
Plasma Exchange Reduces Aβ Levels in Plasma and Decreases Amyloid Plaques in the Brain in a Mouse Model of Alzheimer’s Disease
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
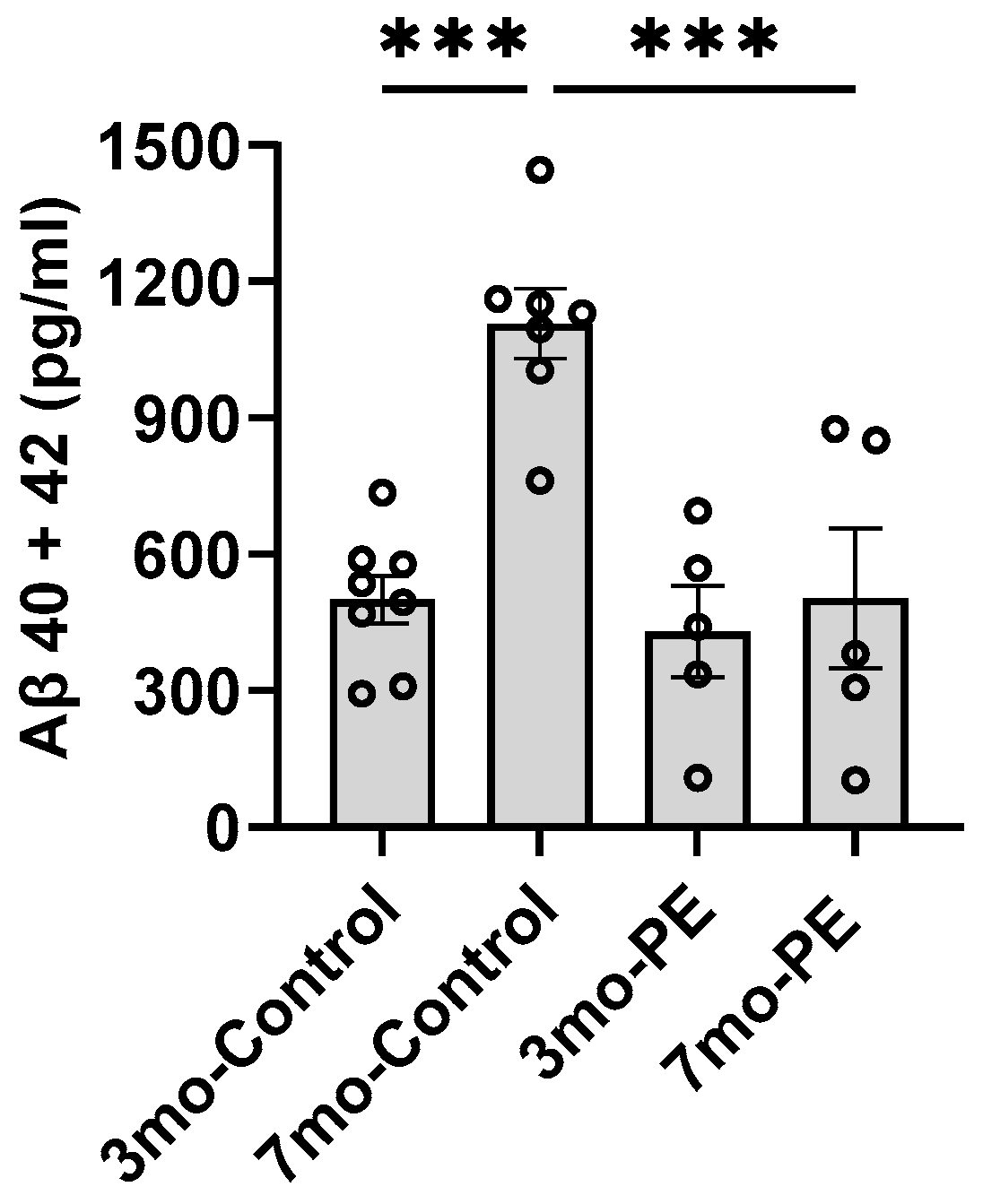
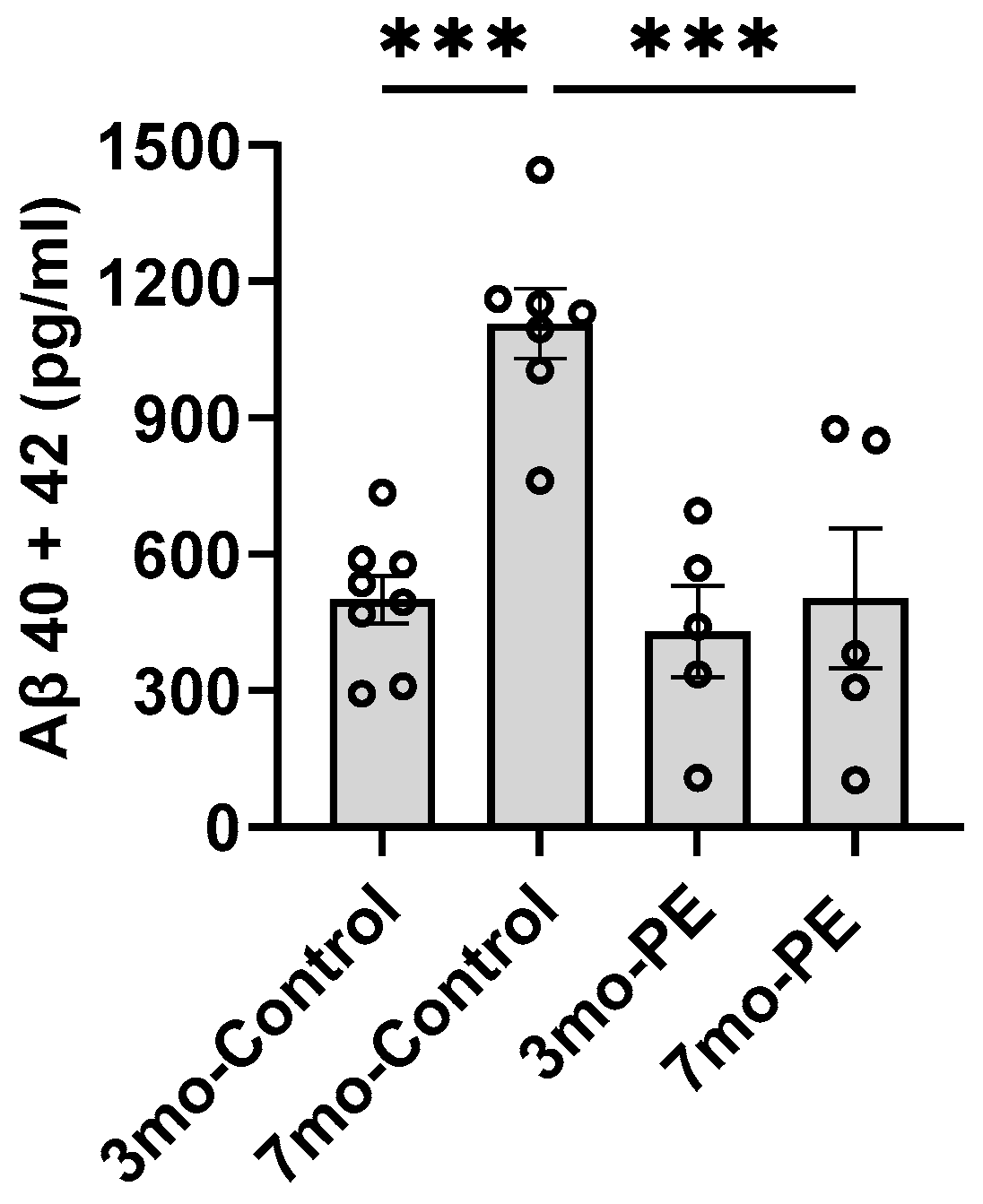
2.1. Plasma Exchange Prevents the Increase in Aβ Levels in Plasma
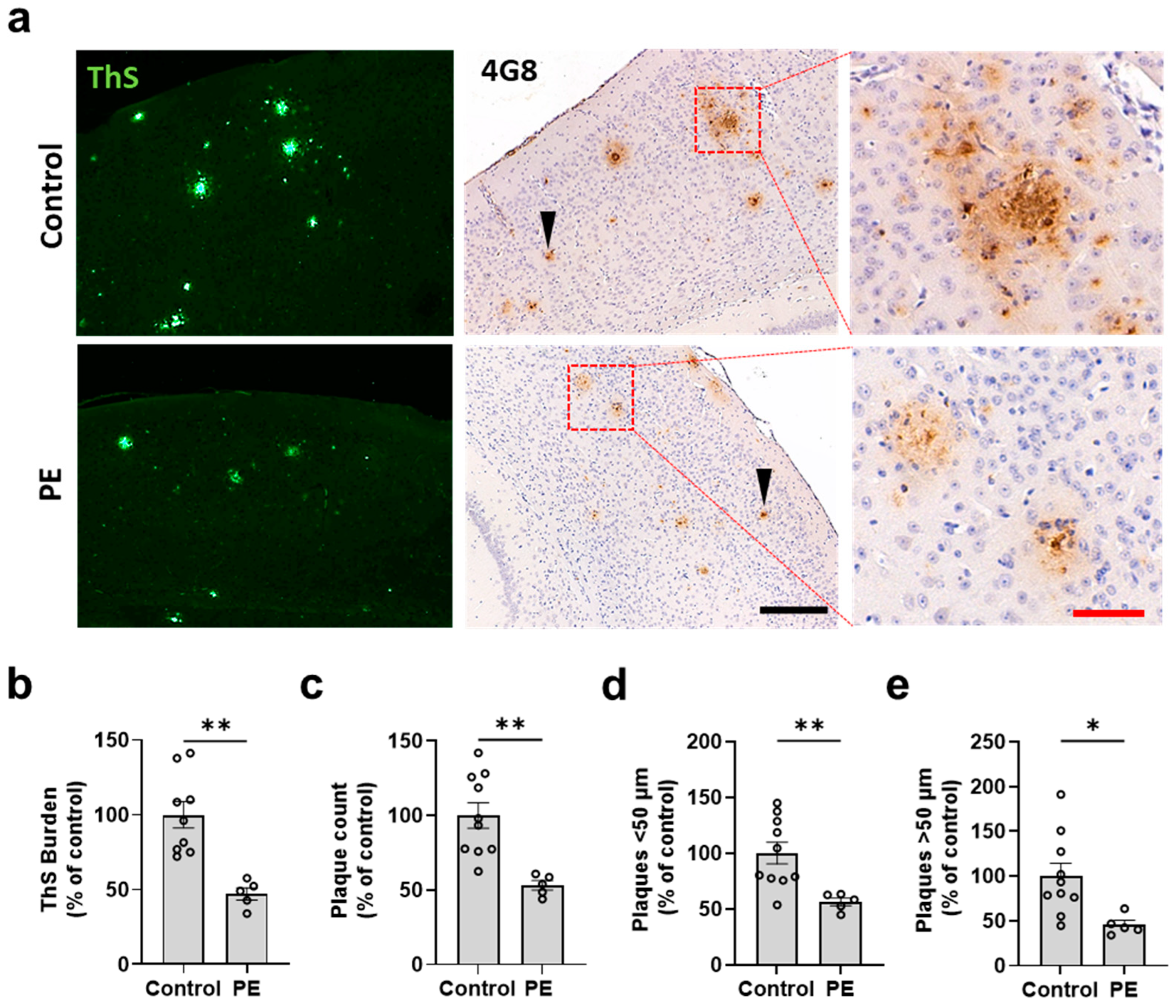
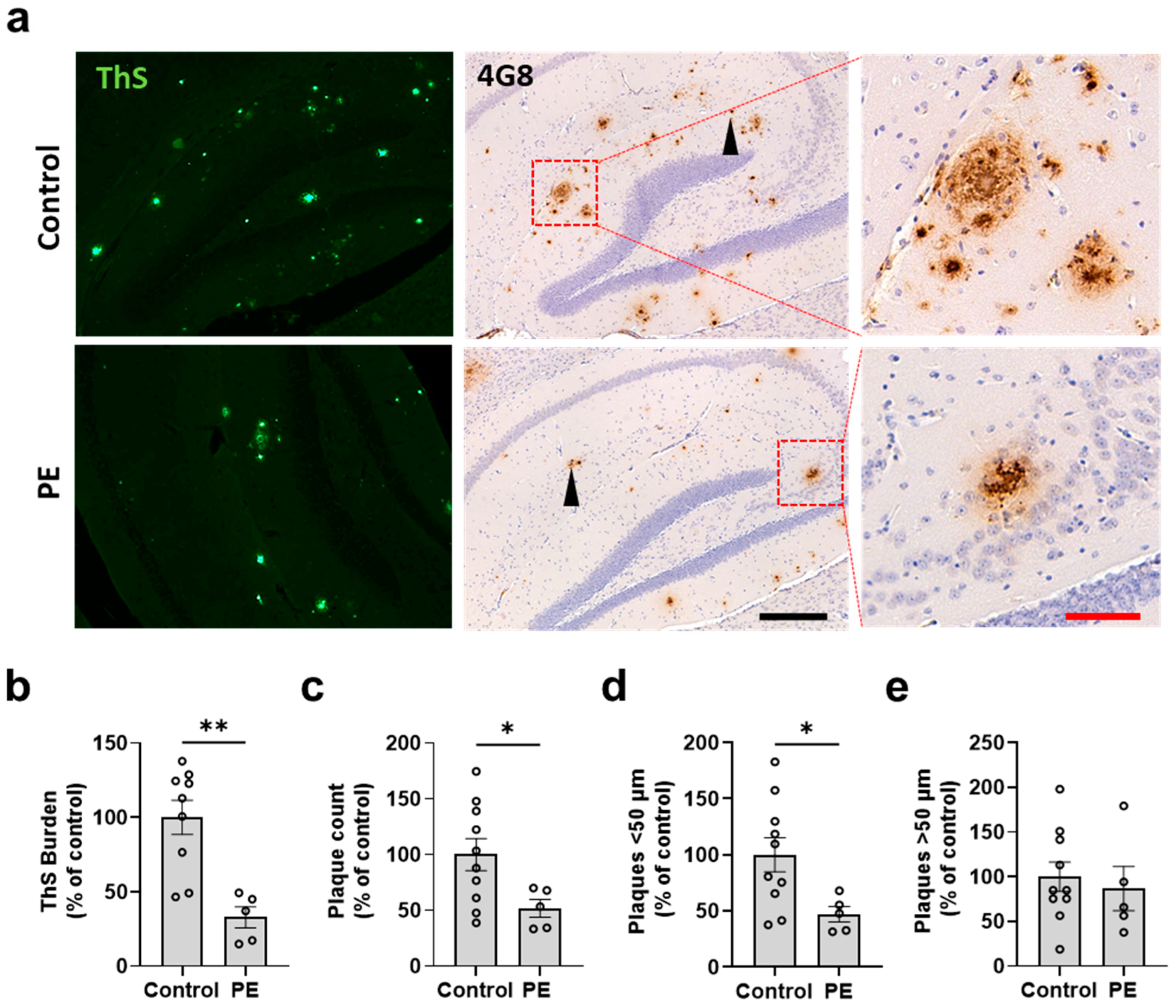
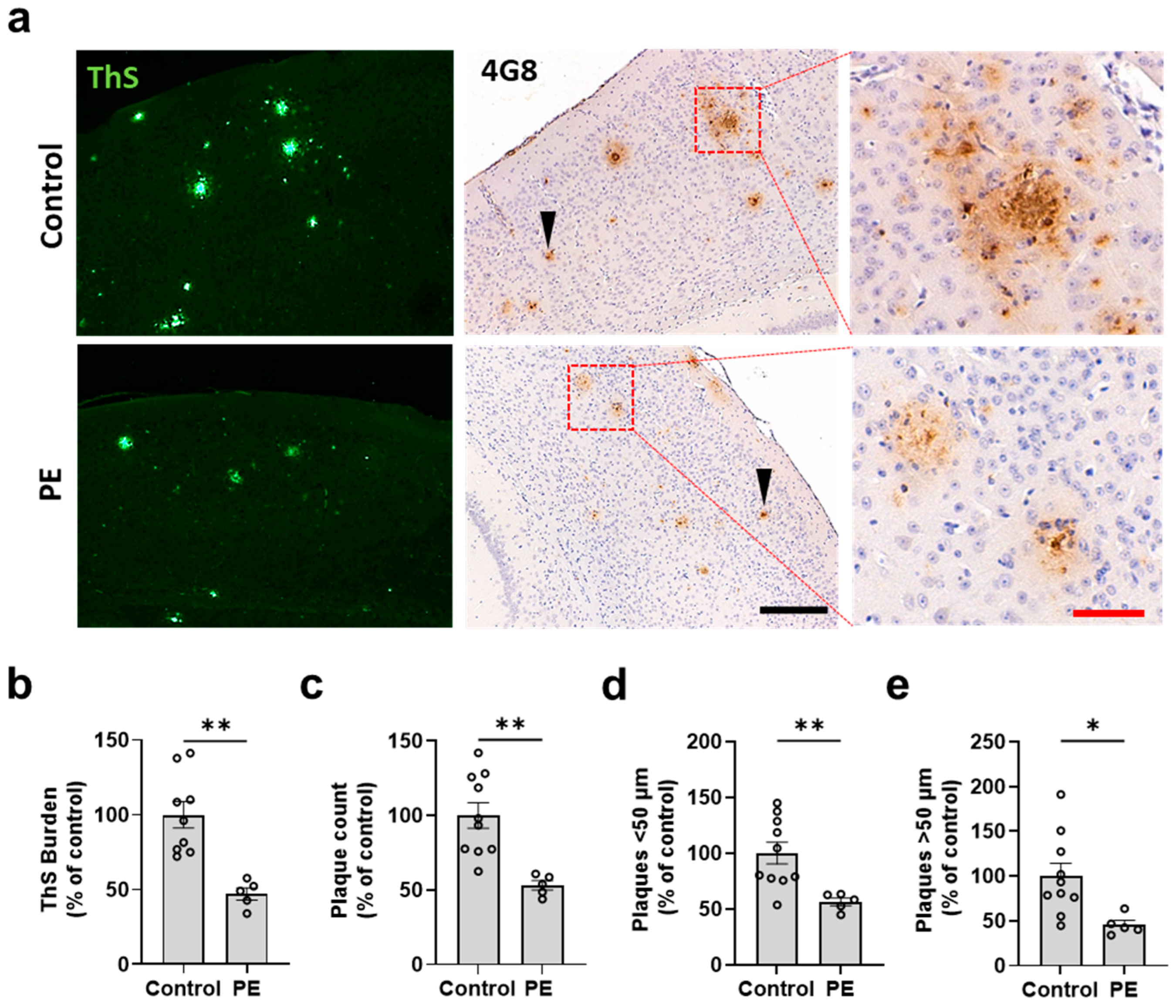
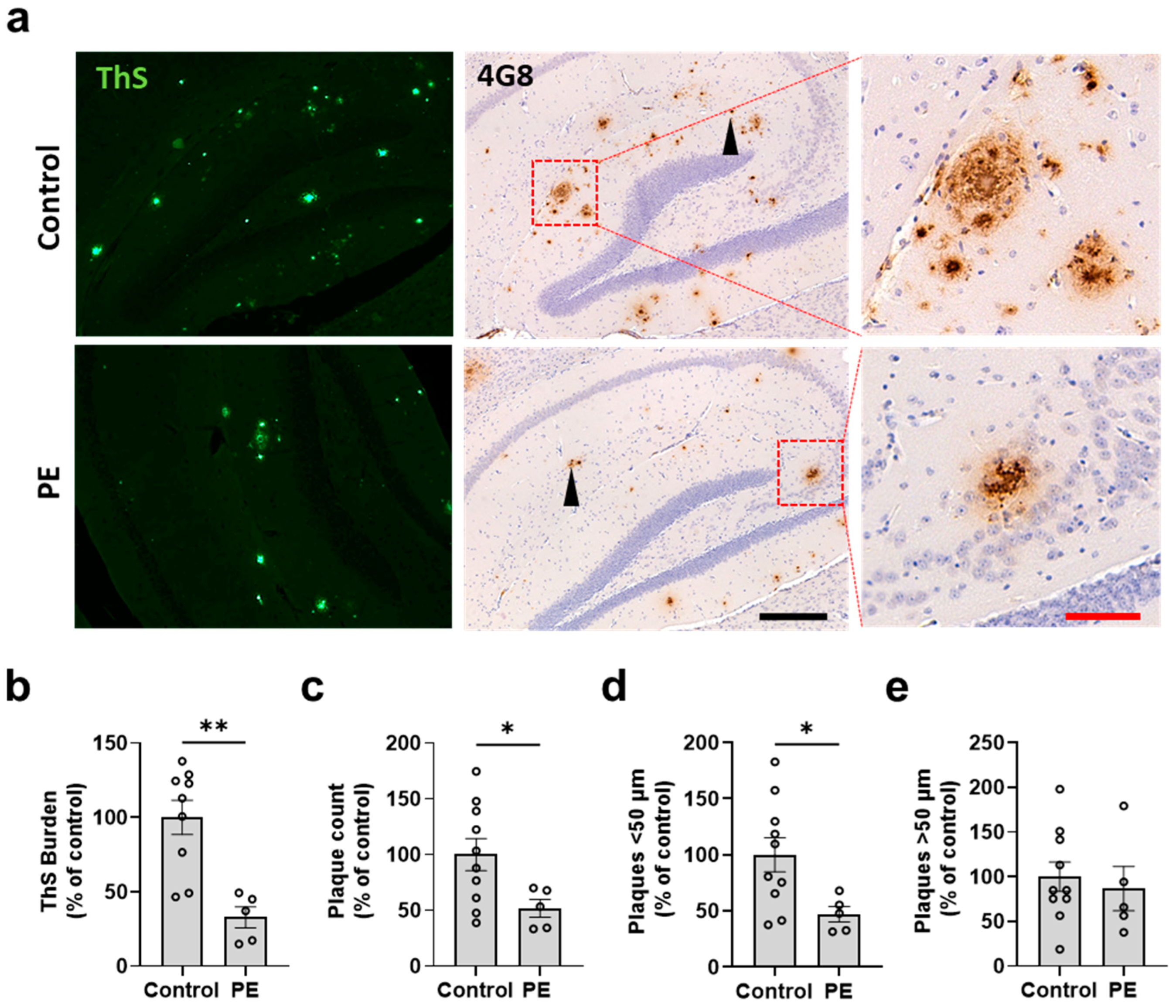
2.2. Plasma Exchange Prevents the Formation of Amyloid Plaques in the Brain
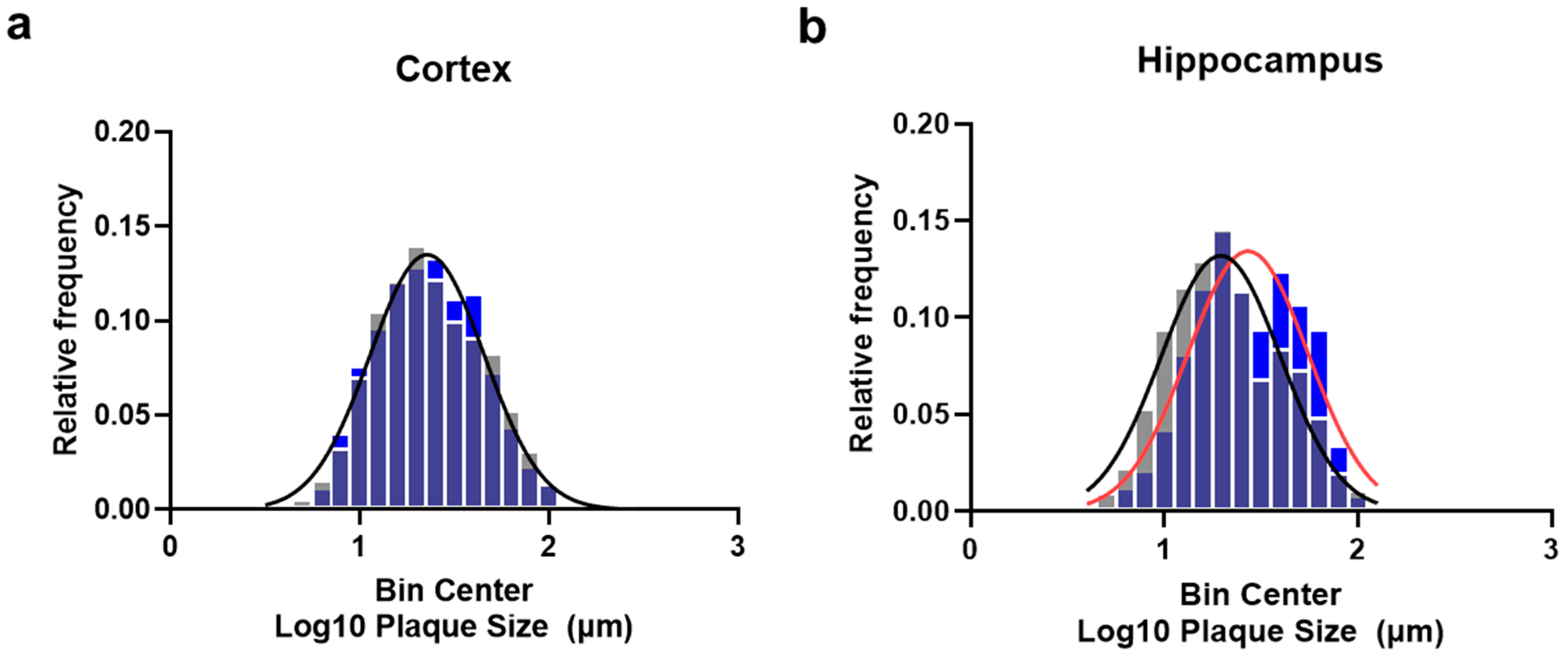
2.3. Plasma Exchange Modifies the Relative Frequencies of Aβ Plaque Size
3. Discussion
4. Materials and Methods
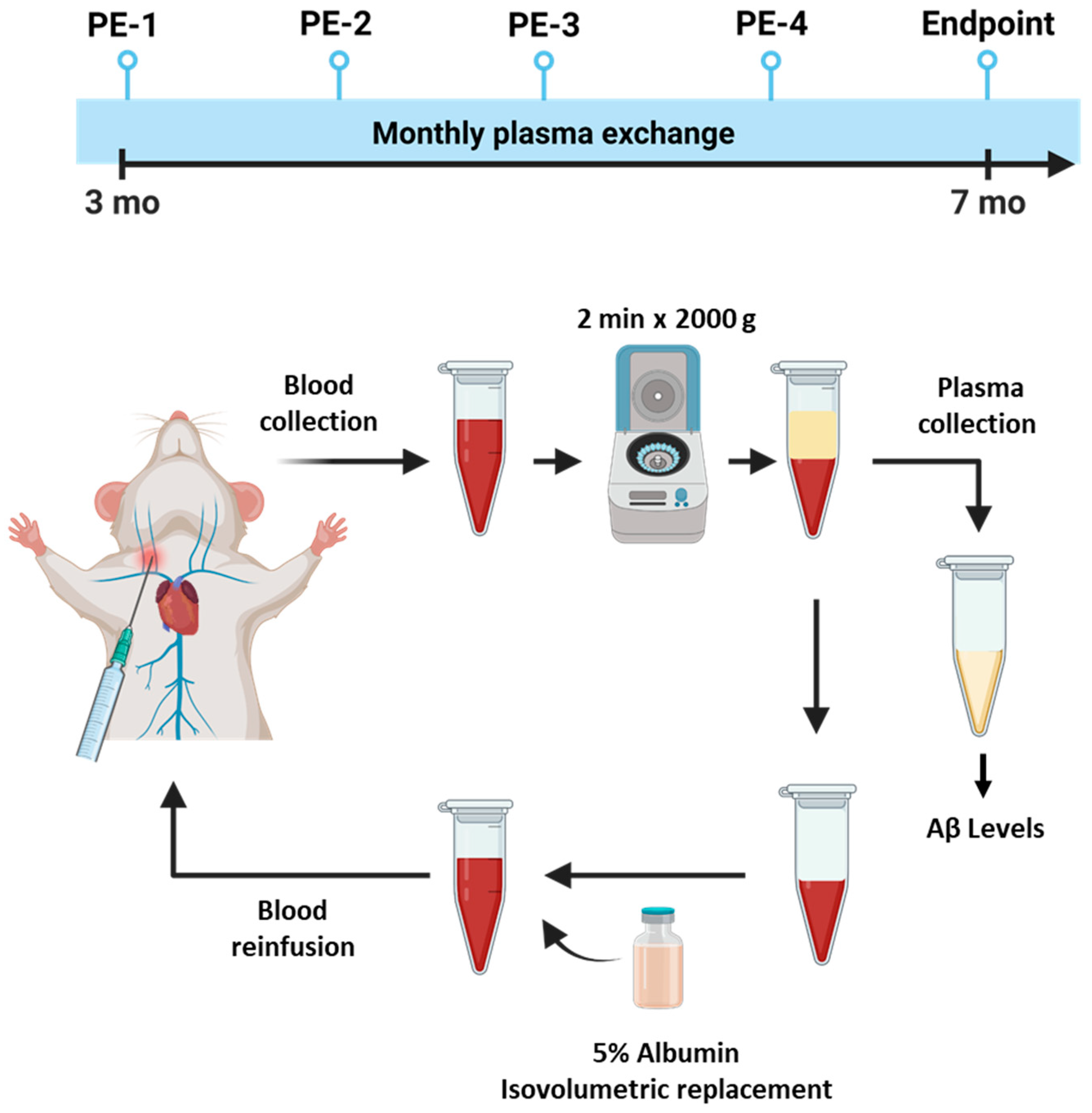
4.1. Animal Experiments
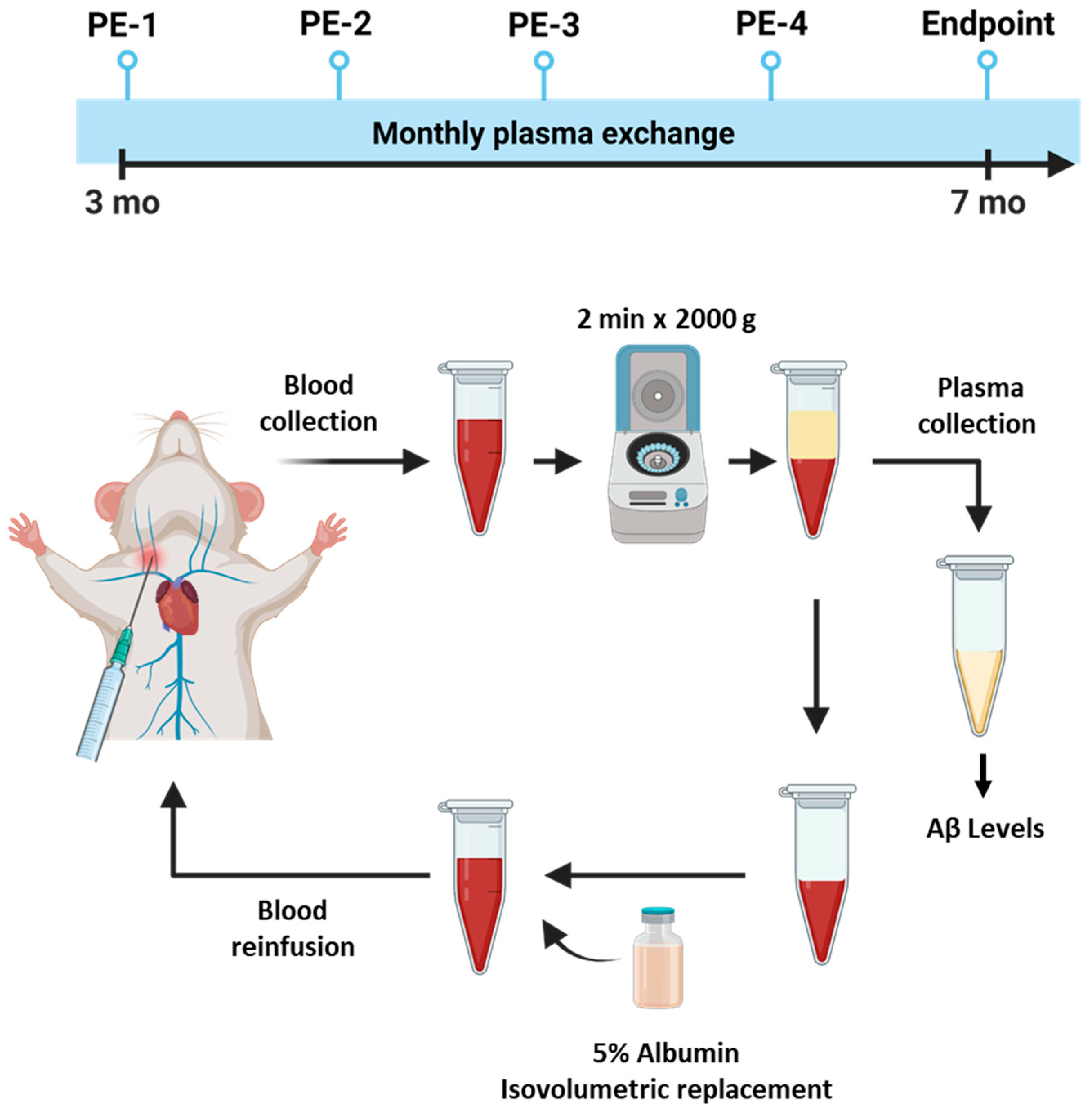
4.2. Plasma Exchange Procedure
4.3. Immunohistochemistry
4.4. Thioflavin-S Staining
4.5. Immunofluorescence
4.6. Image Analysis
4.7. Determination of the Levels of Aβ in Plasma and Brain via Enzyme-Linked Immunoassay (ELISA)
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [PubMed]
- Se Thoe, E.; Fauzi, A.; Tang, Y.Q.; Chamyuang, S.; Chia, A.Y.Y. A review on advances of treatment modalities for Alzheimer’s disease. Life Sci. 2021, 276, 119129. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.B.D.D.F. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar]
- Association, A. 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023, 19, 1598–1695. [Google Scholar]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Bancher, C.; Lassmann, H.; Budka, H.; Grundke-Iqbal, I.; Iqbal, K.; Wiche, G.; Seitelberger, F.; Wisniewski, H.M. Neurofibrillary tangles in Alzheimer’s disease and progressive supranuclear palsy: Antigenic similarities and differences. Microtubule-associated protein tau antigenicity is prominent in all types of tangles. Acta Neuropathol. 