009, 47, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Grindheim, J.M.; Nicetto, D.; Donahue, G.; Zaret, K.S. PRC2 Proteins EZH1 and EZH2 Regulate Timing of Postnatal Hepatocyte Maturation and Fibrosis by Repressing Gene Expression at Promoter Regions in Euchromatin in Mice. Gastroenterology 2019, 156, 1834–1848. [Google Scholar] [CrossRef] [PubMed]
- Bhate, A.; Parker, D.J.; Bebee, T.W.; Ahn, J.; Arif, W.; Rashan, E.H.; Chorghade, S.; Chau, A.; Lee, J.-H.; Anakk, S.; et al. ESRP2 Controls an Adult Splicing Programme in Hepatocytes to Support Postnatal Liver Maturation. Nat. Commun. 2015, 6, 8768. [Google Scholar] [CrossRef] [PubMed]
- Morford, L.A.; Davis, C.; Jin, L.; Dobierzewska, A.; Peterson, M.L.; Spear, B.T. The Oncofetal Gene Glypican 3 Is Regulated in the Postnatal Liver by Zinc Fingers and Homeoboxes 2 and in the Regenerating Liver by Alpha-Fetoprotein Regulator 2. Hepatology 2007, 46, 1541–1547. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Principles of Liver Regeneration and Growth Homeostasis. Compr. Physiol. 2013, 3, 485–513. [Google Scholar] [PubMed]
- Palmes, D.; Spiegel, H.-U. Animal Models of Liver Regeneration. Biomaterials 2004, 25, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Gart, E.; van Duyvenvoorde, W.; Snabel, J.M.; de Ruiter, C.; Attema, J.; Caspers, M.P.M.; Lek, S.; van Heuven, B.J.; Speksnijder, A.G.C.L.; Giera, M.; et al. Translational Characterization of the Temporal Dynamics of Metabolic Dysfunctions in Liver, Adipose Tissue and the Gut during Diet-Induced NASH Development in Ldlr-/-.Leiden Mice. Heliyon 2023, 9, e13985. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, X.; Zhang, Y.-F.; Tian, J.-N.; Fan, S.-C.; Gao, Y.; Li, H.-L.; Cai, C.-H.; Huang, M.; Bi, H.-C. Peroxisome Proliferator-Activated Receptor α Agonist Induces Mouse Hepatomegaly through the Spatial Hepatocyte Enlargement and Proliferation. Acta Pharmacol. Sin. 2023, 44, 2037–2047. [Google Scholar] [CrossRef]
- Comerford, S.A.; Hinnant, E.A.; Chen, Y.; Hammer, R.E. Hepatic Ribosomal Protein S6 (Rps6) Insufficiency Results in Failed Bile Duct Development and Loss of Hepatocyte Viability; a Ribosomopathy-like Phenotype That Is Partially P53-Dependent. PLOS Genet. 2023, 19, e1010595. [Google Scholar] [CrossRef]
- Lu, W.-Y.; Bird, T.G.; Boulter, L.; Tsuchiya, A.; Cole, A.M.; Hay, T.; Guest, R.V.; Wojtacha, D.; Man, T.Y.; Mackinnon, A.; et al. Hepatic Progenitor Cells of Biliary Origin with Liver Repopulation Capacity. Nat. Cell Biol. 2015, 17, 971–983. [Google Scholar] [CrossRef]
- Raven, A.; Lu, W.-Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwyer, B.J.; Thomson, J.P.; Meehan, R.R.; Bogorad, R.; Koteliansky, V.; et al. Cholangiocytes Act as Facultative Liver Stem Cells during Impaired Hepatocyte Regeneration. Nature 2017, 547, 350–354. [Google Scholar] [CrossRef]
- Español-Suñer, R.; Carpentier, R.; Van Hul, N.; Legry, V.; Achouri, Y.; Cordi, S.; Jacquemin, P.; Lemaigre, F.; Leclercq, I.A. Liver Progenitor Cells Yield Functional Hepatocytes in Response to Chronic Liver Injury in Mice. Gastroenterology 2012, 143, 1564–1575.e7. [Google Scholar] [CrossRef]
- Rodrigo-Torres, D.; Affò, S.; Coll, M.; Morales-Ibanez, O.; Millán, C.; Blaya, D.; Alvarez-Guaita, A.; Rentero, C.; Lozano, J.J.; Maestro, M.A.; et al. The Biliary Epithelium Gives Rise to Liver Progenitor Cells. Hepatology 2014, 60, 1367–1377. [Google Scholar] [CrossRef]
