
Socrates: A Novel N-Ethyl-N-nitrosourea-Induced Mouse Mutant with Audiogenic Epilepsy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
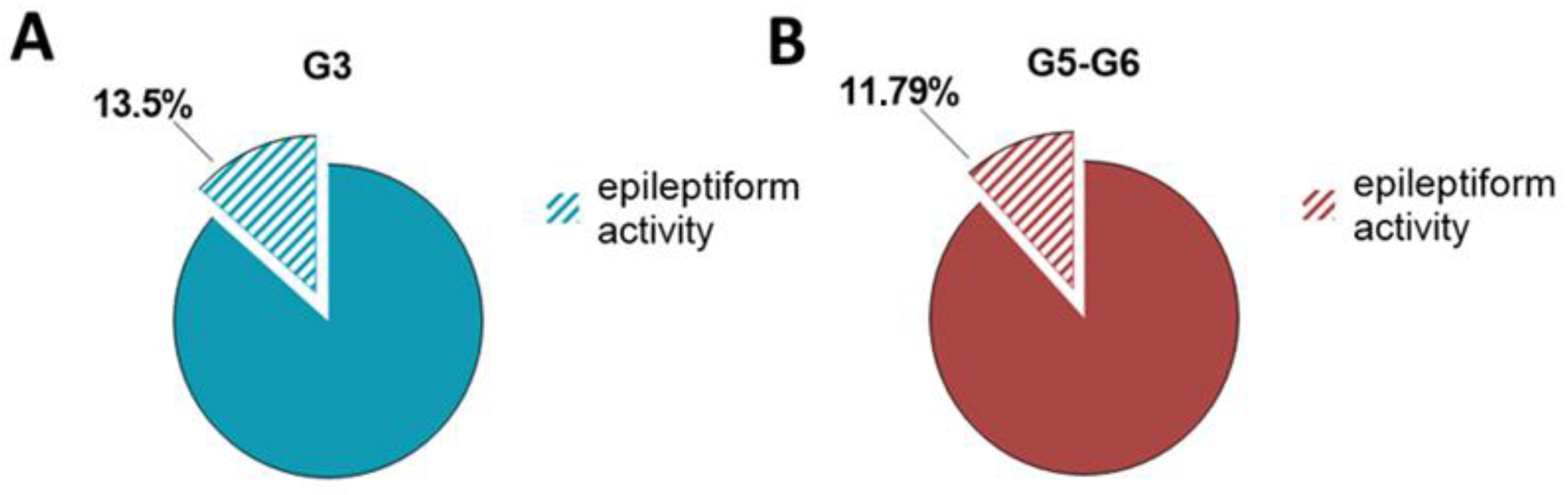
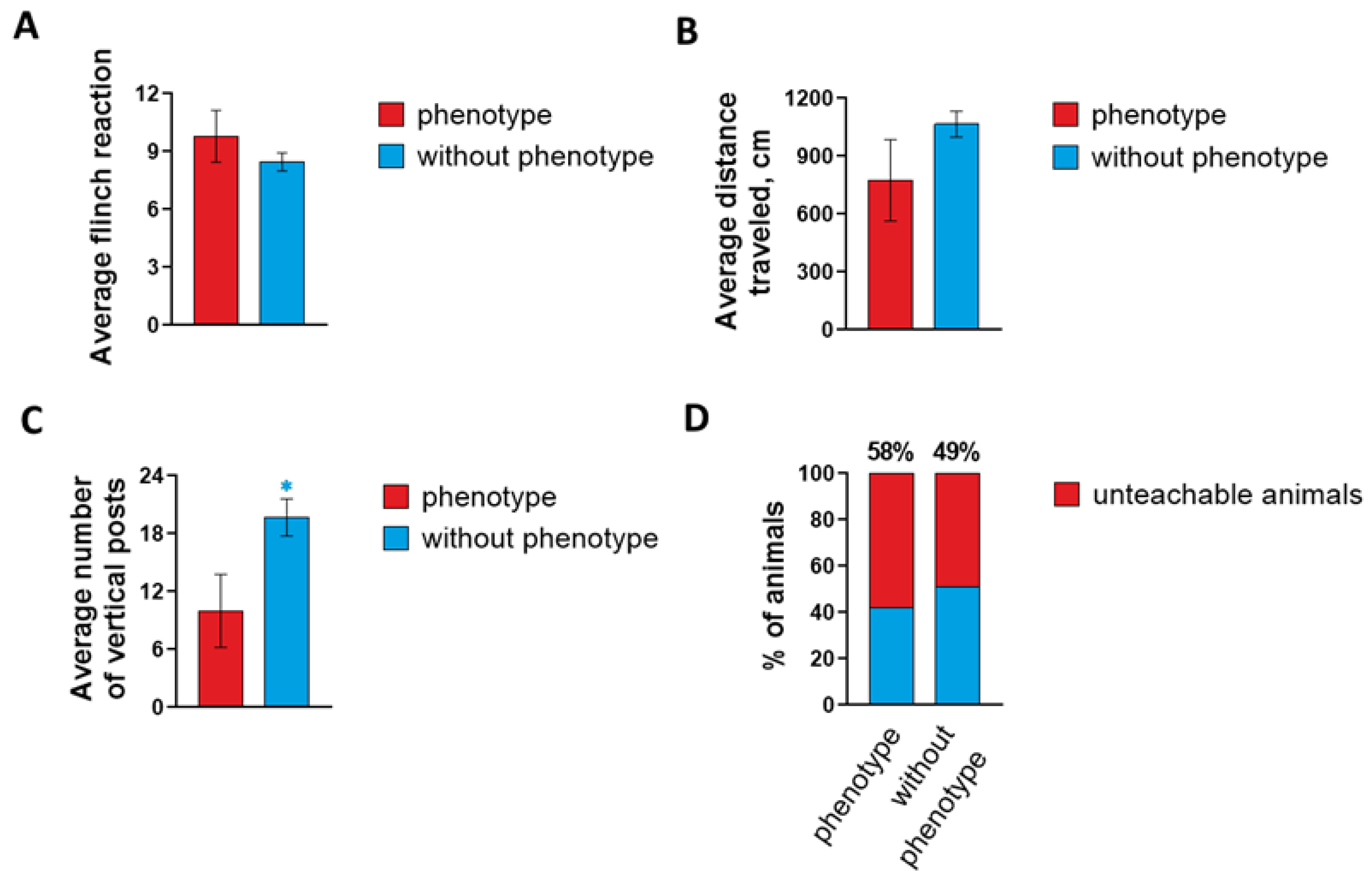
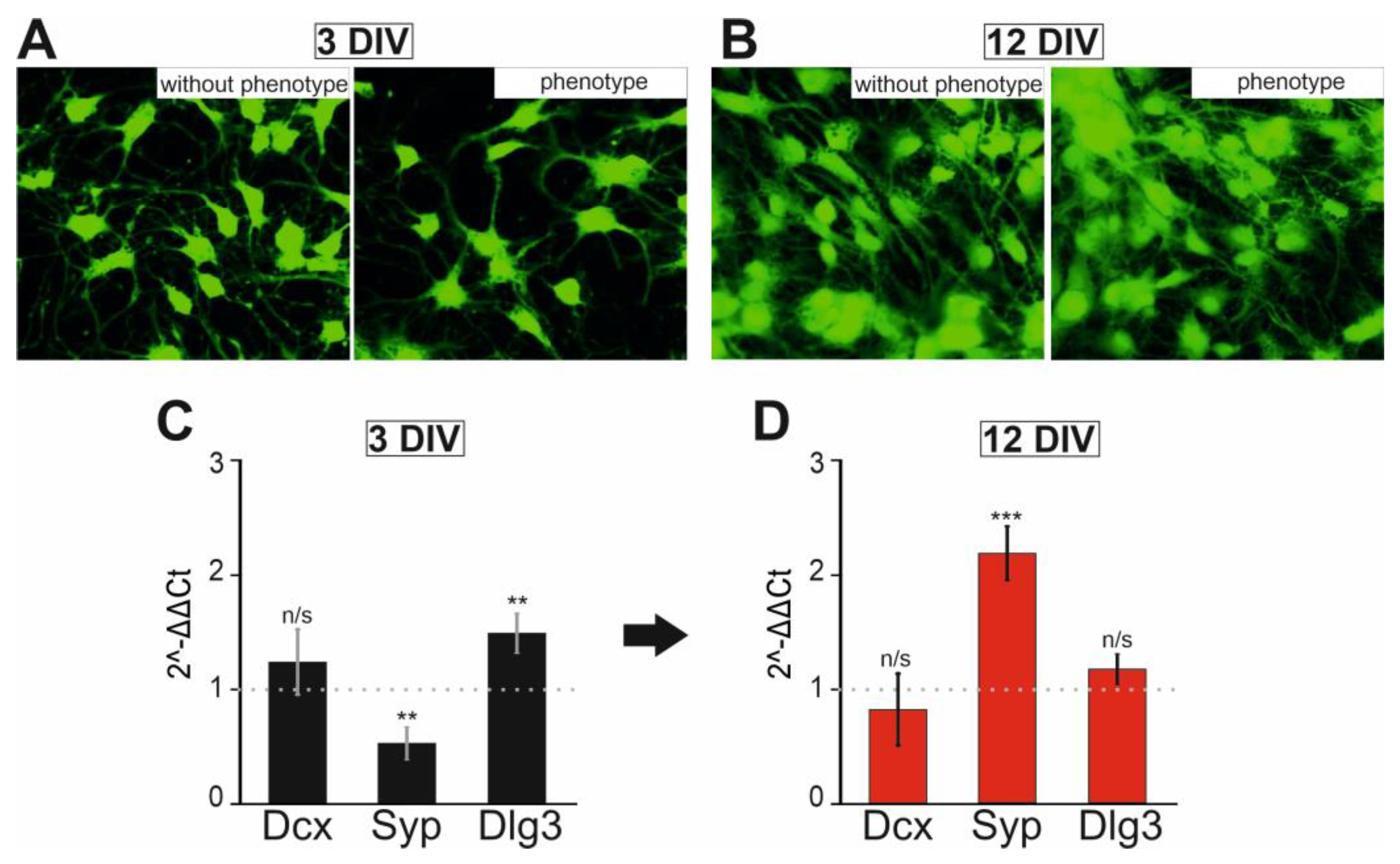
2.1. Characterization of Epileptiform Activity and Behavior in a Novel Mouse Mutant Soc
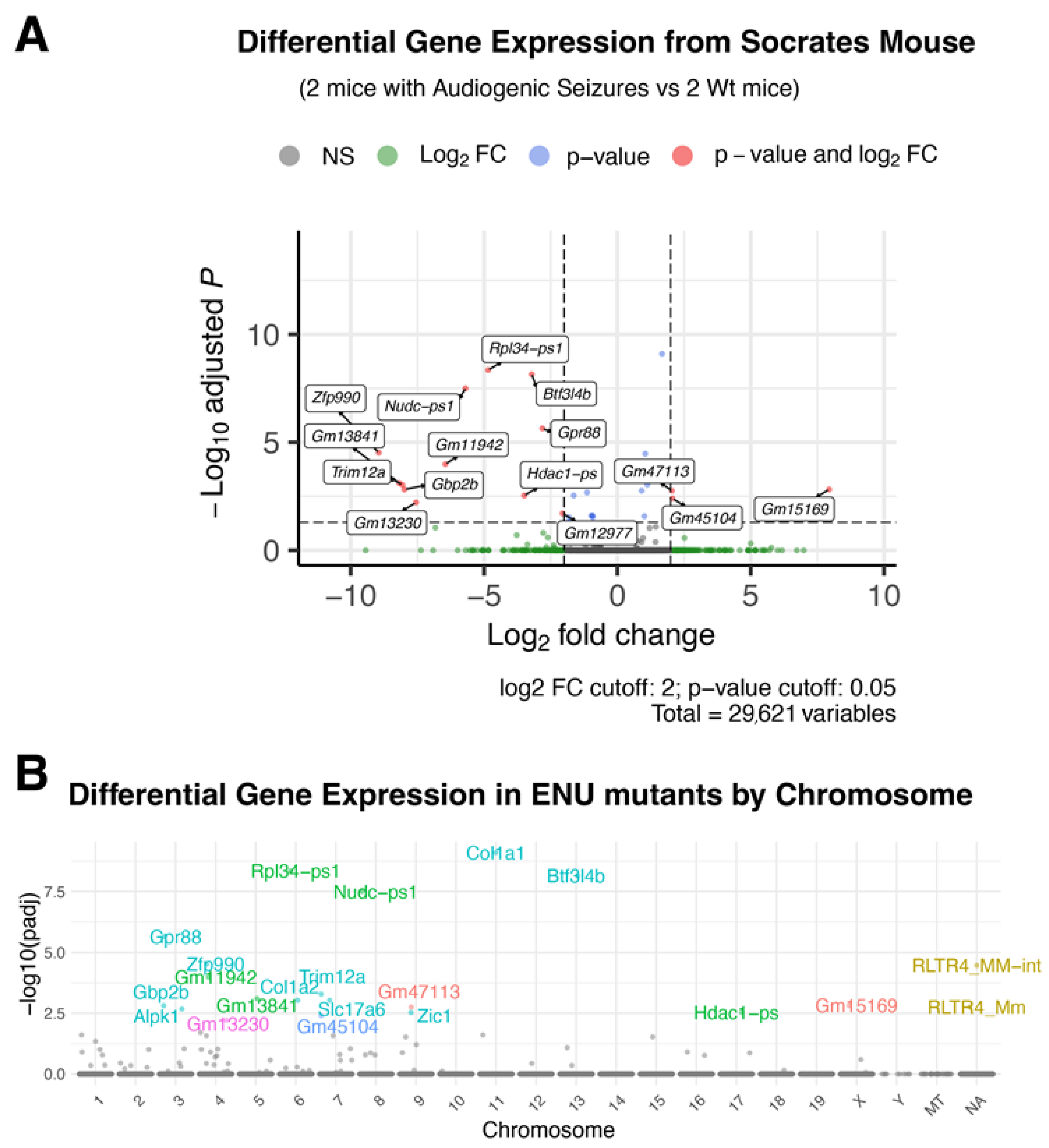
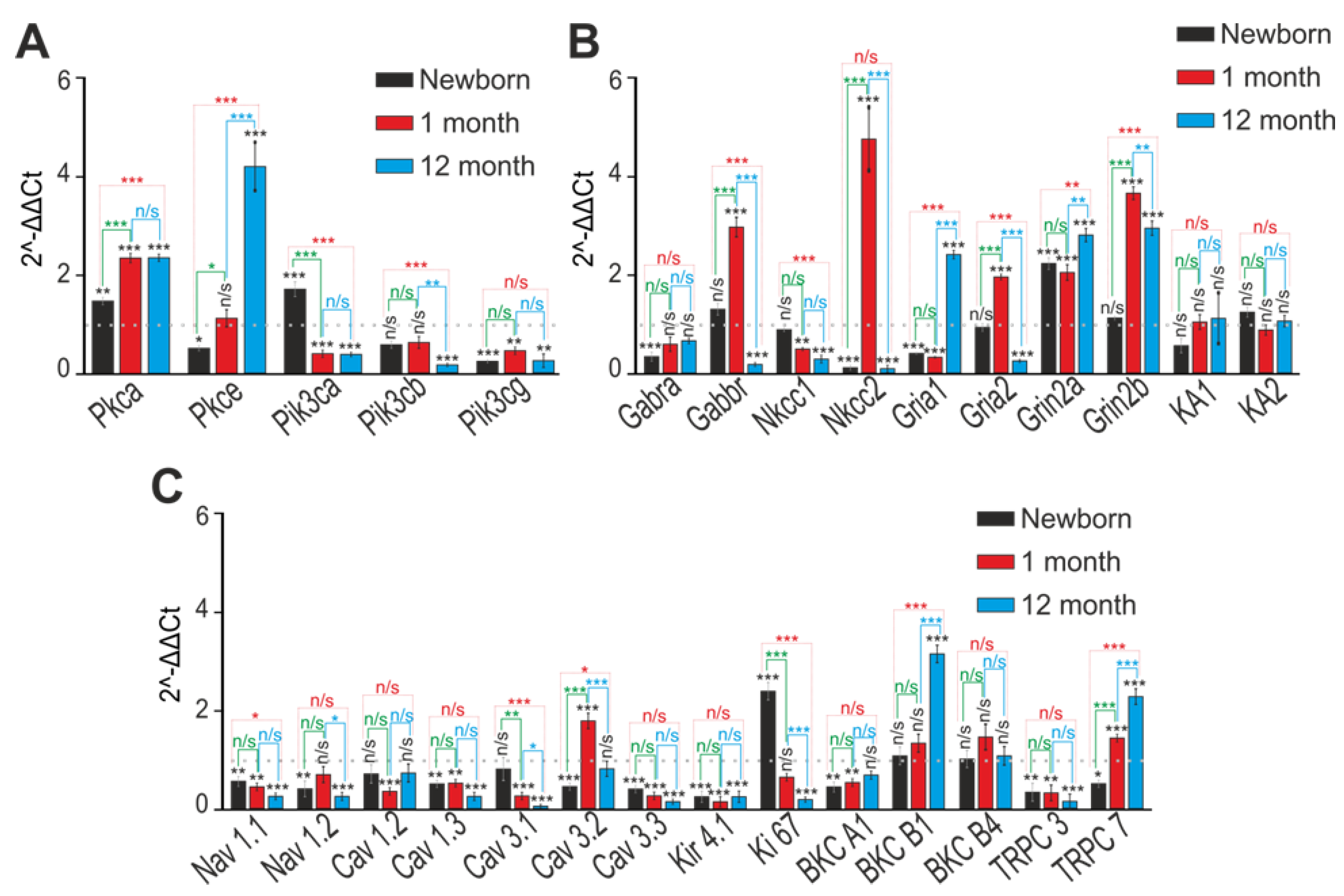
2.2. Soc Mice Show Gene Expression Differences in the Brain
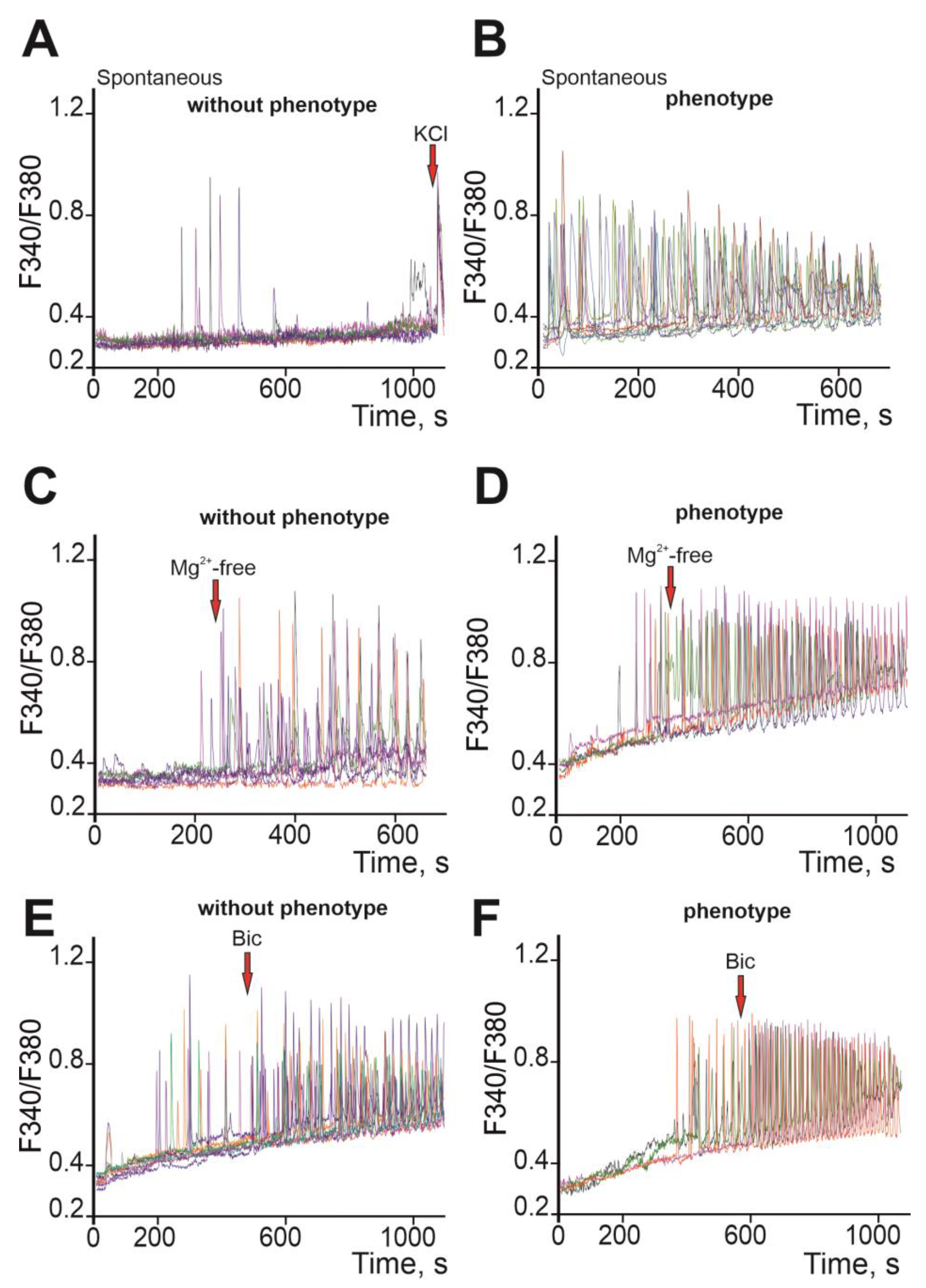