1987, 74, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hong, F.; Yang, S. Amyloidosis in Alzheimer’s Disease: Pathogeny, Etiology, and Related Therapeutic Directions. Molecules 2022, 27, 1210. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D. If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLoS Biol. 2022, 20, e3001694. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 2016, 12, 248. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Bales, K.R.; Parsadanian, M.; O’Dell, M.A.; Foss, E.M.; Paul, S.M.; Holtzman, D.M. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease. J. Neurochem. 2002, 81, 229–236. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Paul, S.M.; Holtzman, D.M. Brain to plasma amyloid-beta efflux: A measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science 2002, 295, 2264–2267. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, D.Y.; Wang, Y.J. Peripheral clearance of brain-derived Abeta in Alzheimer’s disease: Pathophysiology and therapeutic perspectives. Transl. Neurodegener. 2020, 9, 16. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Xiang, Y.; Bu, X.L.; Liu, Y.H.; Zhu, C.; Shen, L.L.; Jiao, S.S.; Zhu, X.Y.; Giunta, B.; Tan, J.; Song, W.H.; et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef]
- Morales, R.; Bravo-Alegria, J.; Moreno-Gonzalez, I.; Duran-Aniotz, C.; Gamez, N.; Edwards III, G.; Soto, C. Transmission of cerebral amyloid pathology by peripheral administration of misfolded Abeta aggregates. Mol. Psychiatry 2021, 26, 5690–5701. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Duran-Aniotz, C.; Bravo-Alegria, J.; Estrada, L.D.; Shahnawaz, M.; Hu, P.P.; Kramm, C.; Morales-Scheihing, D.; Urayama, A.; Soto, C. Infusion of blood from mice displaying cerebral amyloidosis accelerates amyloid pathology in animal models of Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 213. [Google Scholar] [CrossRef]
- Urayama, A.; Moreno-Gonzalez, I.; Morales-Scheihing, D.; Kharat, V.; Pritzkow, S.; Soto, C. Preventive and therapeutic reduction of amyloid deposition and behavioral impairments in a model of Alzheimer’s disease by whole blood exchange. Mol. Psychiatry 2022, 27, 4285–4296. [Google Scholar] [CrossRef] [PubMed]
- Kitaguchi, N.; Kato, T.; Matsunaga, S.; Hirano, K.; Iwata, K.; Kawaguchi, K.; Fujita, K.; Takechi, H.; Hasegawa, M.; Yuzawa, Y.; et al. Removal of blood amyloid-beta with hemodialysis reduced brain amyloid-beta, confirmed by brain imaging: A case report. Neuropsychiatr. Dis. Treat. 2018, 14, 2931–2937. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Sakata, M.; Kobayakawa, M.; Kawachi, H.; Kawaguchi, K.; Hiki, Y.; Kato, M.; Mori, M.; Hasegawa, M.; Ohashi, N.; et al. Removal of Abeta Oligomers from the Blood: A Potential Therapeutic System for Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2020, 16, 607–627. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.S.; Shen, L.L.; Bu, X.L.; Zhang, W.W.; Chen, S.H.; Huang, Z.L.; Xiong, J.X.; Gao, C.Y.; Dong, Z.; He, Y.N.; et al. