
Attenuation of Vanadium-Induced Neurotoxicity in Rat Hippocampal Slices (In Vitro) and Mice (In Vivo) by ZA-II-05, a Novel NMDA-Receptor Antagonist



 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
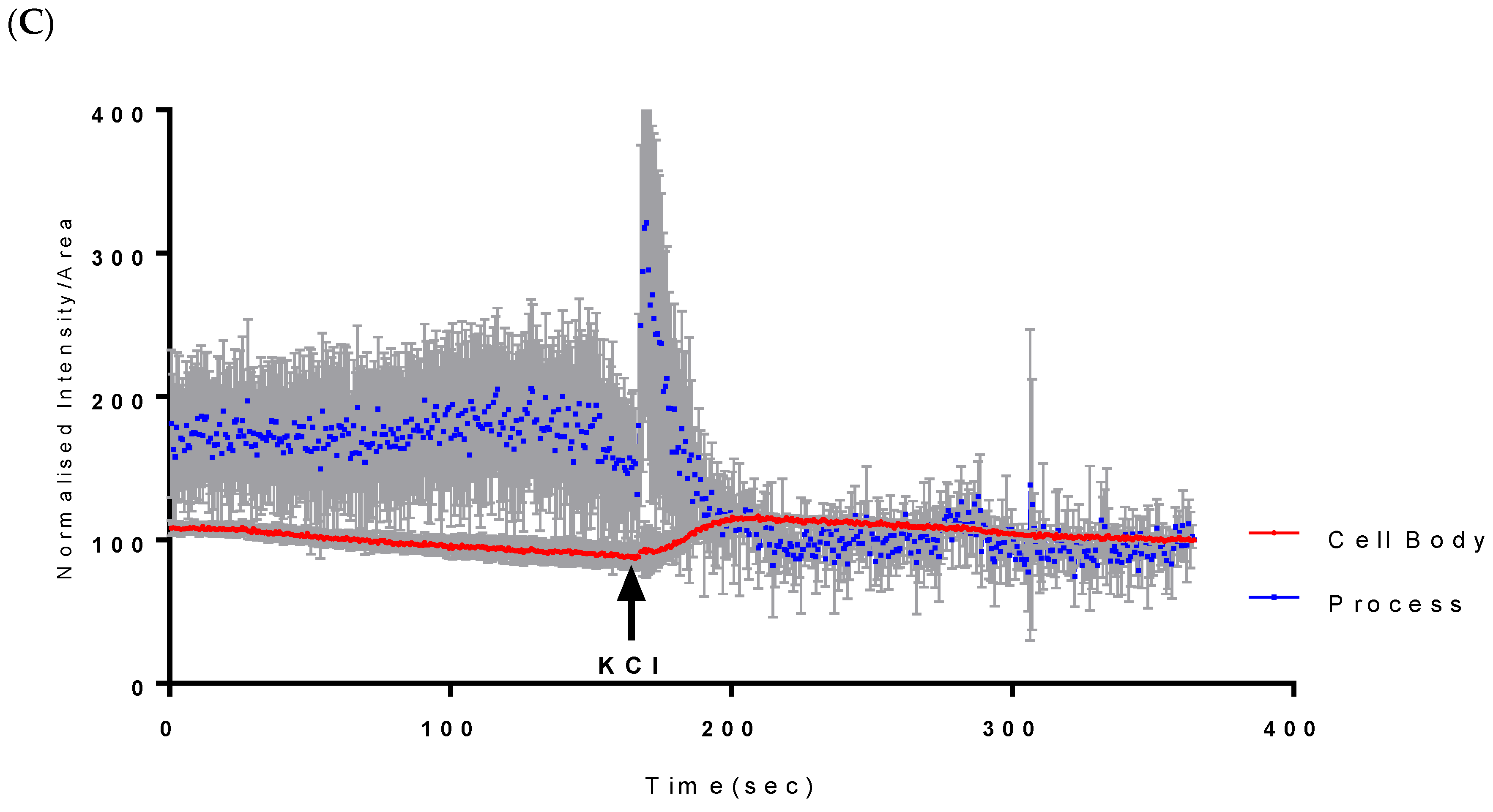
2.1. Calcium Mobilisation on Vanadium-Treated CAD Cells
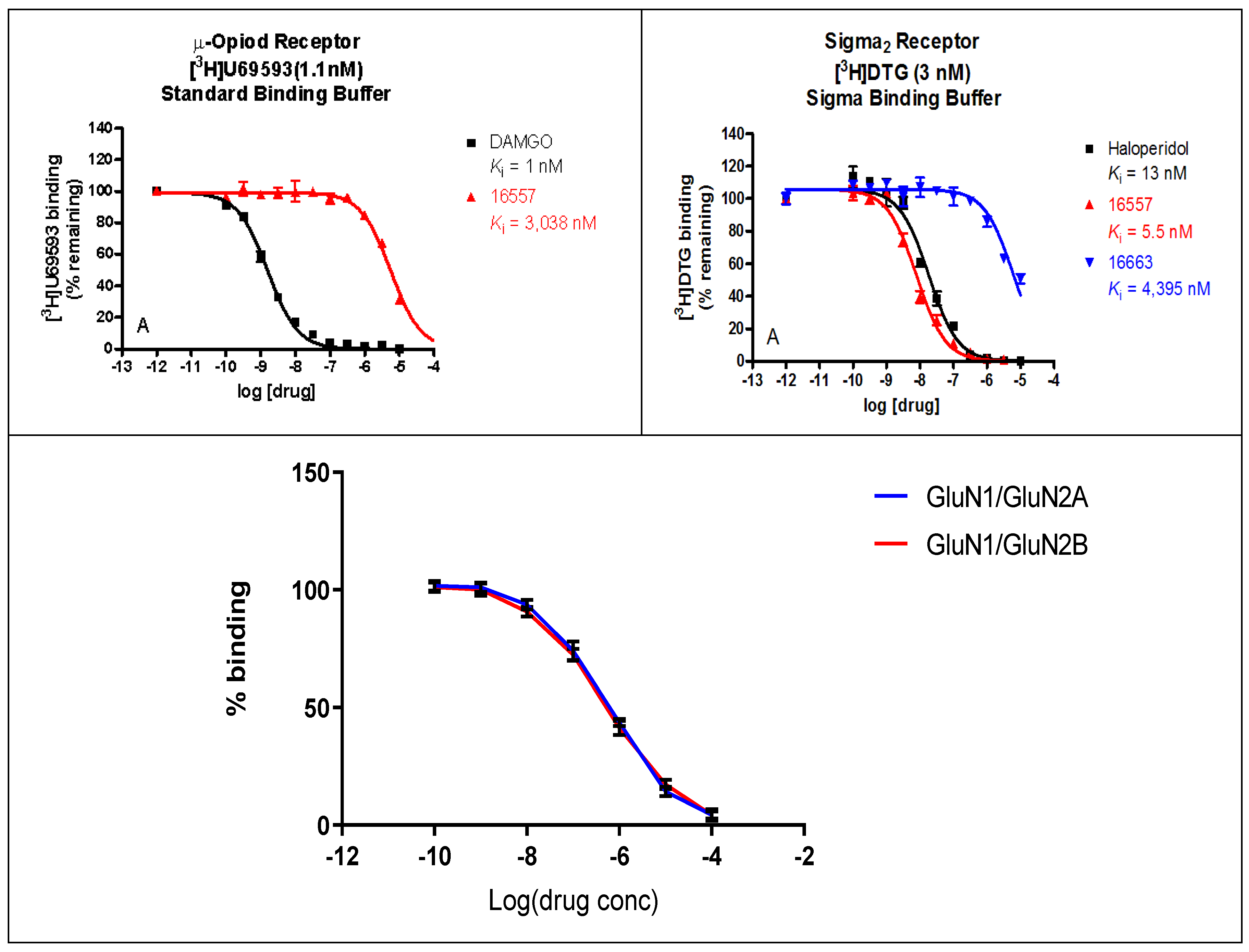
2.2. Binding Profile of ZA-II-05 on Neurotransmitters and Transporters
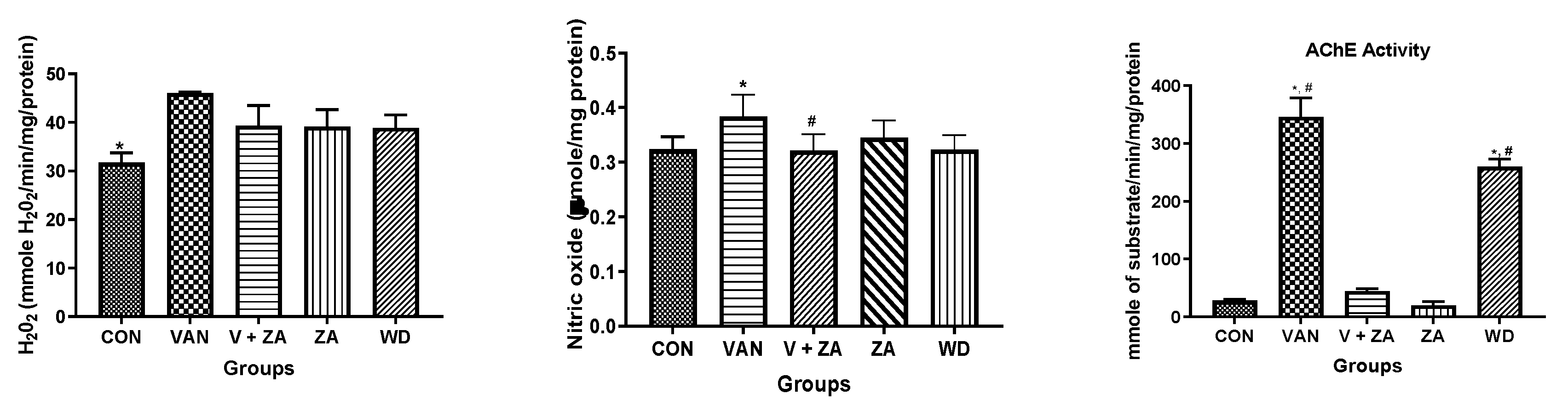
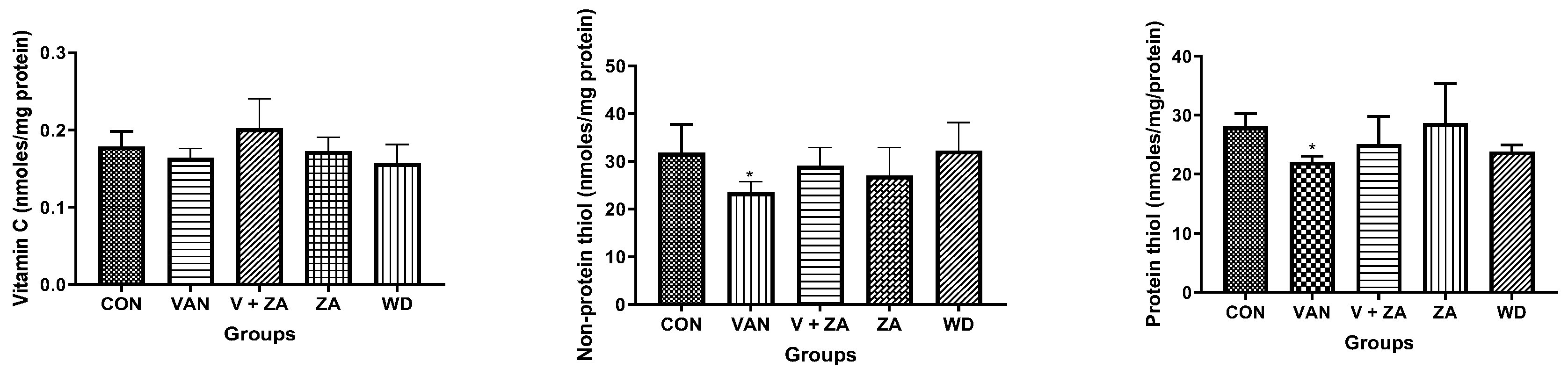
2.3. Biochemical Studies
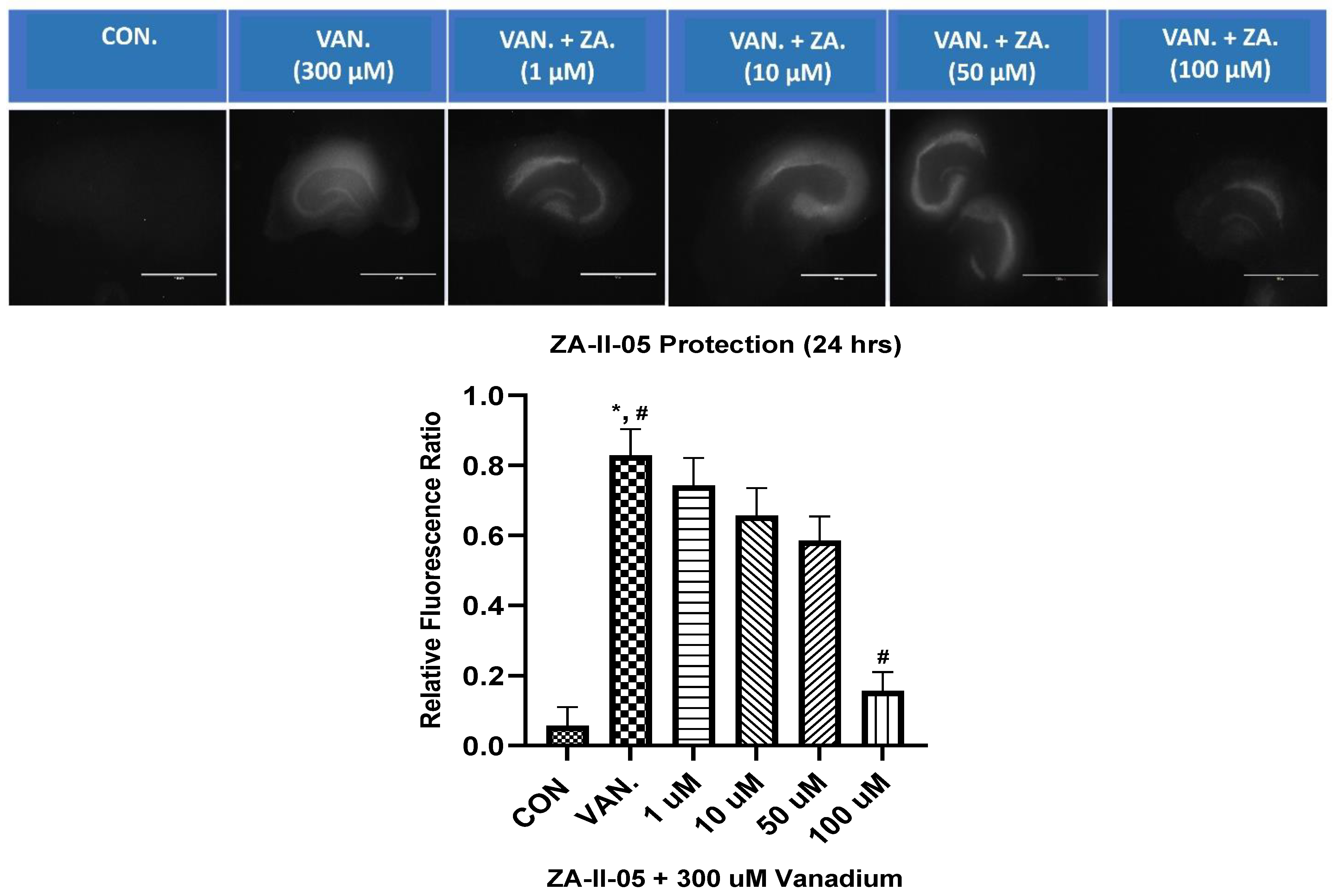
2.4. In Vitro Neuroprotection from Vanadium Toxicity Hippocampal Slices
2.5. Histopathology
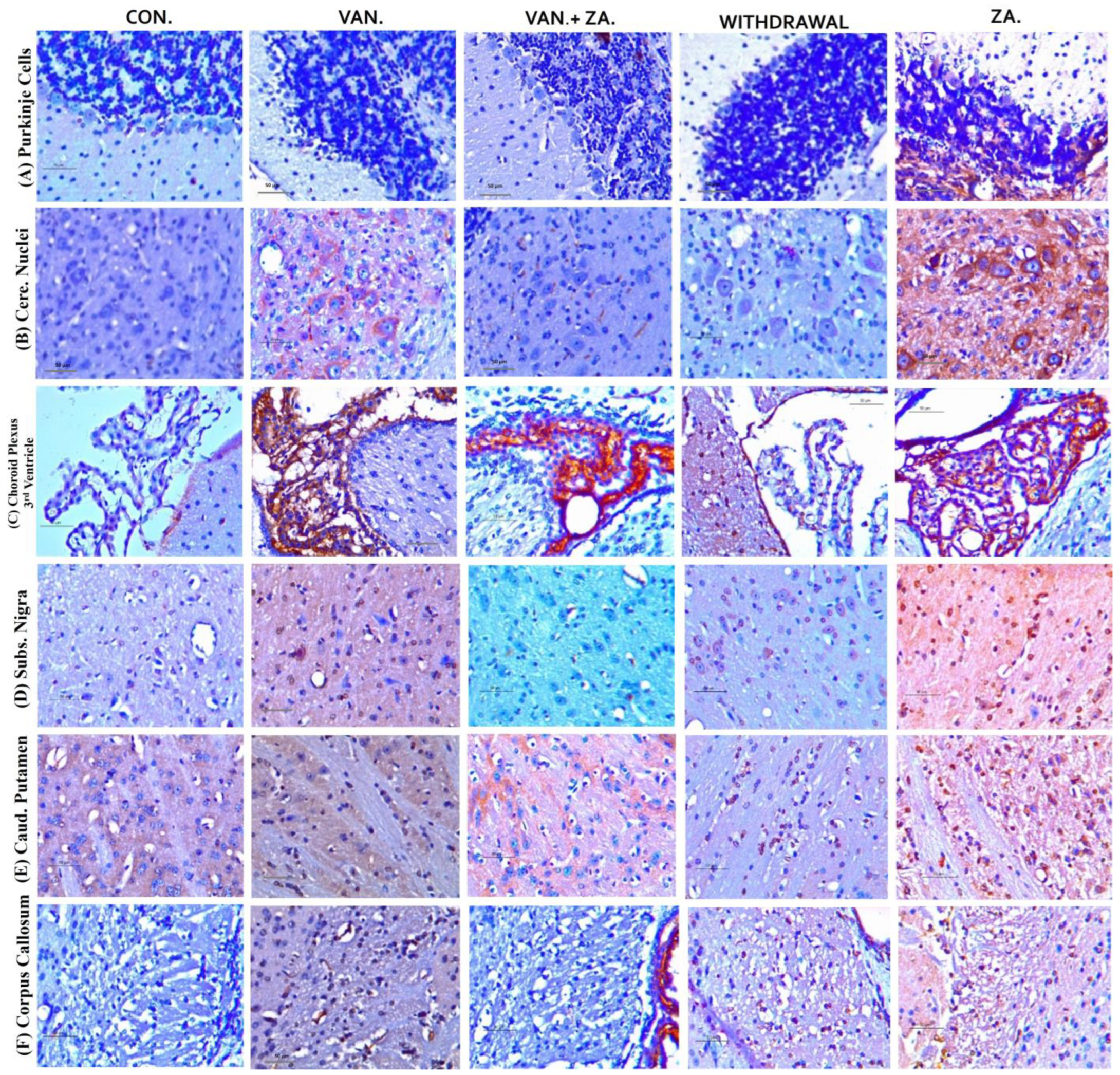
2.5.1. Expression of Neuronal Count/Density (NeuN) in Different Regions of the Brain Treated with Vanadium and ZA-II-05
2.5.2. Quantification of Dendritic Density and Arborizations
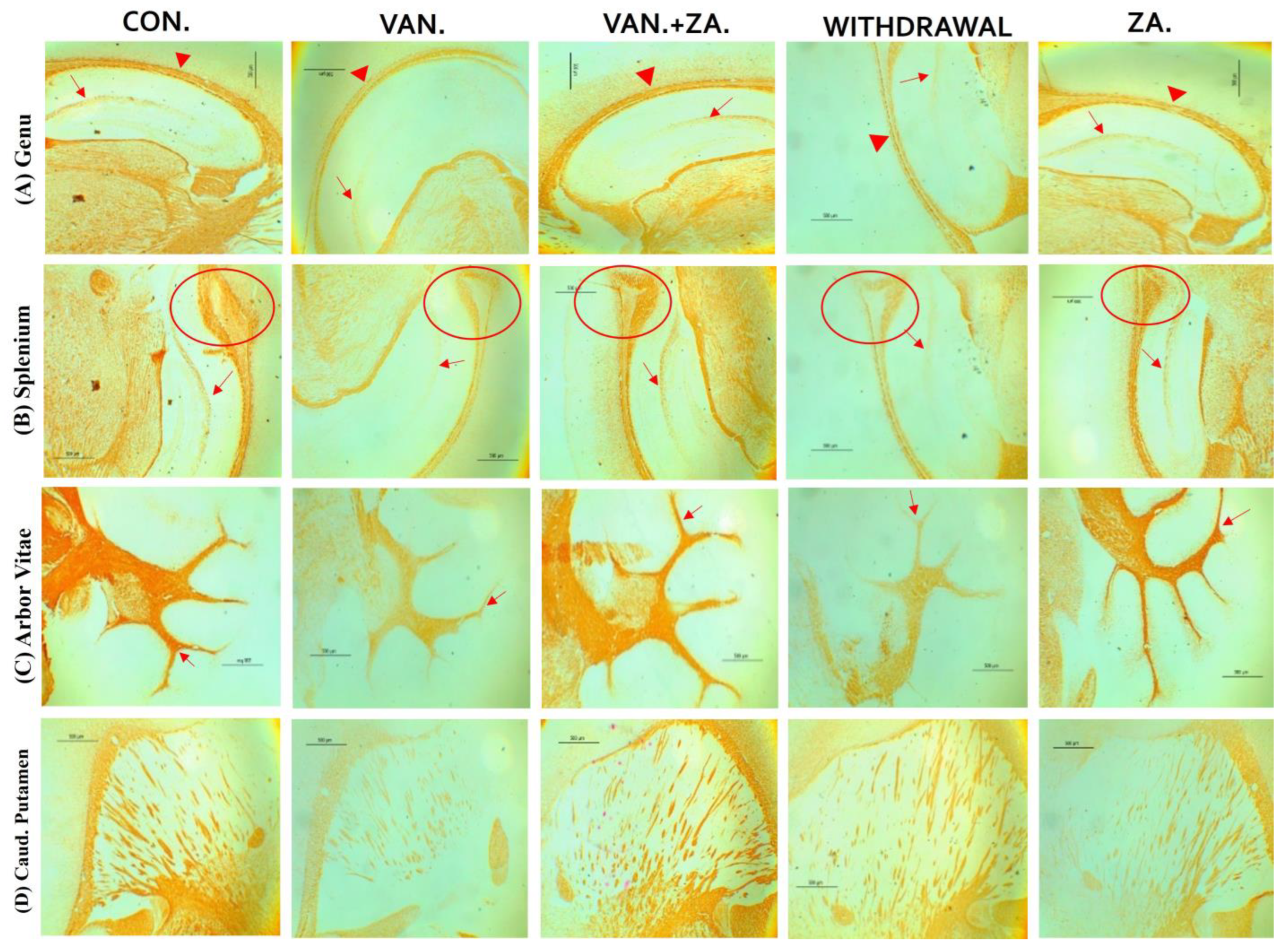
2.5.3. Expression of Myelin Basic Protein (MBP) in Different Regions of the Brain Treated with Vanadium and ZA-II-05
2.5.4. Expression of Glial Fibrillary Protein (GFAP)in Different Regions of the Brain Treated with Vanadium and ZA-II-05