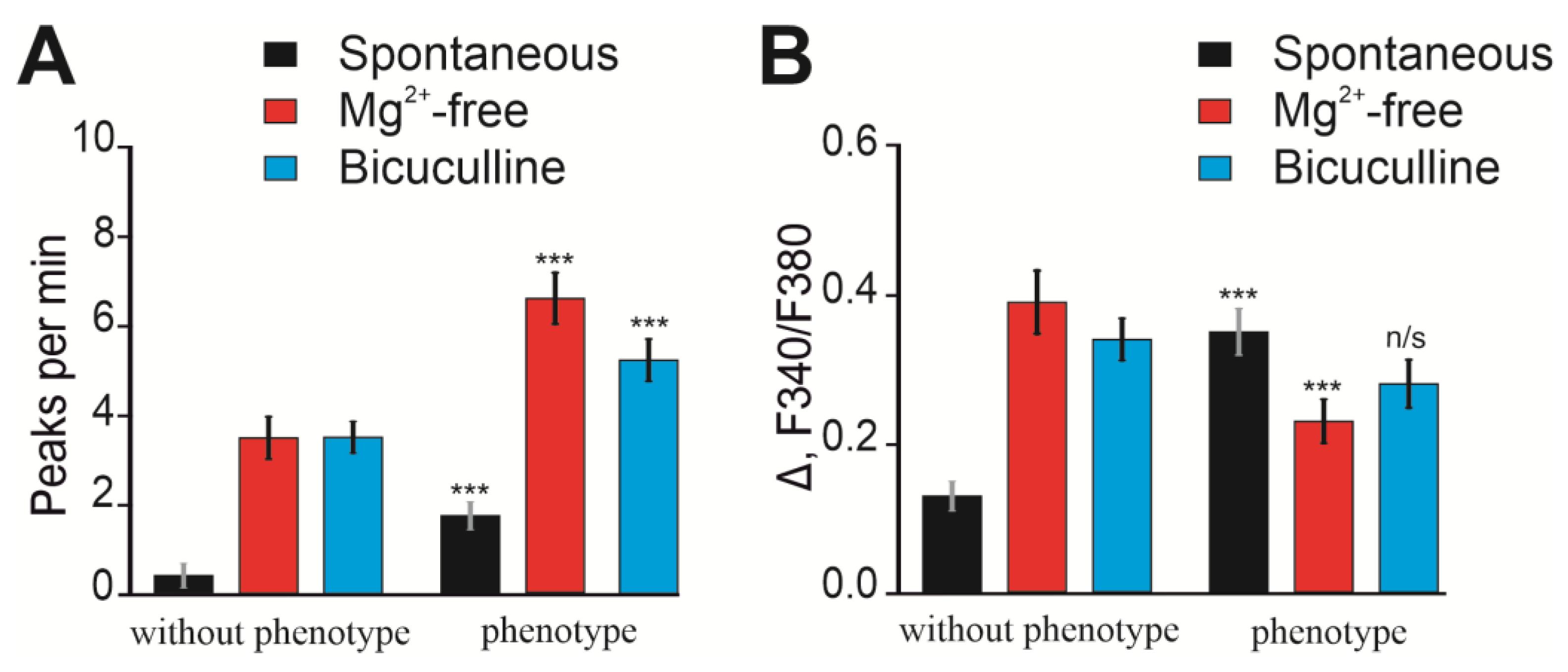
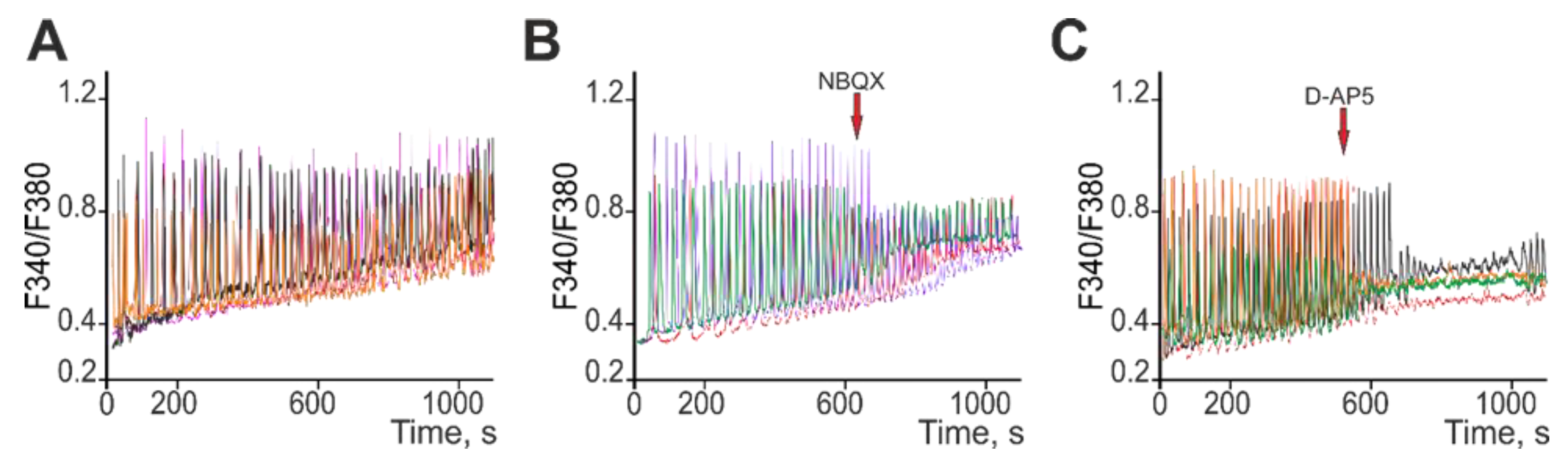
2.3. Ca2+ Activity in Cortical Neurons of Soc Mice
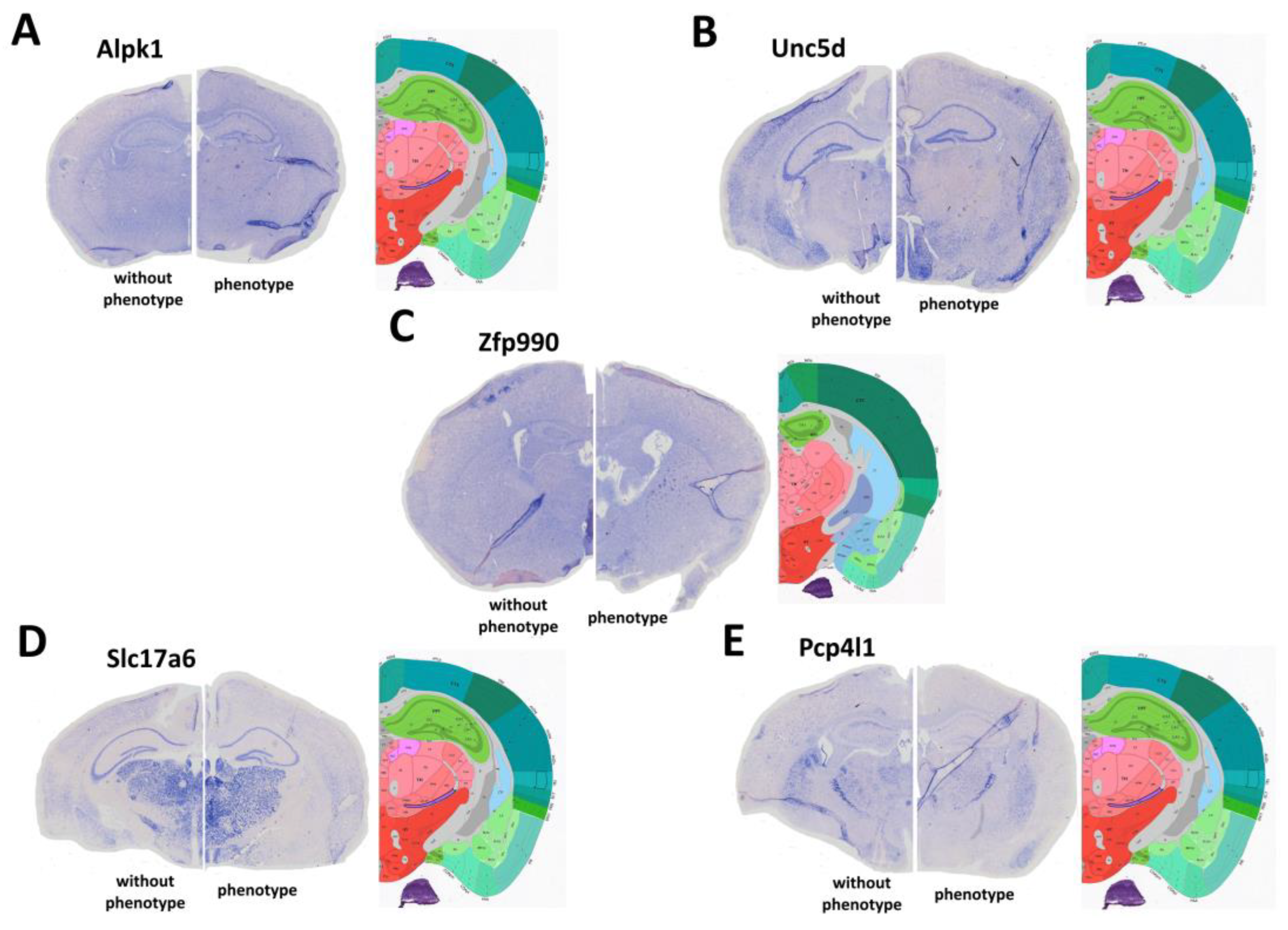
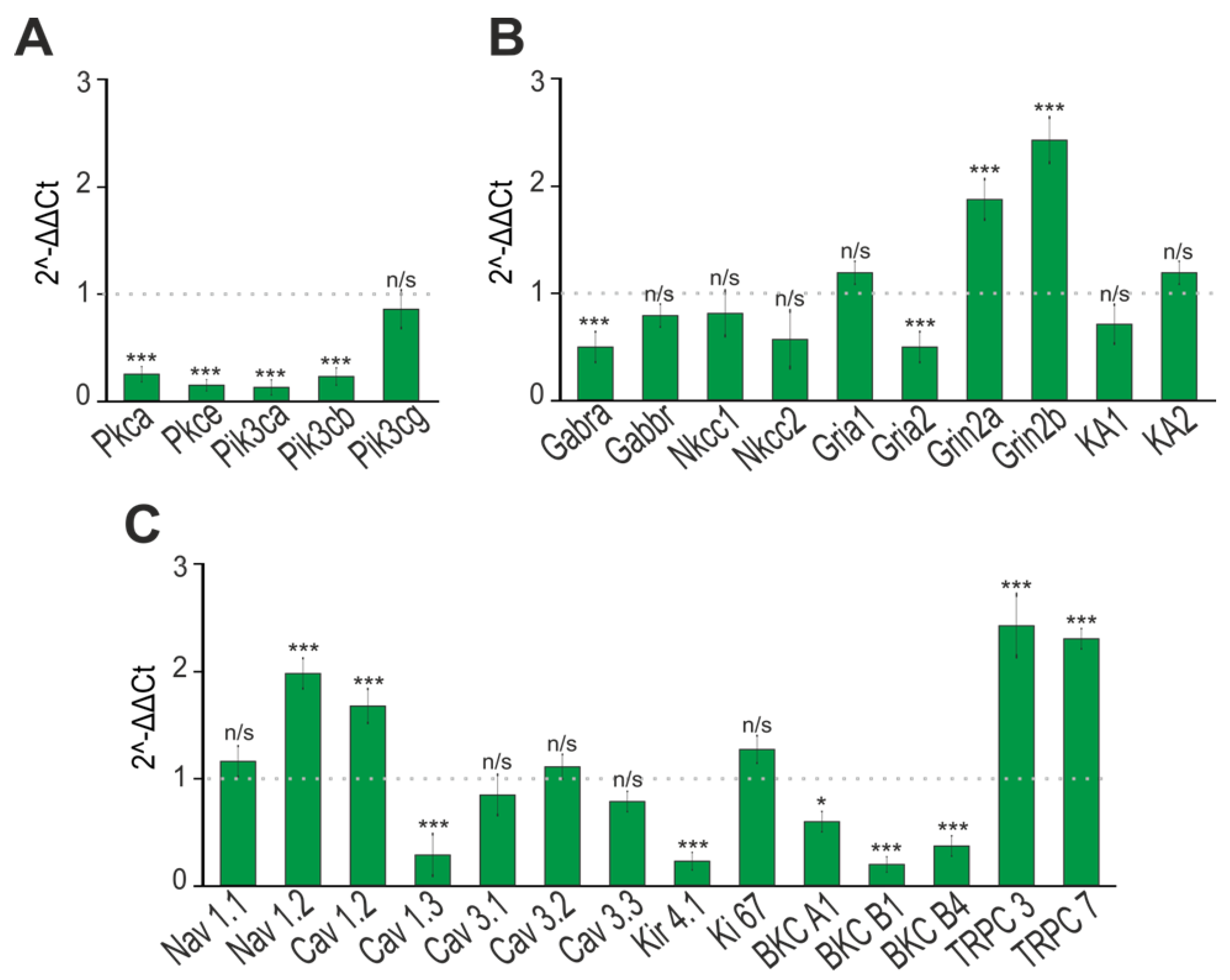
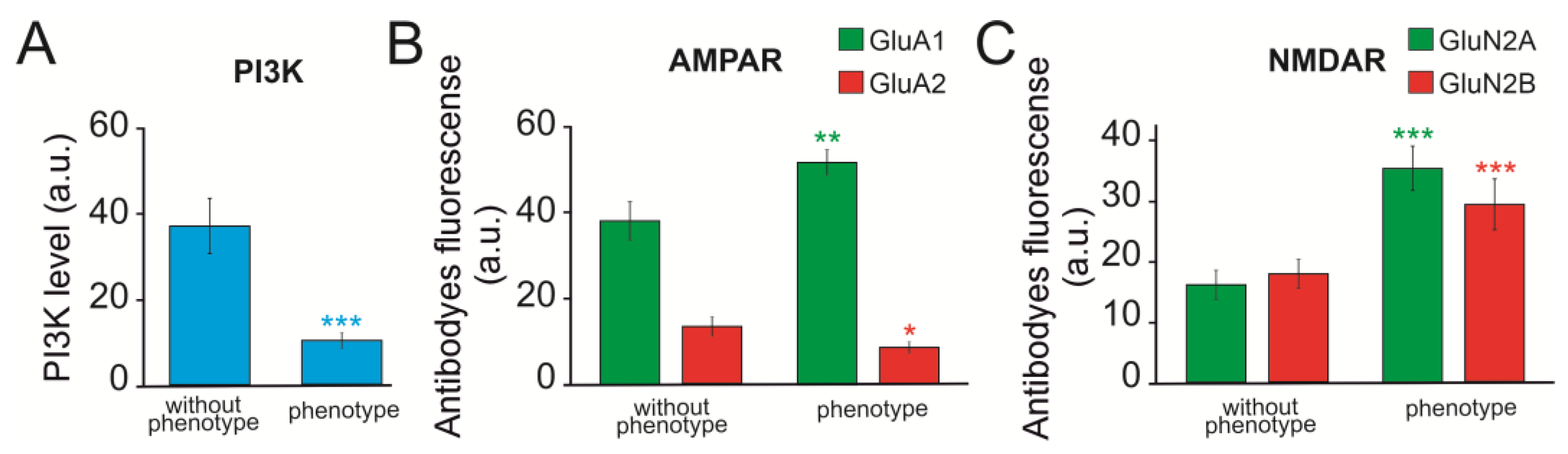
2.4. Changes in the Expression Patterns of Genes That Regulate Neuronal Hyperexcitation
2.5. Changes in Expression Patterns of Genes That Regulate Neuronal Hyperexcitation
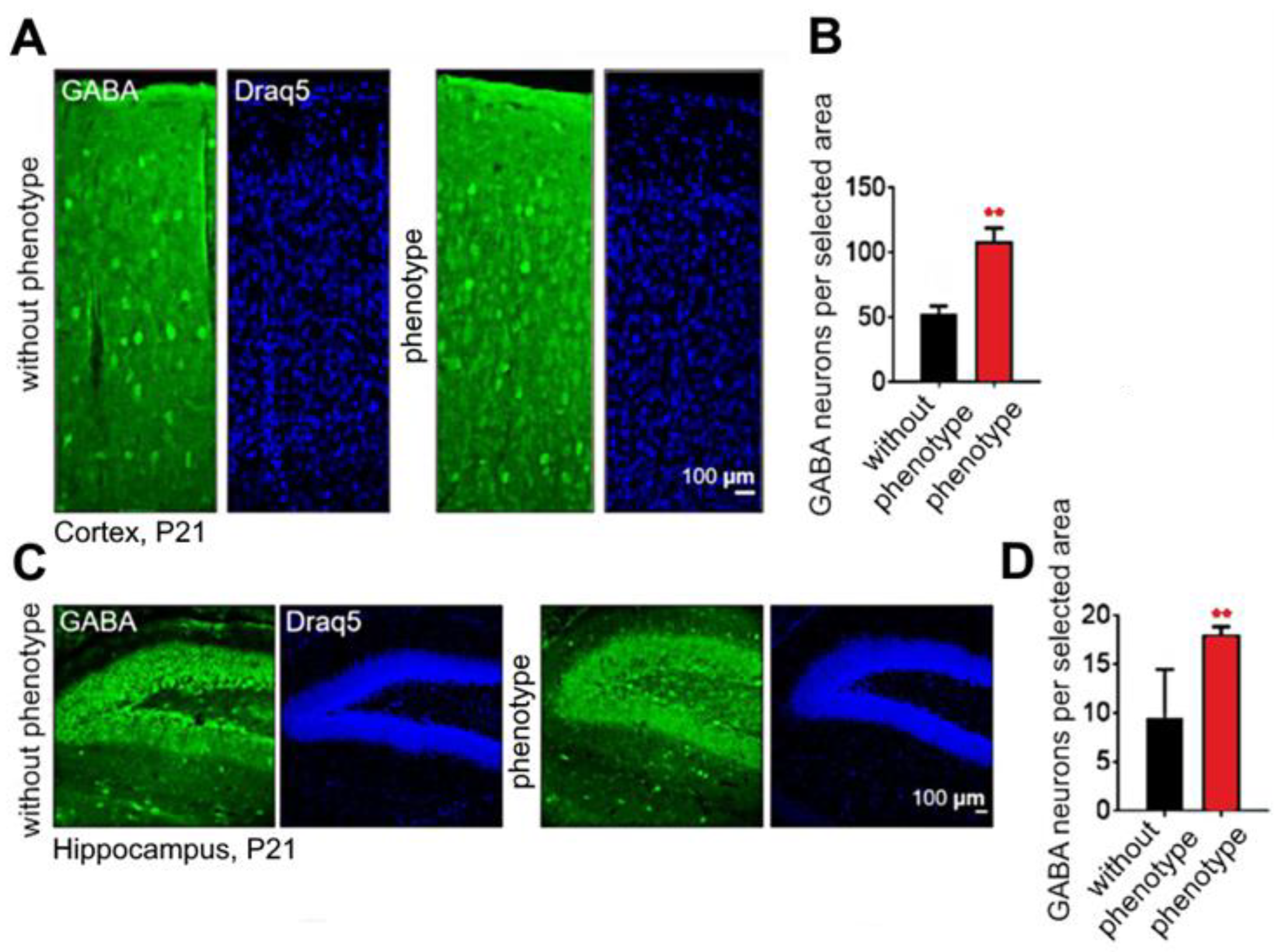
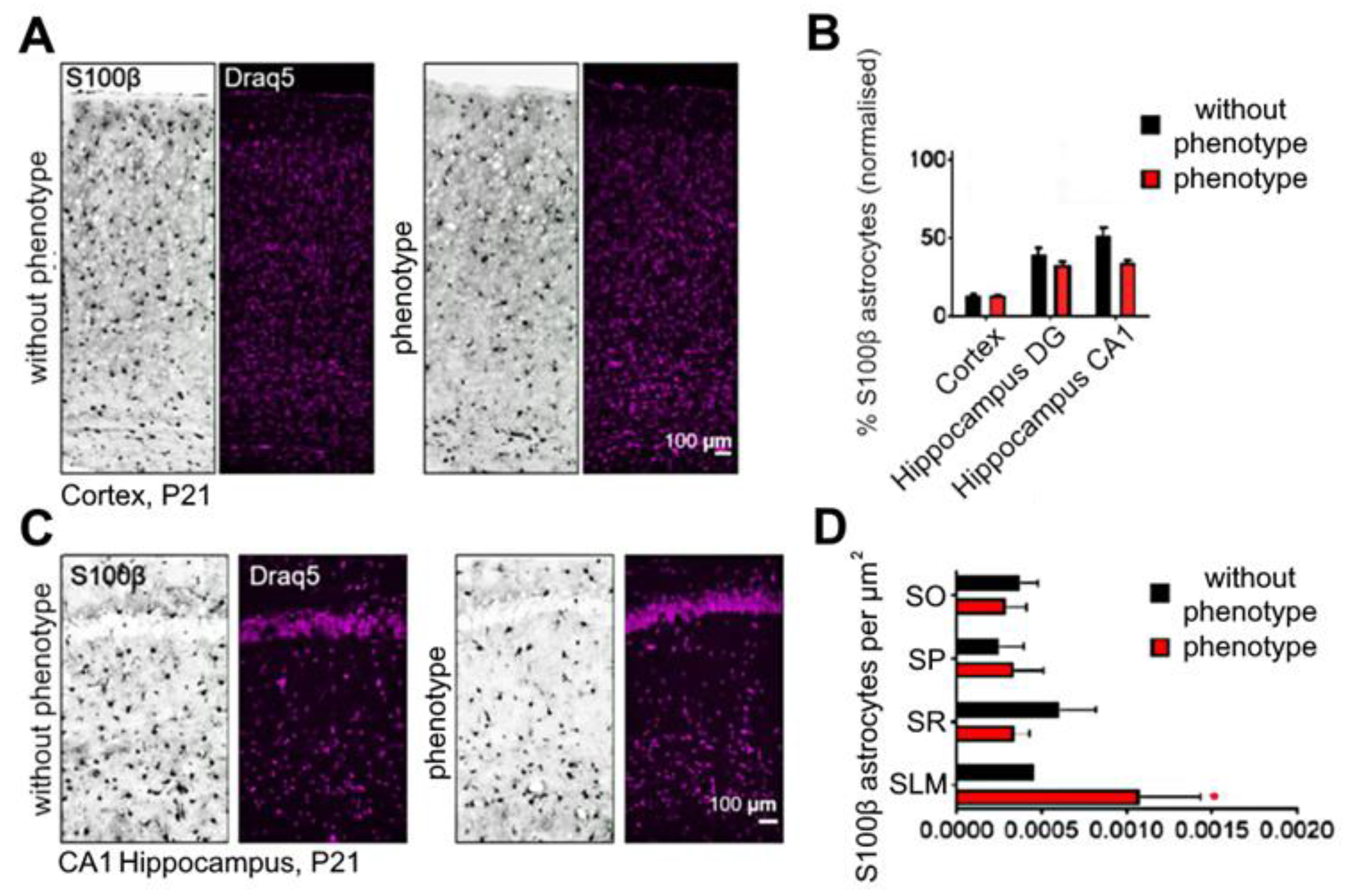
2.6. Soc Mutation Causes Increased Numbers of Interneurons