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017, 134, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Boada, M.; Anaya, F.; Ortiz, P.; Olazaran, J.; Shua-Haim, J.R.; Obisesan, T.O.; Hernandez, I.; Munoz, J.; Buendia, M.; Alegret, M.; et al. Efficacy and Safety of Plasma Exchange with 5% Albumin to Modify Cerebrospinal Fluid and Plasma Amyloid-beta Concentrations and Cognition Outcomes in Alzheimer’s Disease Patients: A Multicenter, Randomized, Controlled Clinical Trial. J. Alzheimers Dis. 2017, 56, 129–143. [Google Scholar] [CrossRef]
- Kiprov, D.D. Therapeutic apheresis delivery systems in the US. Transfus. Apher. Sci. 2003, 28, 163–164. [Google Scholar] [CrossRef]
- Basic-Jukic, N.; Brunetta, B.; Kes, P. Plasma exchange in elderly patients. Ther. Apher. Dial. 2010, 14, 161–165. [Google Scholar] [CrossRef]
- Abel, J.J.; Rowntree, L.G.; Turner, B.B. Plasma removal with return of corpuscles (plasmaphaeresis), J Pharmacol Exp Ther Vol. V. No. 6, July, 1914. Transfus. Sci. 1990, 11, 166–177. [Google Scholar]
- Fernandez-Zarzoso, M.; Gomez-Segui, I.; de la Rubia, J. Therapeutic plasma exchange: Review of current indications. Transfus. Apher. Sci. 2019, 58, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Connelly-Smith, L.; Aqui, N.; Balogun, R.A.; Klingel, R.; Meyer, E.; Pham, H.P.; Schneiderman, J.; Witt, V.; Wu, Y.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice—Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J. Clin. Apher. 2019, 34, 171–354. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Ippati, S.; Ceravolo, F.; Watling, M. Perspective: Is therapeutic plasma exchange a viable option for treating Alzheimer’s disease? Alzheimers Dement. 2020, 6, e12004. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, L.; Yunce, M.; Montine, T.J.; Shan, H. Plasma Exchange in Alzheimer’s Disease. Transfus. Med. Rev. 2023, 37, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Bero, A.W.; Cirrito, J.R.; Xiao, Q.; Hu, X.; Wang, Y.; Gonzales, E.; Holtzman, D.M.; Lee, J.M. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J. Neurosci. 2009, 29, 10706–10714. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- McClure, D.E. Clinical pathology and sample collection in the laboratory rodent. Vet. Clin. N. Am. Exot. Anim. Pract. 1999, 2, 565–590. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, J.D.; Buckley, A.R.; Knox, J.E.; Kuan, L.; Graddis, N.; Pelos, A.; Mukora, A.; Wakeman, W.; Bohn, P.; Ho, A.; et al. Whole brain imaging reveals distinct spatial patterns of amyloid beta deposition in three mouse models of Alzheimer’s disease. J. Comp. Neurol. 2019, 527, 2122–2145. [Google Scholar] [CrossRef]
- Sarma, J.V.; Ward, P.A. The complement system. Cell Tissue Res. 2011, 343, 227–235. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef]
- Citron, M.; Vigo-Pelfrey, C.; Teplow, D.B.; Miller, C.; Schenk, D.; Johnston, J.; Winblad, B.; Venizelos, N.; Lannfelt, L.; Selkoe, D.J. Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc. Natl. Acad. Sci. USA 1994, 91, 11993–11997. [Google Scholar] [CrossRef] [PubMed]
- Gardella, J.E.; Gorgone, G.A.; Munoz, P.C.; Ghiso, J.; Frangione, B.; Gorevic, P.D. Beta protein precursor expression in human platelets and a megakaryocyte cell line. Possible implications for the origin of cerebral amyloidosis in Alzheimer’s disease. Lab. Investig. 1992, 67, 303–313. [Google Scholar] [PubMed]