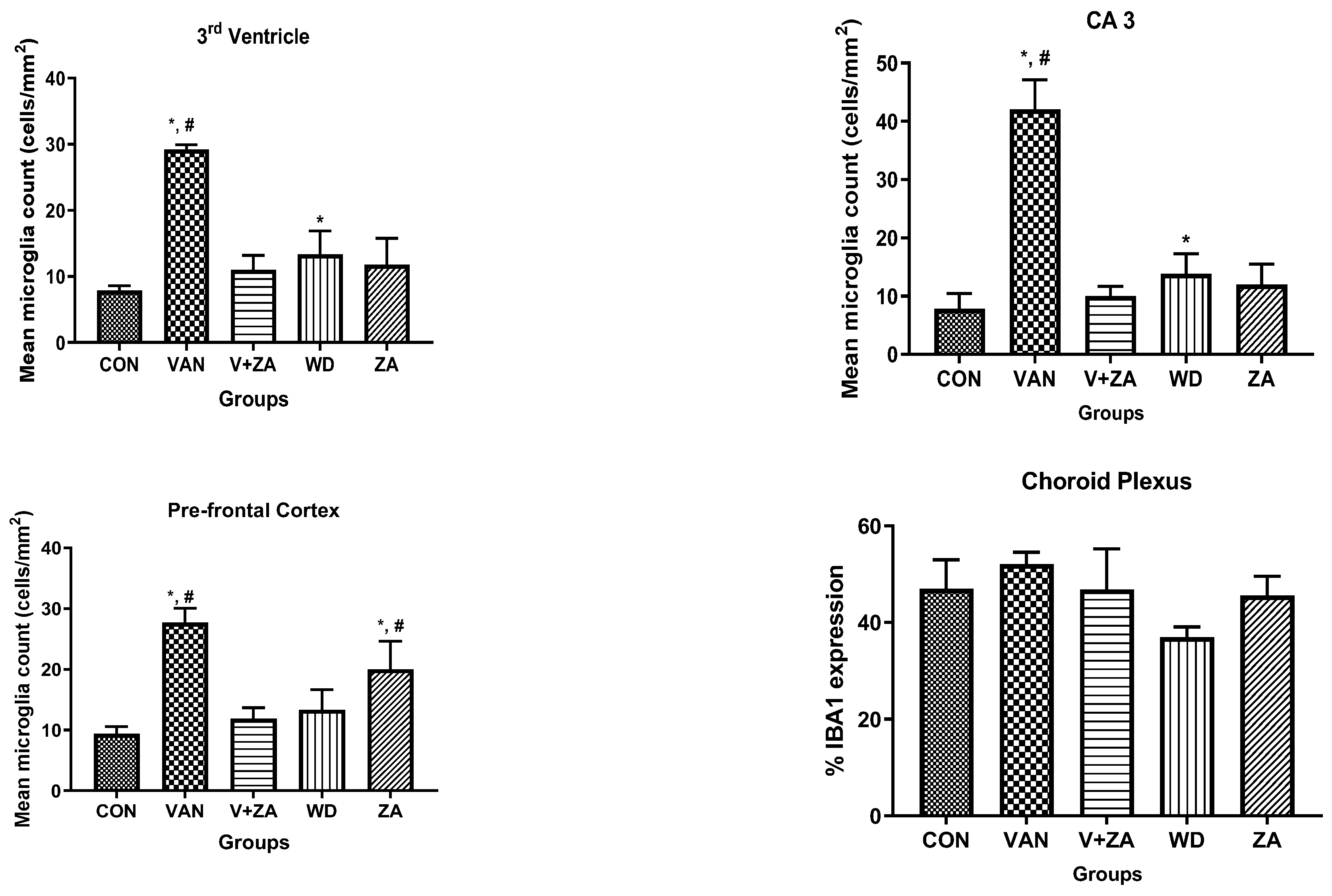
2.5.5. Expression of IBA1 in Different Regions of the Brain Treated with Vanadium and ZA-II-05
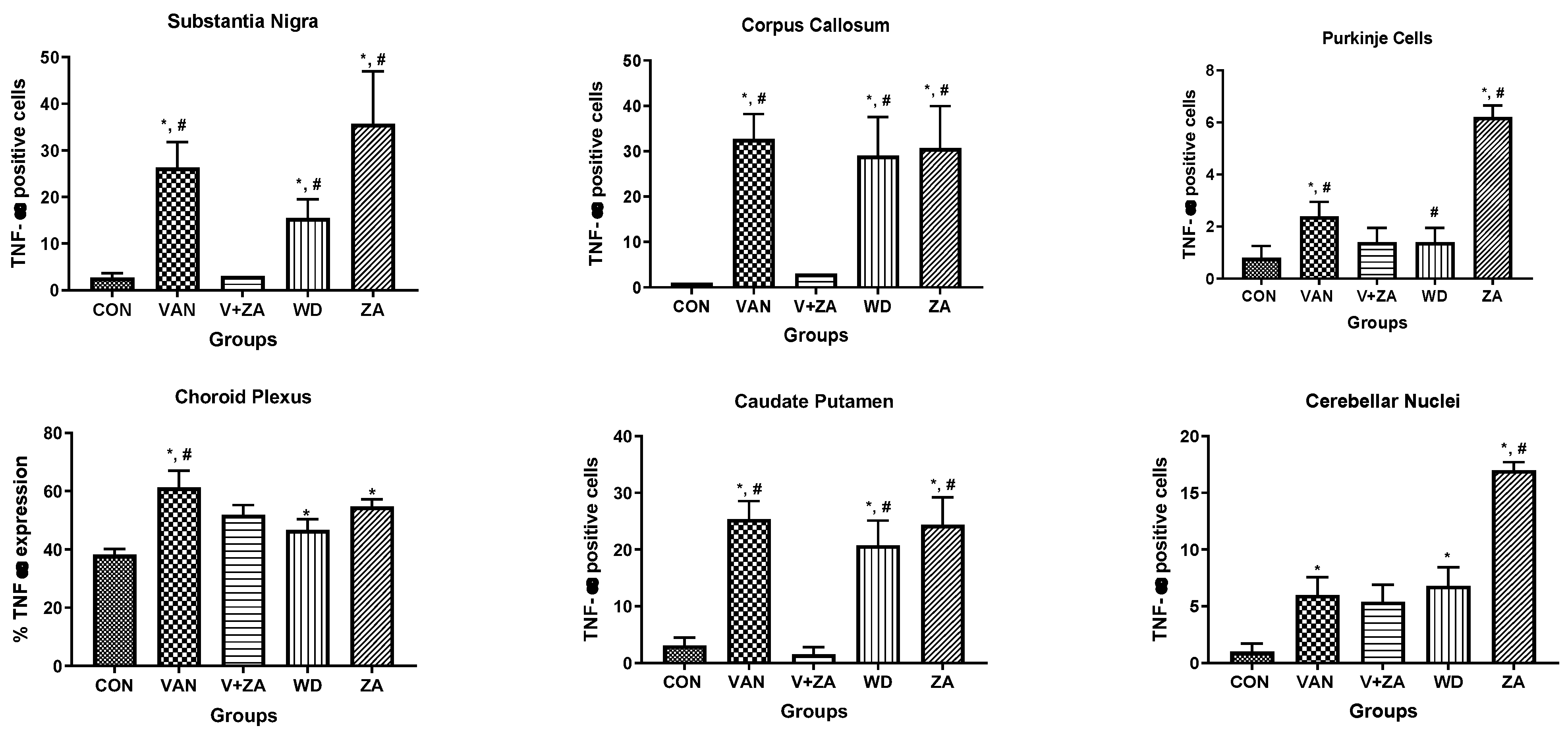
2.5.6. Expression of Tumor Necrosis Factor (TNF) in Different Regions of the Brain Treated with Vanadium and ZA-II-05
3. Discussion
3.1. Calcium Dyshomeostasis following Treatment of CAD Cells with Vanadium
3.1.1. ZA-II-05’s Binding Affinity Studies
3.1.2. Sub-Type Selectivity of ZA-II-05 for GluN2A and GluN2B
3.2. Acetylcholinesterase Activity
3.3. Protective Effect of ZA-II-05 in Hippocampal Slices
3.4. Dendritic Morphology
3.5. Glia Cells in Neurotoxicity Progression
3.5.1. Demyelination
3.5.2. Astrocytic Activation
3.5.3. Microglial Activation
3.5.4. Tumour Necrosis Factor Expression
4. Materials and Methods
4.1. Chemicals
4.2. Neuronal Calcium Mobilization
4.3. Pharmacological Characterization of ZA-II-05
4.3.1. Radioligand Binding Assay
4.3.2. Subtype Ligand Binding Assay
4.4. Biochemical Tests
4.5. Measurement of Protective Effect of ZA-II-05 In Vitro in Hippocampal Slices