3. Discussion
4. Materials and Methods
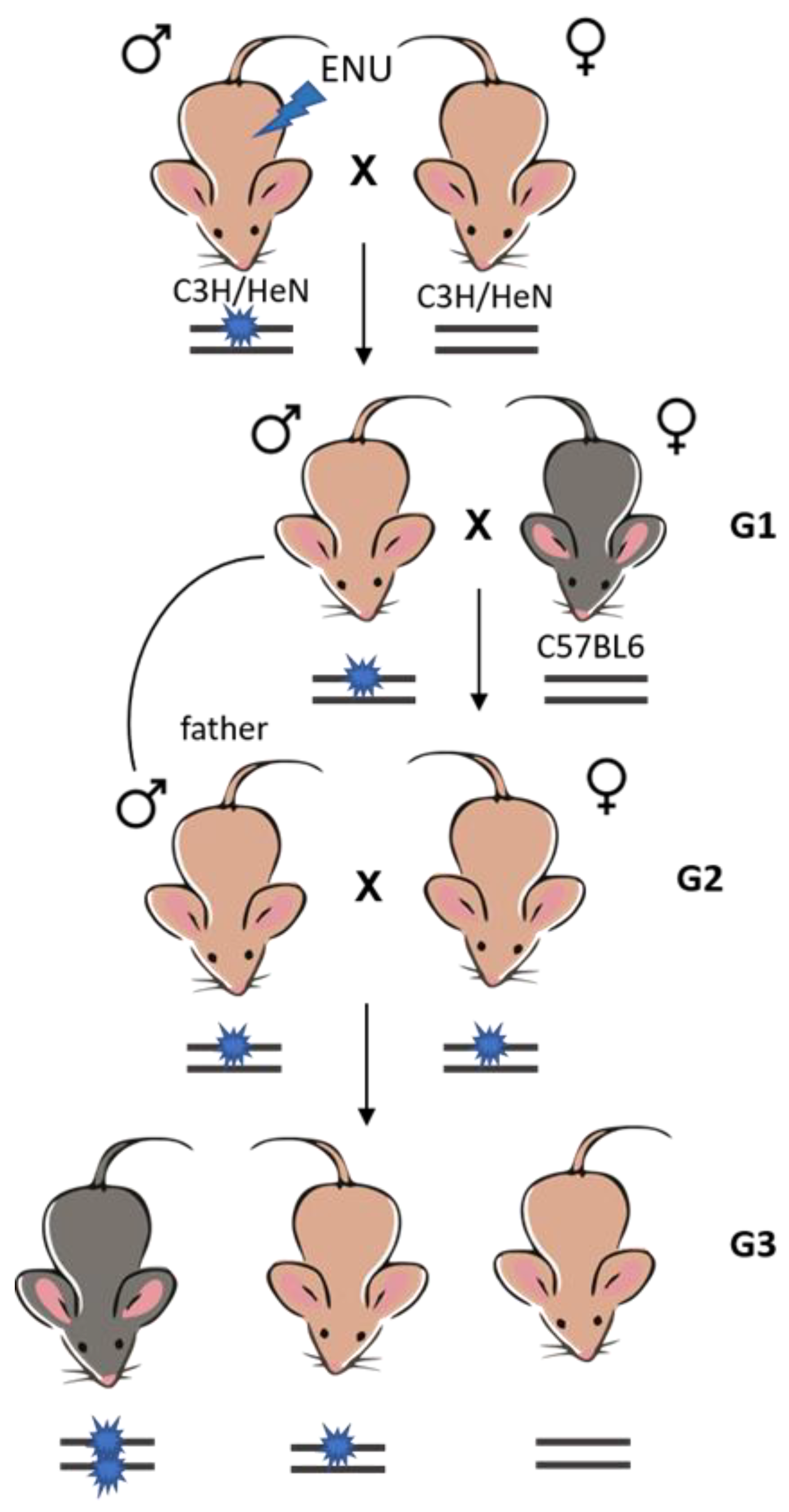
4.1. Generation of the Soc Mouse Strain Using ENU-Directed Mutagenesis and Breeding Scheme—Identifying Signs of Epilepsy
4.2. Basic Behavioral Phenotyping
4.2.1. Acoustic Startle Response
- Adjustment period—7 min;
- Stimulus (110 dB)—10 times;
- Prestimulus-1 (70 dB)—10 times;
- Prestimulus-2 (80 dB)—10 times;
- Prestimulus-3 (85 dB)—10 times;
- Prestimulus-4 (90 dB)—10 times;
- No sound stimulus—10 times;
- Signals are sent in randomly at an interval of 100 ms 10 times.