- Matsubara, E.; Soto, C.; Governale, S.; Frangione, B.; Ghiso, J. Apolipoprotein J and Alzheimer’s amyloid beta solubility. Biochem. J. 1996, 316 Pt 2, 671–679. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Kokjohn, T.A.; Kalback, W.; Luehrs, D.; Galasko, D.R.; Chevallier, N.; Koo, E.H.; Emmerling, M.R.; Roher, A.E. Amyloid-beta peptides interact with plasma proteins and erythrocytes: Implications for their quantitation in plasma. Biochem. Biophys. Res. Commun. 2000, 268, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Biere, A.L.; Ostaszewski, B.; Stimson, E.R.; Hyman, B.T.; Maggio, J.E.; Selkoe, D.J. Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J. Biol. Chem. 1996, 271, 32916–32922. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Clerici, M.; Giustarini, D.; Rossi, R.; Milzani, A.; Dalle-Donne, I. Redox albuminomics: Oxidized albumin in human diseases. Antioxid. Redox Signal. 2012, 17, 1515–1527. [Google Scholar] [CrossRef]
- Costa, M.; Paez, A. Emerging insights into the role of albumin with plasma exchange in Alzheimer’s disease management. Transfus. Apher. Sci. 2021, 60, 103164. [Google Scholar] [CrossRef]
- Llewellyn, D.J.; Langa, K.M.; Friedland, R.P.; Lang, I.A. Serum albumin concentration and cognitive impairment. Curr. Alzheimer Res. 2010, 7, 91–96. [Google Scholar] [CrossRef]
- Chang, D.C.; Xu, X.; Ferrante, A.W., Jr.; Krakoff, J. Reduced plasma albumin predicts type 2 diabetes and is associated with greater adipose tissue macrophage content and activation. Diabetol. Metab. Syndr. 2019, 11, 14. [Google Scholar] [CrossRef]
- Greenblatt, D.J. Reduced serum albumin concentration in the elderly: A report from the Boston Collaborative Drug Surveillance Program. J. Am. Geriatr. Soc. 1979, 27, 20–22. [Google Scholar] [CrossRef]