4.6. Experimental Design and In Vivo Drug Administration
Immunohistochemistry
4.7. Golgi Staining
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, W.; Guo, H.; Cui, H. Vanadium toxicity in the thymic development. Oncotarget 2015, 6, 28661–28677. [Google Scholar] [CrossRef] [PubMed]
- Olopade, J.O.; Connor, J.R. Vanadium and neurotoxicity: A review. Curr. Top. Toxicol. 2011, 7, 33–38. [Google Scholar]
- Ifukibot, L.U.; Olopade, J.O.; Emikpe, B.O.; Oyagbemi, A.A.; Adedapo, A.A. Oxidative stress changes observed in selected organs of African giant rats (Cricetomys gambianus) exposed to sodium metavanadate. Int. J. Vet. Sci. Med. 2018, 6, 80–89. [Google Scholar] [CrossRef]
- Haider, S.S.; Abdel-Gayoum, A.A.; El-Fakhri, M. Effect of selenium on vanadium toxicity in different regions of rat brain. Hum. Exp. Toxicol. 1998, 17, 23–28. [Google Scholar] [CrossRef]
- Garcia, B.; Biancardi, E.; Quiroga, D. Vanadium (V)-induced neurotoxicity in the rat central nervous system: A histoimmunohistochemical study. Drug Chem Toxicol. 2005, 28, 329–344. [Google Scholar] [CrossRef]