4.2.2. Mouse Open Field Test and Recording the Number of Vertical Stands
- Total mileage, including mileage in the central sector and the periphery (in cm);
- Number of vertical stands.
4.2.3. Conditioned Passive Avoidance Response
4.3. Cortical Neuroglial Culture Preparation
4.4. Immunocytochemical Method
4.5. Immunohistochemistry
4.6. In Situ Hybridization (ISH)
- -
- Zfp990 (ISH-Zfp990_fw 5′-GACCTCGAGTGGAGGAATGGGAATGTCTC-3′; ISH-Zfp990_rv-T3: 5′-AATTAACCCTCACTAAAGGGCGGCCGCAGATGGATCTGATGGGTAA GG-3′);
- -
- Pcp4l1 (ISH-Pcp4l1_fw5′-GACCTCGAGGCGAGCTTAACACCAAAACA-3; ISH-Pcp4l1_rv-T3: 5′-AATTAACCCTCACTAAAGGGCGGCCGCGGAGCTGGAATCCTTTTTC C-3);
- -
- Slc17a6 (ISH-Slc17a6_fw 5′-GACCTCGAGAAGAAGCAGGACAACCGAGA-3′; ISH-Slc17a6_rv-T3: 5′-AATTAACCCTCACTAAAGGGCGGCCGCGCAATGACTGCTCCAGCA TA-3′);
- -
- Alpk1 (ISH-Alpk1_fw 5′-GACCTCGAGTGCTGGATCGTCTCTTGTTG-3′; ISH-Alpk1_rv-T3: 5′-AATTAACCCTCACTAAAGGGCGGCCGCCAGTATGCCCAGTGATGT GG-3′);
- -
- Unc5d (ISH-Unc5d_fw 5′-GACCTCGAGTCCCGACTCTATCCCATCTG-3′; ISH-Unc5d_rv-T3: 5′-AATTAACCCTCACTAAAGGGCGGCCGCTCCATTCACGTAGACCACC A-3′).