- Kim, J.W.; Byun, M.S.; Lee, J.H.; Yi, D.; Jeon, S.Y.; Sohn, B.K.; Lee, J.Y.; Shin, S.A.; Kim, Y.K.; Kang, K.M.; et al. Serum albumin and beta-amyloid deposition in the human brain. Neurology 2020, 95, e815–e826. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Gonzalez, M.; Gasparovic, C. Albumin Exchange in Alzheimer’s Disease: Might CSF Be an Alternative Route to Plasma? Front. Neurol. 2019, 10, 1036. [Google Scholar] [CrossRef] [PubMed]
- Tholen, S.; Schmaderer, C.; Chmielewski, S.; Forstl, H.; Heemann, U.; Baumann, M.; Steubl, D.; Grimmer, T. Reduction of Amyloid-beta Plasma Levels by Hemodialysis: An Anti-Amyloid Treatment Strategy? J. Alzheimers Dis. 2016, 50, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kiprov, D.D.; Luellen, C.; Lieb, M.; Liu, C.; Watanabe, E.; Mei, X.; Cassaleto, K.; Kramer, J.; Conboy, M.J.; et al. Old plasma dilution reduces human biological age: A clinical study. Geroscience 2022, 44, 2701–2720. [Google Scholar] [CrossRef]
- Mehdipour, M.; Skinner, C.; Wong, N.; Lieb, M.; Liu, C.; Etienne, J.; Kato, C.; Kiprov, D.; Conboy, M.J.; Conboy, I.M. Rejuvenation of three germ layers tissues by exchanging old blood plasma with saline-albumin. Aging 2020, 12, 8790–8819. [Google Scholar] [CrossRef]
- Ramirez, S.; Mukherjee, A.; Sepulveda, S.; Becerra-Calixto, A.; Bravo-Vasquez, N.; Gherardelli, C.; Chavez, M.; Soto, C. Modeling Traumatic Brain Injury in Human Cerebral Organoids. Cells 2021, 10, 2683. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, I.; Edwards, G., 3rd; Morales, R.; Duran-Aniotz, C.; Escobedo, G., Jr.; Marquez, M.; Pumarola, M.; Soto, C. Aged Cattle Brain Displays Alzheimer’s Disease-Like Pathology and Promotes Brain Amyloidosis in a Transgenic Animal Model. Front. Aging Neurosci. 2021, 13, 815361. [Google Scholar] [CrossRef]