- Azeez, I.A.; Olopade, F.; Laperchia, C.; Andrioli, A.; Scambi, I.; Onwuka, S.K.; Bentivoglio, M.; Olopade, J.O. Regional Myelin and Axon Damage and Neuroinflammation in the Adult Mouse Brain After Long-Term Postnatal Vanadium Exposure. J. Neuropathol. Exp. Neurol. 2016, 75, 843–854. [Google Scholar] [CrossRef]
- Mustapha, O.; Oke, B.; Offen, N. Neurobehavioral and cytotoxic effects of vanadium during oligodendrocyte maturation: A protective role for erythropoietin. Environ. Toxicol. Pharmacol. 2014, 38, 98–111. [Google Scholar] [CrossRef]
- Wang, D.C.; Lin, Y.Y.; Lin, H.T. Recovery of motor coordination after exercise is correlated to enhancement of brain-derived neurotrophic factor in lactational vanadium-exposed rats. Neurosci. Lett. 2015, 600, 232–237. [Google Scholar] [CrossRef]
- Todorich, B.; Olopade, J.O.; Surguladze, N. The mechanism of vanadium-mediated developmental hypomyelination is related to destruction of oligodendrocyte progenitors through a relationship with ferritin and iron. Neurotox. Res. 2011, 19, 361–373. [Google Scholar] [CrossRef]