4.7. Fluorescent Ca2+ Measurements
4.8. Extraction of RNA
4.9. RNA Sequencing
4.10. Real-Time Polymerase Chain Reaction (RT-qPCR)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, R.S.; Walsh, C.A. Molecular Insights into Human Brain Evolution. Nature 2005, 437, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Dobyns, W.B. Malformations of Cortical Development: Clinical Features and Genetic Causes. Lancet Neurol. 2014, 13, 710–726. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E. The Epidemiology of Epilepsy. Neuroepidemiology 2020, 54, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Saviuk, M.; Sleptsova, E.; Redkin, T.; Turubanova, V. Unexplained Causes of Glioma-Associated Epilepsies: A Review of Theories and an Area for Research. Cancers 2023, 15, 5539. [Google Scholar] [CrossRef]
- Symonds, J.D.; Zuberi, S.M.; Stewart, K.; McLellan, A.; O’Regan, M.; MacLeod, S.; Jollands, A.; Joss, S.; Kirkpatrick, M.; Brunklaus, A.; et al. Incidence and Phenotypes of Childhood-Onset Genetic Epilepsies: A Prospective Population-Based National Cohort. Brain 2019, 142, 2303–2318. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Z.-J.; Liu, L.; Xu, H.-Q.; Shi, Y.-W.; Yi, Y.-H.; He, N.; Liao, W.-P. Epilepsy-Associated Genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef]
- Chen, T.; Giri, M.; Xia, Z.; Subedi, Y.N.; Li, Y. Genetic and Epigenetic Mechanisms of Epilepsy: A Review. Neuropsychiatr. Dis. Treat. 2017, 13, 1841–1859. [Google Scholar] [CrossRef]
- Nolan, P.M.; Peters, J.; Strivens, M.; Rogers, D.; Hagan, J.; Spurr, N.; Gray, I.C.; Vizor, L.; Brooker, D.; Whitehill, E.; et al. A Systematic, Genome-Wide, Phenotype-Driven Mutagenesis Programme for Gene Function Studies in the Mouse. Nat. Genet. 2000, 25, 440–443. [Google Scholar] [CrossRef]
- de Angelis, M.H.; Flaswinkel, H.; Fuchs, H.; Rathkolb, B.; Soewarto, D.; Marschall, S.; Heffner, S.; Pargent, W.; Wuensch, K.; Jung, M.; et al. Genome-Wide, Large-Scale Production of Mutant Mice by ENU Mutagenesis. Nat. Genet. 2000, 25, 444–447. [Google Scholar] [CrossRef]
- Bućan, M.; Abel, T. The Mouse: Genetics Meets Behaviour. Nat. Rev. Genet. 2002, 3, 114–123. [Google Scholar] [CrossRef]
- Shimada, T.; Yamagata, K. Pentylenetetrazole-Induced Kindling Mouse Model. J. Vis. Exp. 2018, 12, e56573. [Google Scholar] [CrossRef]
- Abdelbasset, W.K.; Jasim, S.A.; Rudiansyah, M.; Huldani, H.; Margiana, R.; Jalil, A.T.; Mohammad, H.J.; Ridha, H.S.; Yasin, G. Treatment of Pilocarpine-Induced Epileptic Seizures in Adult Male Mice. Braz. J. Biol. 2022, 84, e260091. [Google Scholar] [CrossRef] [PubMed]
- Falcón-Moya, R.; Sihra, T.S.; Rodríguez-Moreno, A. Kainate Receptors: Role in Epilepsy. Front. Mol. Neurosci. 2018, 11, 217. [Google Scholar] [CrossRef] [PubMed]
- Garbuz, D.G.; Davletshin, A.A.; Litvinova, S.A.; Fedotova, I.B.; Surina, N.M.; Poletaeva, I.I. Rodent Models of Audiogenic Epilepsy: Genetic Aspects, Advantages, Current Problems and Perspectives. Biomedicines 2022, 10, 2934. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Li, Z.-N.; Huang, T.-Z.; Li, J.-H.; Yang, L.; Wei, J.-K. Threshold for Maximal Electroshock Seizures (MEST) at Three Developmental Stages in Young Mice. Zool. Res. 2019, 40, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Borisova, E.V.; Epifanova, E.A.; Tutukova, S.A.; Belousova, I.I.; Zhidkova, N.M.; Rusanova, A.M.; Salina, V.A.; Turovsky, E.A.; Turovskaya, M.V.; Tarabykin, V.S.; et al. Identification of Novel Mutations Controlling Cerebral Cortex Malformations Caused by ENU-Induced Mutagenesis in the Mouse. Sovrem. Tehnol. V Med. 2018, 10, 70–76. [Google Scholar] [CrossRef]
- Borisova, E.V.; Turovsky, E.A.; Turovskaya, M.V.; Tomilin, A.N.; Nedospasov, S.A.; Tarabykin, V.S. Enu Mutagenesis as a Tool for Identifying Novel Mouse Models of Epilepsy. Opera Med. Physiol. 2021, 8, 5–11. [Google Scholar] [CrossRef]
- Chen, S.; Xu, D.; Fan, L.; Fang, Z.; Wang, X.; Li, M. Roles of N-Methyl-D-Aspartate Receptors (NMDARs) in Epilepsy. Front. Mol. Neurosci. 2022, 14, 797253. [Google Scholar] [CrossRef]
- Tipton, A.E.; Russek, S.J. Regulation of Inhibitory Signaling at the Receptor and Cellular Level; Advances in Our Understanding of GABAergic Neurotransmission and the Mechanisms by Which It Is Disrupted in Epilepsy. Front. Synaptic Neurosci. 2022, 14, 914374. [Google Scholar] [CrossRef]
- Hanada, T. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef]
- Borisova, E. Studies on the Formation of Cortical Circuits the Role of Ire1α in the Developing Neocortex Identification of Epilepsy-Associated Genes by ENU-Induced Mutagenesis in Mice. Ph.D. Thesis, Humboldt-Universität zu Berlin, Berlin, Germany, 2022. [Google Scholar]
- Elsea, S.H.; Lucas, R.E. The Mousetrap: What We Can Learn When the Mouse Model Does Not Mimic the Human Disease. ILAR J. 2002, 43, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.; Bocchini, C.; Hamosh, A. A New Face and New Challenges for Online Mendelian Inheritance in Man (OMIM®). Hum. Mutat. 2011, 32, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Prabantu, V.M.; Naveenkumar, N.; Srinivasan, N. Influence of Disease-Causing Mutations on Protein Structural Networks. Front. Mol. Biosci. 2021, 7, 620554. [Google Scholar] [CrossRef] [PubMed]
- Bras, J.; Guerreiro, R.; Hardy, J. Use of Next-Generation Sequencing and Other Whole-Genome Strategies to Dissect Neurological Disease. Nat. Rev. Neurosci. 2012, 13, 453–464. [Google Scholar] [CrossRef]
- Justice, M.J.; Noveroske, J.K.; Weber, J.S.; Zheng, B.; Bradley, A. Mouse ENU Mutagenesis. Hum. Mol. Genet. 1999, 8, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Groszer, M.; Keays, D.A.; Deacon, R.M.J.; de Bono, J.P.; Prasad-Mulcare, S.; Gaub, S.; Baum, M.G.; French, C.A.; Nicod, J.; Coventry, J.A.; et al. Impaired Synaptic Plasticity and Motor Learning in Mice with a Point Mutation Implicated in Human Speech Deficits. Curr. Biol. 2008, 18, 354–362. [Google Scholar] [CrossRef]
- Achilli, F.; Bros-Facer, V.; Williams, H.P.; Banks, G.T.; AlQatari, M.; Chia, R.; Tucci, V.; Groves, M.; Nickols, C.D.; Seburn, K.L.; et al. An ENU-Induced Mutation in Mouse Glycyl-TRNA Synthetase (GARS) Causes Peripheral Sensory and Motor Phenotypes Creating a Model of Charcot-Marie-Tooth Type 2D Peripheral Neuropathy. Dis. Model. Mech. 2009, 2, 359–373. [Google Scholar] [CrossRef]
- Furuse, T.; Yamada, I.; Kushida, T.; Masuya, H.; Miura, I.; Kaneda, H.; Kobayashi, K.; Wada, Y.; Yuasa, S.; Wakana, S. Behavioral and Neuromorphological Characterization of a Novel Tuba1 Mutant Mouse. Behav. Brain Res. 2012, 227, 167–174. [Google Scholar] [CrossRef]
- Clapcote, S.J.; Lipina, T.V.; Millar, J.K.; Mackie, S.; Christie, S.; Ogawa, F.; Lerch, J.P.; Trimble, K.; Uchiyama, M.; Sakuraba, Y.; et al. Behavioral Phenotypes of Disc1 Missense Mutations in Mice. Neuron 2007, 54, 387–402. [Google Scholar] [CrossRef]
- Liu, X.; Dobbie, M.; Tunningley, R.; Whittle, B.; Zhang, Y.; Ittner, L.M.; Götz, J. ENU Mutagenesis Screen to Establish Motor Phenotypes in Wild-Type Mice and Modifiers of a Pre-Existing Motor Phenotype in Tau Mutant Mice. J. Biomed. Biotechnol. 2011, 2011, 130947. [Google Scholar] [CrossRef]
- Ittner, L.M.; Fath, T.; Ke, Y.D.; Bi, M.; van Eersel, J.; Li, K.M.; Gunning, P.; Götz, J. Parkinsonism and Impaired Axonal Transport in a Mouse Model of Frontotemporal Dementia. Proc. Natl. Acad. Sci. USA 2008, 105, 15997–16002. [Google Scholar] [CrossRef]
- Clapcote, S.J.; Duffy, S.; Xie, G.; Kirshenbaum, G.; Bechard, A.R.; Rodacker Schack, V.; Petersen, J.; Sinai, L.; Saab, B.J.; Lerch, J.P.; et al. Mutation I810N in the A3 Isoform of Na+, K+-ATPase Causes Impairments in the Sodium Pump and Hyperexcitability in the CNS. Proc. Natl. Acad. Sci. USA 2009, 106, 14085–14090. [Google Scholar] [CrossRef]
- Goldstein, I.; Lerer, E.; Laiba, E.; Mallet, J.; Mujaheed, M.; Laurent, C.; Rosen, H.; Ebstein, R.P.; Lichtstein, D. Association Between Sodium- and Potassium-Activated Adenosine Triphosphatase α Isoforms and Bipolar Disorders. Biol. Psychiatry 2009, 65, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Einholm, A.P.; Toustrup-Jensen, M.S.; Holm, R.; Andersen, J.P.; Vilsen, B. The Rapid-Onset Dystonia Parkinsonism Mutation D923N of the Na+, K+-ATPase A3 Isoform Disrupts Na+ Interaction at the Third Na+ Site. J. Biol. Chem. 2010, 285, 26245–26254. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, P.; Yan, F.; Luo, Y.; Zhao, G. Animal Models of Epilepsy: A Phenotype-Oriented Review. Aging Dis. 2022, 13, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Mitina, N.N.; Kondakova, E.V.; Tarabykin, V.S.; Babaev, A.A. ANIMAL MODELS OF EPILEPSY. Genes Cells 2023, 4, 568026. [Google Scholar] [CrossRef]
- Löscher, W.; White, H.S. Animal Models of Drug-Resistant Epilepsy as Tools for Deciphering the Cellular and Molecular Mechanisms of Pharmacoresistance and Discovering More Effective Treatments. Cells 2023, 12, 1233. [Google Scholar] [CrossRef] [PubMed]
- Marshall, G.F.; Gonzalez-Sulser, A.; Abbott, C.M. Modelling Epilepsy in the Mouse: Challenges and Solutions. Dis. Model. Mech. 2021, 14, 047449. [Google Scholar] [CrossRef]
- Pitkänen, A.; Löscher, W.; Vezzani, A.; Becker, A.J.; Simonato, M.; Lukasiuk, K.; Gröhn, O.; Bankstahl, J.P.; Friedman, A.; Aronica, E.; et al. Advances in the Development of Biomarkers for Epilepsy. Lancet Neurol. 2016, 15, 843–856. [Google Scholar] [CrossRef]