- Nilsson, L.N.; Bales, K.R.; DiCarlo, G.; Gordon, M.N.; Morgan, D.; Paul, S.M.; Potter, H. Alpha-1-antichymotrypsin promotes beta-sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 1444–1451. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, L.; Xia, C.; Tao, Y. Complications of therapeutic plasma exchange: A retrospective study of 1201 procedures in 435 children. Medicine 2019, 98, e18308. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez, S.; Koerich, S.; Astudillo, N.; De Gregorio, N.; Al-Lahham, R.; Allison, T.; Rocha, N.P.; Wang, F.; Soto, C. Plasma Exchange Reduces Aβ Levels in Plasma and Decreases Amyloid Plaques in the Brain in a Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 17087. https://doi.org/10.3390/ijms242317087
Ramirez S, Koerich S, Astudillo N, De Gregorio N, Al-Lahham R, Allison T, Rocha NP, Wang F, Soto C. Plasma Exchange Reduces Aβ Levels in Plasma and Decreases Amyloid Plaques in the Brain in a Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(23):17087. https://doi.org/10.3390/ijms242317087
Chicago/Turabian StyleRamirez, Santiago, Suelyn Koerich, Natalia Astudillo, Nicole De Gregorio, Rabab Al-Lahham, Tyler Allison, Natalia Pessoa Rocha, Fei Wang, and Claudio Soto. 2023. "Plasma Exchange Reduces Aβ Levels in Plasma and Decreases Amyloid Plaques in the Brain in a Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 23: 17087. https://doi.org/10.3390/ijms242317087
APA StyleRamirez, S., Koerich, S., Astudillo, N., De Gregorio, N., Al-Lahham, R., Allison, T., Rocha, N. P., Wang, F., & Soto, C. (2023). Plasma Exchange Reduces Aβ Levels in Plasma and Decreases Amyloid Plaques in the Brain in a Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 24(23), 17087. https://doi.org/10.3390/ijms242317087