- Avila-Costa, M.R.; Fortoul, T.I.; Nino-Cabrera, G. Hippocampal cell alterations induced by the inhalation of vanadium pentoxide (V2O5) promote memory deterioration. Neurotoxicology 2006, 27, 1007–1215. [Google Scholar] [CrossRef]
- Avila-Costa, M.R.; Colın-Barenque, L.; Zepeda-Rodrıguez, A. Ependymal epithelium disruption after vanadium pentoxide inhalation. A mice experimental model. Neurosci. Lett. 2005, 381, 21–516. [Google Scholar] [CrossRef]
- Colin-Barenque, L.; Pedraza-Chaverri, J.; Medina-Campos, O. Functional and morphological olfactory bulb modifications in mice after vanadium inhalation. Toxicol. Pathol. 2014, 43, 282–289. [Google Scholar] [CrossRef]
- Li, H.; Zhou, D.; Zhang, Q. Vanadium exposure-induced neurobehavioral alterations among Chinese workers. Neurotoxicology 2013, 36, 49–5413. [Google Scholar] [CrossRef] [PubMed]
- Igado, O.; Olopade, J.O.; Adesida, A.; Aina, O.O.; Farombi, E.O. Morphological and biochemical investigation into the possible neuroprotective effects of kolaviron (Garcinia kola bioflavonoid) on the brains of rats exposed to vanadium. Drug Chem. Toxicol. 