- Shearn, C.T.; Anderson, A.L.; Devereux, M.W.; Orlicky, D.J.; Michel, C.; Petersen, D.R.; Miller, C.G.; Harpavat, S.; Schmidt, E.E.; Sokol, R.J. The Autophagic Protein P62 Is a Target of Reactive Aldehydes in Human and Murine Cholestatic Liver Disease. PLoS ONE 2022, 17, e0276879. [Google Scholar] [CrossRef]
- Gouw, A.S.H.; Clouston, A.D.; Theise, N.D. Ductular Reactions in Human Liver: Diversity at the Interface. Hepatology 2011, 54, 1853–1863. [Google Scholar] [CrossRef]
- Bamgbose, G.; Johnson, S.; Tulin, A. Cooperative Targeting of PARP-1 Domains to Regulate Metabolic and Developmental Genes. Front. Endocrinol. 2023, 14, 1152570. [Google Scholar] [CrossRef] [PubMed]
- Bordet, G.; Karpova, I.; Tulin, A.V. Poly(ADP-Ribosyl)Ating Enzymes Cooperate to Coordinate Development. Sci. Rep. 2022, 12, 22120. [Google Scholar] [CrossRef] [PubMed]
- Kotova, E.; Jarnik, M.; Tulin, A.V. Poly (ADP-Ribose) Polymerase 1 Is Required for Protein Localization to Cajal Body. PLoS Genet. 2009, 5, e1000387. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Dmitrieva-Posocco, O.; Dzutsev, A.; Posocco, D.F.; Hou, V.; Yuan, W.; Thovarai, V.; Mufazalov, I.A.; Gunzer, M.; Shilovskiy, I.P.; Khaitov, M.R.; et al. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 2019, 50, 166–180.e7. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-kappaB: Linking Inflammation and Immunity to Cancer Development and Progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, Z.; Zhou, Q.; Zhang, B.; Huang, J.; Jin, L.; Zhou, B.; Liu, S.; Yan, J.; Li, X.; et al. PARG Inhibition Limits HCC Progression and Potentiates the Efficacy of Immune Checkpoint Therapy. J. Hepatol. 2022, 77, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Bamgbose, G.; Tulin, A. PARP-1 Is a Transcriptional Rheostat of Metabolic and Bivalent Genes during Development. Life Sci. Alliance 2023, 7, e202302369. [Google Scholar] [CrossRef]
- Bordet, G.; Bamgbose, G.; Tulin, A.V. Poly(ADP-Ribosyl)Ating Enzymes Coordinate Changes in the Expression of Metabolic Genes with Developmental Progression. Sci. Rep. 2023, 13, 20320. [Google Scholar] [CrossRef]
- Karpova, Y.; Tulin, A.V. TaqMan Multiplex qPCR Method to Genotype PARG Knockout Mice. Methods Mol. Biol. 2023, 2609, 363–371. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpova, Y.; Orlicky, D.J.; Schmidt, E.E.; Tulin, A.V. Disrupting Poly(ADP-ribosyl)ating Pathway Creates Premalignant Conditions in Mammalian Liver. Int. J. Mol. Sci. 2023, 24, 17205. https://doi.org/10.3390/ijms242417205
Karpova Y, Orlicky DJ, Schmidt EE, Tulin AV. Disrupting Poly(ADP-ribosyl)ating Pathway Creates Premalignant Conditions in Mammalian Liver. International Journal of Molecular Sciences. 2023; 24(24):17205. https://doi.org/10.3390/ijms242417205
Chicago/Turabian StyleKarpova, Yaroslava, David J. Orlicky, Edward E. Schmidt, and Alexei V. Tulin. 2023. "Disrupting Poly(ADP-ribosyl)ating Pathway Creates Premalignant Conditions in Mammalian Liver" International Journal of Molecular Sciences 24, no. 24: 17205. https://doi.org/10.3390/ijms242417205
APA StyleKarpova, Y., Orlicky, D. J., Schmidt, E. E., & Tulin, A. V. (2023). Disrupting Poly(ADP-ribosyl)ating Pathway Creates Premalignant Conditions in Mammalian Liver. International Journal of Molecular Sciences, 24(24), 17205. https://doi.org/10.3390/ijms242417205