- Goodspeed, K.; Bailey, R.M.; Prasad, S.; Sadhu, C.; Cardenas, J.A.; Holmay, M.; Bilder, D.A.; Minassian, B.A. Gene Therapy: Novel Approaches to Targeting Monogenic Epilepsies. Front. Neurol. 2022, 13, 805007. [Google Scholar] [CrossRef]
- Ramzan, F.; Vickers, M.H.; Mithen, R.F. Epigenetics, MicroRNA and Metabolic Syndrome: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 5047. [Google Scholar] [CrossRef]
- Couillard-Despres, S.; Quehl, E.; Altendorfer, K.; Karl, C.; Ploetz, S.; Bogdahn, U.; Winkler, J.; Aigner, L. Human in Vitro Reporter Model of Neuronal Development and Early Differentiation Processes. BMC Neurosci. 2008, 9, 31. [Google Scholar] [CrossRef]
- Verstraelen, P.; Van Dyck, M.; Verschuuren, M.; Kashikar, N.D.; Nuydens, R.; Timmermans, J.-P.; De Vos, W.H. Image-Based Profiling of Synaptic Connectivity in Primary Neuronal Cell Culture. Front. Neurosci. 2018, 12, 389. [Google Scholar] [CrossRef]
- Bonansco, C.; Fuenzalida, M. Plasticity of Hippocampal Excitatory-Inhibitory Balance: Missing the Synaptic Control in the Epileptic Brain. Neural Plast. 2016, 2016, 8607038. [Google Scholar] [CrossRef]
- Carlson, H.; Ronne-Engström, E.; Ungerstedt, U.; Hillered, L. Seizure Related Elevations of Extracellular Amino Acids in Human Focal Epilepsy. Neurosci. Lett. 1992, 140, 30–32. [Google Scholar] [CrossRef]
- Sarlo, G.L.; Holton, K.F. Brain Concentrations of Glutamate and GABA in Human Epilepsy: A Review. Seizure 2021, 91, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-S.; Huang, T.-H.; Lai, M.-C.; Huang, C.-W. The Role of Glutamate Receptors in Epilepsy. Biomedicines 2023, 11, 783. [Google Scholar] [CrossRef]
- Huang, T.-H.; Lai, M.-C.; Chen, Y.-S.; Huang, C.-W. The Roles of Glutamate Receptors and Their Antagonists in Status Epilepticus, Refractory Status Epilepticus, and Super-Refractory Status Epilepticus. Biomedicines 2023, 11, 686. [Google Scholar] [CrossRef] [PubMed]
- Wesnes, K.A.; Edgar, C.; Dean, A.D.P.; Wroe, S.J. The Cognitive and Psychomotor Effects of Remacemide and Carbamazepine in Newly Diagnosed Epilepsy. Epilepsy Behav. 2009, 14, 522–528. [Google Scholar] [CrossRef]
- Lavu, A.; Aboulatta, L.; Abou-Setta, A.M.; Aloud, B.; Askin, N.; Rabbani, R.; Shouman, W.; Zarychanski, R.; Eltonsy, S. Efficacy and Safety of Perampanel in Epilepsy: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Seizure Eur. J. Epilepsy 2022, 102, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Gazaryan, L.M.; Selyanina, N.V.; Karakulova, Y.V.; Sosnin, D.Y. Role of the <I>GRIN1</I> Gene Polymorphism in the Formation of Post-Traumatic Epilepsy. I.P. Pavlov. Russ. Med. Biol. Her. 2021, 29, 449–456. [Google Scholar] [CrossRef]
- Vieira, M.; Yong, X.L.H.; Roche, K.W.; Anggono, V. Regulation of NMDA Glutamate Receptor Functions by the GluN2 Subunits. J. Neurochem. 2020, 154, 121–143. [Google Scholar] [CrossRef]
- Collins, C.; Duff, C.; Duncan, A.M.V.; Planells-Cases, R.; Sun, W.; Norremolle, A.; Michaelis, E.; Montal, M.; Worton, R.; Hayden, M.R. Mapping of the Human NMDA Receptor Subunit (NMDAR1) and the Proposed NMDA Receptor Glutamate-Binding Subunit (NMDARA1) to Chromosomes 9q34.3 and Chromosome 8, Respectively. Genomics 1993, 17, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Regan, B.M.; Yendle, S.C.; O’Roak, B.J.; Lozovaya, N.; Bruneau, N.; Burnashev, N.; Khan, A.; Cook, J.; Geraghty, E.; et al. GRIN2A Mutations Cause Epilepsy-Aphasia Spectrum Disorders. Nat. Genet. 2013, 45, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- DeGiorgio, L.A.; Konstantinov, K.N.; Lee, S.C.; Hardin, J.A.; Volpe, B.T.; Diamond, B. A Subset of Lupus Anti-DNA Antibodies Cross-Reacts with the NR2 Glutamate Receptor in Systemic Lupus Erythematosus. Nat. Med. 2001, 7, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Lesca, G.; Rudolf, G.; Bruneau, N.; Lozovaya, N.; Labalme, A.; Boutry-Kryza, N.; Salmi, M.; Tsintsadze, T.; Addis, L.; Motte, J.; et al. GRIN2A Mutations in Acquired Epileptic Aphasia and Related Childhood Focal Epilepsies and Encephalopathies with Speech and Language Dysfunction. Nat. Genet. 2013, 45, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.R.; Lal, D.; Reinthaler, E.M.; Steiner, I.; Nothnagel, M.; Alber, M.; Geider, K.; Laube, B.; Schwake, M.; Finsterwalder, K.; et al. Mutations in GRIN2A Cause Idiopathic Focal Epilepsy with Rolandic Spikes. Nat. Genet. 2013, 45, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Nunes, J.; Sousa, J.M.; Fonseca, J.; Melo, C.; Alves, D.; Sampaio, M.; Sousa, R. Successful Treatment of a Child with Epileptic Encephalopathy with Spike-Wave Activation in Sleep and GRIN2A Variant Using Sulthiame. Cureus 2023, 15, e34686. [Google Scholar] [CrossRef]
- Deshmukh, A.; Leichner, J.; Bae, J.; Song, Y.; Valdés-Hernández, P.A.; Lin, W.-C.; Riera, J.J. Histological Characterization of the Irritative Zones in Focal Cortical Dysplasia Using a Preclinical Rat Model. Front. Cell. Neurosci. 2018, 12, 52. [Google Scholar] [CrossRef]
- Terunuma, M.; Vargas, K.J.; Wilkins, M.E.; Ramírez, O.A.; Jaureguiberry-Bravo, M.; Pangalos, M.N.; Smart, T.G.; Moss, S.J.; Couve, A. Prolonged Activation of NMDA Receptors Promotes Dephosphorylation and Alters Postendocytic Sorting of GABA B Receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 13918–13923. [Google Scholar] [CrossRef]
- Yu, S.P.; Jiang, M.Q.; Shim, S.S.; Pourkhodadad, S.; Wei, L. Extrasynaptic NMDA Receptors in Acute and Chronic Excitotoxicity: Implications for Preventive Treatments of Ischemic Stroke and Late-Onset Alzheimer’s Disease. Mol. Neurodegener. 2023, 18, 43. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Hansen, K.B.; Zhang, J.; Mark Pierson, T.; Markello, T.C.; Fajardo, K.V.F.; Holloman, C.M.; Golas, G.; Adams, D.R.; Boerkoel, C.F.; et al. Functional Analysis of a de Novo GRIN2A Missense Mutation Associated with Early-Onset Epileptic Encephalopathy. Nat. Commun. 2014, 5, 3251. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-R.; Xu, X.-X.; Lin, S.-M.; Fan, C.-Y.; Ye, T.-T.; Tang, B.; Shi, Y.-W.; Su, T.; Li, B.-M.; Yi, Y.-H.; et al. GRIN2A Variants Associated With Idiopathic Generalized Epilepsies. Front. Mol. Neurosci. 2021, 14, 720984. [Google Scholar] [CrossRef] [PubMed]
- Zweier, C. Characterisation of Deletions of the ZFHX1B Region and Genotype-Phenotype Analysis in Mowat-Wilson Syndrome. J. Med. Genet. 2003, 40, 601–605. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett Syndrome Is Caused by Mutations in X-Linked MECP2, Encoding Methyl-CpG-Binding Protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Fish, J.L. SATB2-associated Syndrome: Mechanisms, Phenotype, and Practical Recommendations. Am. J. Med. Genet. A 2017, 173, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Turovsky, E.; Karagiannis, A.; Abdala, A.P.; Gourine, A.V. Impaired CO2 Sensitivity of Astrocytes in a Mouse Model of Rett Syndrome. J. Physiol. 2015, 593, 3159–3168. [Google Scholar] [CrossRef]
- Turovsky, E.A.; Turovskaya, M.V.; Fedotova, E.I.; Babaev, A.A.; Tarabykin, V.S.; Varlamova, E.G. Role of Satb1 and Satb2 Transcription Factors in the Glutamate Receptors Expression and Ca2+ Signaling in the Cortical Neurons In Vitro. Int. J. Mol. Sci. 2021, 22, 5968. [Google Scholar] [CrossRef]
- Turovskaya, M.V.; Epifanova, E.A.; Tarabykin, V.S.; Babaev, A.A.; Turovsky, E.A. Interleukin-10 Restores Glutamate Receptor-Mediated Ca2+-Signaling in Brain Circuits under Loss of Sip1 Transcription Factor. Int. J. Neurosci. 2022, 132, 114–125. [Google Scholar] [CrossRef]
- Rakhade, S.N.; Zhou, C.; Aujla, P.K.; Fishman, R.; Sucher, N.J.; Jensen, F.E. Early Alterations of AMPA Receptors Mediate Synaptic Potentiation Induced by Neonatal Seizures. J. Neurosci. 2008, 28, 7979–7990. [Google Scholar] [CrossRef]
- Lippman-Bell, J.J.; Rakhade, S.N.; Klein, P.M.; Obeid, M.; Jackson, M.C.; Joseph, A.; Jensen, F.E. AMPA Receptor Antagonist NBQX Attenuates Later-life Epileptic Seizures and Autistic-like Social Deficits Following Neonatal Seizures. Epilepsia 2013, 54, 1922–1932. [Google Scholar] [CrossRef]
- Twele, F.; Bankstahl, M.; Klein, S.; Römermann, K.; Löscher, W. The AMPA Receptor Antagonist NBQX Exerts Anti-Seizure but Not Antiepileptogenic Effects in the Intrahippocampal Kainate Mouse Model of Mesial Temporal Lobe Epilepsy. Neuropharmacology 2015, 95, 234–242. [Google Scholar] [CrossRef]
- Chen, W.; Li, Y.-S.; Gao, J.; Lin, X.-Y.; Li, X.-H. AMPA Receptor Antagonist NBQX Decreased Seizures by Normalization of Perineuronal Nets. PLoS ONE 2016, 11, e0166672. [Google Scholar] [CrossRef] [PubMed]
- Loddenkemper, T.; Talos, D.M.; Cleary, R.T.; Joseph, A.; Sánchez Fernández, I.; Alexopoulos, A.; Kotagal, P.; Najm, I.; Jensen, F.E. Subunit Composition of Glutamate and Gamma-Aminobutyric Acid Receptors in Status Epilepticus. Epilepsy Res. 2014, 108, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Cossart, R. Kainate, a Double Agent That Generates Seizures: Two Decades of Progress. Trends Neurosci. 2000, 23, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Crépel, V.; Mulle, C. Physiopathology of Kainate Receptors in Epilepsy. Curr. Opin. Pharmacol. 2015, 20, 83–88. [Google Scholar] [CrossRef]
- Frerking, M.; Nicoll, R.A. Synaptic Kainate Receptors. Curr. Opin. Neurobiol. 2000, 10, 342–351. [Google Scholar] [CrossRef]
- Lerma, J.; Marques, J.M. Kainate Receptors in Health and Disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef]