2012, 35, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Olopade, J.O.; Fatola, I.O.; Olopade, F.E. Vertical Administration of vanadium through lactation induces behavioural and neuromorphological changes: Protective role of vitamin E. Niger. J. Physiol. Sci. 2011, 26, 55–60. [Google Scholar]
- Adeboye, A. Drug Discovery Approaches for the Treatment of Neurodegenerative Disorders: Alzheimer’s Disease; Academic Press: Cambridge, MA, USA; Elsevier: Cambridge, MA, USA, 2017; ISBN 9780128028100. [Google Scholar]
- Plutino, S.; Sciaccaluga, M.; Fucile, S. Extracellular mild acidosis decreases the Ca2þ permeability of the human NMDA receptors. Cell Calcium. 2019, 80, 63–70. [Google Scholar] [CrossRef]
- Zheng, L.; Xiaoling, Q.; Shinghung, M.; Baojian, G.; Shengquan, H.; Jiajun, W.; Fangcheng, L.; Daping, X.; Yewei, S.; Gaoxiao, Z.; et al. Multifunctional memantine nitrate significantly protects against glutamate-induced excitotoxicity via inhibiting calcium influx and attenuating PI3K/Akt/GSK3beta pathway. Chem.-Biological. Interact. 2020, 325, 109020. [Google Scholar] [CrossRef]