- Christensen, J.K.; Paternain, A.V.; Selak, S.; Ahring, P.K.; Lerma, J. A Mosaic of Functional Kainate Receptors in Hippocampal Interneurons. J. Neurosci. 2004, 24, 8986–8993. [Google Scholar] [CrossRef]
- Fritsch, B.; Reis, J.; Gasior, M.; Kaminski, R.M.; Rogawski, M.A. Role of GluK1 Kainate Receptors in Seizures, Epileptic Discharges, and Epileptogenesis. J. Neurosci. 2014, 34, 5765–5775. [Google Scholar] [CrossRef]
- Nakamura, M.; Sekino, Y.; Manabe, T. GABAergic Interneurons Facilitate Mossy Fiber Excitability in the Developing Hippocampus. J. Neurosci. 2007, 27, 1365–1373. [Google Scholar] [CrossRef]
- Peret, A.; Christie, L.A.; Ouedraogo, D.W.; Gorlewicz, A.; Epsztein, J.; Mulle, C.; Crépel, V. Contribution of Aberrant GluK2-Containing Kainate Receptors to Chronic Seizures in Temporal Lobe Epilepsy. Cell Rep. 2014, 8, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.M.Y.; Polygalov, D.; Wintzer, M.E.; Chiang, M.-C.; McHugh, T.J. CA3 Synaptic Silencing Attenuates Kainic Acid-Induced Seizures and Hippocampal Network Oscillations. eNeuro 2016, 3, ENEURO.0003-16.2016. [Google Scholar] [CrossRef] [PubMed]
- Grosenbaugh, D.K.; Ross, B.M.; Wagley, P.; Zanelli, S.A. The Role of Kainate Receptors in the Pathophysiology of Hypoxia-Induced Seizures in the Neonatal Mouse. Sci. Rep. 2018, 8, 7035. [Google Scholar] [CrossRef]
- Galanopoulou, A. GABAA Receptors in Normal Development and Seizures: Friends or Foes? Curr. Neuropharmacol. 2008, 6, 1–20. [Google Scholar] [CrossRef]
- Chuang, S.-H.; Reddy, D.S. Genetic and Molecular Regulation of Extrasynaptic GABA-A Receptors in the Brain: Therapeutic Insights for Epilepsy. J. Pharmacol. Exp. Ther. 2018, 364, 180–197. [Google Scholar] [CrossRef]
- Fu, X.; Wang, Y.-J.; Kang, J.-Q.; Mu, T.-W. GABAA Receptor Variants in Epilepsy. In Epilepsy; Exon Publications: Brisbane City, Australia, 2022; pp. 95–118. [Google Scholar]
- Bazyan, A.S.; van Luijtelaar, G. Neurochemical and Behavioral Features in Genetic Absence Epilepsy and in Acutely Induced Absence Seizures. ISRN Neurol. 2013, 2013, 875834. [Google Scholar] [CrossRef] [PubMed]
- Luscher, B.; Fuchs, T.; Kilpatrick, C.L. GABAA Receptor Trafficking-Mediated Plasticity of Inhibitory Synapses. Neuron 2011, 70, 385–409. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Plotnikov, E.Y.; Turovsky, E.A. Neuronal Calcium Sensor-1 Protects Cortical Neurons from Hyperexcitation and Ca2+ Overload during Ischemia by Protecting the Population of GABAergic Neurons. Int. J. Mol. Sci. 2022, 23, 15675. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Seizures Beget Seizures: The Quest for GABA as a Key Player. Crit. Rev. Neurobiol. 2006, 18, 135–144. [Google Scholar] [CrossRef]
- Boerma, R.S.; Braun, K.P.; van de Broek, M.P.H.; van Berkestijn, F.M.C.; Swinkels, M.E.; Hagebeuk, E.O.; Lindhout, D.; van Kempen, M.; Boon, M.; Nicolai, J.; et al. Remarkable Phenytoin Sensitivity in 4 Children with SCN8A-Related Epilepsy: A Molecular Neuropharmacological Approach. Neurotherapeutics 2016, 13, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Kaneda, M.; Sugawara, T.; Mazaki, E.; Okamura, N.; Montal, M.; Makita, N.; Tanaka, M.; Fukushima, K.; Fujiwara, T.; et al. A Nonsense Mutation of the Sodium Channel Gene SCN2A in a Patient with Intractable Epilepsy and Mental Decline. J. Neurosci. 2004, 24, 2690–2698. [Google Scholar] [CrossRef]
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; et al. A Novel Potassium Channel Gene, KCNQ2, Is Mutated in an Inherited Epilepsy of Newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A Potassium Channel Mutation in Neonatal Human Epilepsy. Science 1979 1998, 279, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Chand, P.; Angez, M.; Hameed, A.N.; Kirmani, S. Potassium Channel Subfamily T Member 1 (KCNT1) Pathological Variant Causing Epilepsy of Infancy with Migrating Focal Seizures: A Case Report. J. Pak. Med. Assoc. 2023, 73, 1720–1722. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Balobaid, A.; Grottesi, A.; Dabbagh, O.; Cenciarini, M.; Rawashdeh, R.; Al-Sagheir, A.; Bove, C.; Macchioni, L.; Pessia, M.; et al. Lethal Digenic Mutations in the K+ Channels Kir4.1 (KCNJ10) and SLACK (KCNT1) Associated with Severe-Disabling Seizures and Neurodevelopmental Delay. J. Neurophysiol. 2017, 118, 2402–2411. [Google Scholar] [CrossRef] [PubMed]
- Ostendorf, A.P.; Wong, M. MTOR Inhibition in Epilepsy: Rationale and Clinical Perspectives. CNS Drugs 2015, 29, 91–99. [Google Scholar] [CrossRef]
- Hodges, S.L.; Lugo, J.N. Therapeutic Role of Targeting MTOR Signaling and Neuroinflammation in Epilepsy. Epilepsy Res. 2020, 161, 106282. [Google Scholar] [CrossRef]
- Mirzaa, G.; Roy, A.; Dobyns, W.B.; Millen, K.; Hevner, R.F. Hemimegalencephaly and Dysplastic Megalencephaly. In Developmental Neuropathology; Wiley: Hoboken, NJ, USA, 2018; pp. 55–61. [Google Scholar]
- Kim, J.K.; Lee, J.H. Mechanistic Target of Rapamycin Pathway in Epileptic Disorders. J. Korean Neurosurg. Soc. 2019, 62, 272–287. [Google Scholar] [CrossRef]
- Berdichevsky, Y.; Dryer, A.M.; Saponjian, Y.; Mahoney, M.M.; Pimentel, C.A.; Lucini, C.A.; Usenovic, M.; Staley, K.J. PI3K-Akt Signaling Activates MTOR-Mediated Epileptogenesis in Organotypic Hippocampal Culture Model of Post-Traumatic Epilepsy. J. Neurosci. 2013, 33, 9056–9067. [Google Scholar] [CrossRef]
- Roy, A.; Skibo, J.; Kalume, F.; Ni, J.; Rankin, S.; Lu, Y.; Dobyns, W.B.; Mills, G.B.; Zhao, J.J.; Baker, S.J.; et al. Mouse Models of Human PIK3CA-Related Brain Overgrowth Have Acutely Treatable Epilepsy. eLife 2015, 4, e12703. [Google Scholar] [CrossRef]
- Brandt, C.; Hillmann, P.; Noack, A.; Römermann, K.; Öhler, L.A.; Rageot, D.; Beaufils, F.; Melone, A.; Sele, A.M.; Wymann, M.P.; et al. The Novel, Catalytic MTORC1/2 Inhibitor PQR620 and the PI3K/MTORC1/2 Inhibitor PQR530 Effectively Cross the Blood-Brain Barrier and Increase Seizure Threshold in a Mouse Model of Chronic Epilepsy. Neuropharmacology 2018, 140, 107–120. [Google Scholar] [CrossRef]
- Chang, P.; Walker, M.C.; Williams, R.S.B. Seizure-Induced Reduction in PIP3 Levels Contributes to Seizure-Activity and Is Rescued by Valproic Acid. Neurobiol. Dis. 2014, 62, 296–306. [Google Scholar] [CrossRef]
- Wei, H.; Duan, G.; He, J.; Meng, Q.; Liu, Y.; Chen, W.; Meng, Y. Geniposide Attenuates Epilepsy Symptoms in a Mouse Model through the PI3K/Akt/GSK-3β Signaling Pathway. Exp. Ther. Med. 2017, 15, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Yang, G.; Liu, H.; Liu, X.; Lin, S.; Sun, D.; Wang, Y. Aged Black Garlic Extract Inhibits HT29 Colon Cancer Cell Growth via the PI3K/Akt Signaling Pathway. Biomed. Rep. 2014, 2, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Han, V.Z.; Bard, A.M.; Wehle, D.T.; Smith, P.S.E.; Ramirez, J.-M.; Kalume, F.; Millen, K.J. Suppression of PIK3CA-Driven Epileptiform Activity by Acute Pathway Control. bioRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Turovsky, E.A.; Tarabykin, V.S.; Varlamova, E.G. Deletion of the Neuronal Transcription Factor Satb1 Induced Disturbance of the Kinome and Mechanisms of Hypoxic Preconditioning. Biology 2023, 12, 1207. [Google Scholar] [CrossRef] [PubMed]
- Krushinsky, L.V. Some Stages of Integration in the Formation of Behavior in Animals. Usp. Sovrem. Biol. 1948, 26, 737–754. [Google Scholar]
- Turovsky, E.A.; Zinchenko, V.P.; Gaidin, S.G.; Turovskaya, M.V. Calcium-Binding Proteins Protect GABAergic Neurons of the Hippocampus from Hypoxia and Ischemia in Vitro. Biochem. (Moscow) Suppl. Ser. A Membr. Cell Biol. 2018, 12, 74–84. [Google Scholar] [CrossRef]
- Zinchenko, V.P.; Turovskaya, M.V.; Teplov, I.Y.; Berezhnov, A.V.; Turovsky, E.A. The Role of Parvalbumin-Containing Interneurons in the Regulation of Spontaneous Synchronous Activity of Brain Neurons in Culture. Biophysics 2016, 61, 85–93. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Tam, O.H.; Paniagua, E.; Hammell, M. TEtranscripts: A Package for Including Transposable Elements in Differential Expression Analysis of RNA-Seq Datasets. Bioinformatics 2015, 31, 3593–3599. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varlamova, E.G.; Borisova, E.V.; Evstratova, Y.A.; Newman, A.G.; Kuldaeva, V.P.; Gavrish, M.S.; Kondakova, E.V.; Tarabykin, V.S.; Babaev, A.A.; Turovsky, E.A. Socrates: A Novel N-Ethyl-N-nitrosourea-Induced Mouse Mutant with Audiogenic Epilepsy. Int. J. Mol. Sci. 2023, 24, 17104. https://doi.org/10.3390/ijms242317104
Varlamova EG, Borisova EV, Evstratova YA, Newman AG, Kuldaeva VP, Gavrish MS, Kondakova EV, Tarabykin VS, Babaev AA, Turovsky EA. Socrates: A Novel N-Ethyl-N-nitrosourea-Induced Mouse Mutant with Audiogenic Epilepsy. International Journal of Molecular Sciences. 2023; 24(23):17104. https://doi.org/10.3390/ijms242317104
Chicago/Turabian StyleVarlamova, Elena G., Ekaterina V. Borisova, Yuliya A. Evstratova, Andrew G. Newman, Vera P. Kuldaeva, Maria S. Gavrish, Elena V. Kondakova, Victor S. Tarabykin, Alexey A. Babaev, and Egor A. Turovsky. 2023. "Socrates: A Novel N-Ethyl-N-nitrosourea-Induced Mouse Mutant with Audiogenic Epilepsy" International Journal of Molecular Sciences 24, no. 23: 17104. https://doi.org/10.3390/ijms242317104
APA StyleVarlamova, E. G., Borisova, E. V., Evstratova, Y. A., Newman, A. G., Kuldaeva, V. P., Gavrish, M. S., Kondakova, E. V., Tarabykin, V. S., Babaev, A. A., & Turovsky, E. A. (2023). Socrates: A Novel N-Ethyl-N-nitrosourea-Induced Mouse Mutant with Audiogenic Epilepsy. International Journal of Molecular Sciences, 24(23), 17104. https://doi.org/10.3390/ijms242317104