- Boyce, S.; Wyatt, A.; Webb, J.K.R.; O’donnell, G.; Mason, M.; Sirinathsinghji, R.G.; Hill, N.M.; Rupniak, N.M.J. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: Correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology 1999, 38, 611–623. [Google Scholar] [CrossRef]
- Wessell, R.H.; Ahmed, S.M.; Menniti, F.S.; Dunbar, G.L.; Chase, T.N. NR2B selective NMDA receptor antagonist CP-101,606 prevents levodopa-induced motor response alterations in hemi-parkinsonian rats. Neuropharmacology 2004, 47, 184–194. [Google Scholar] [CrossRef]
- Chandra, A.K.; Ghosh, R.; Chatterjee, A.; Sarkar, M. Amelioration of vanadium-induced testicular toxicity and adrenocortical hyperactivity by vitamin E acetate in rats. Mol. Cell. Biochem. 2007, 306, 189–200. [Google Scholar] [CrossRef]
- Shukla, R.; Padhye, S.; Modak, M.; Ghaskadbi, S.S.; Bhonde, R.R. Bis (quercetinato) oxovanadium IV reverses metabolic changes in streptozotocin-induced diabetic mice. Rev. Diabet. Stud. 2007, 4, 33–43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohiomokhare, S.; Olaolorun, F.; Ladagu, A.; Olopade, F.; Howes, M.-J.R.; Okello, E.; Olopade, J.; Chazot, P.L. The Pathopharmacological Interplay between Vanadium and Iron in Parkinson’s Disease Models. Int. J. Mol. Sci. 2020, 21, 6719. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Caro, M.; Hadzimichalis, N. NMDA receptor activity in neuropsychiatric disorders. Front. Psychiatry 2013, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium. 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Gomes, S.; Sousa, N.; Pinto, L.; Oliveira, J.F. Functional roles of astrocyte calcium elevations: From synapses to behavior. Front. Cell. Neurosci. 2018, 11, 427. [Google Scholar] [CrossRef] [PubMed]
- Ladagu, A.D.; Olopade, F.E.; Folarin, O.R.; Elufioye, T.O.; Wallach, J.V.; Dybek, M.B.; Olopade, J.O.; Adejare, A. Novel NMDA-receptor antagonists ameliorate vanadium neurotoxicity. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1729–1738. [Google Scholar] [CrossRef]
- Thayer, S.A.; Miller, R.J. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J. Physiol. 1990, 425, 85–115. [Google Scholar] [CrossRef]
- Tymianski, M.; Charlton, M.P.; Carlen, P.L.; Tator, C.H. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J. Neurosci. 1993, 13, 2085–2104. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Minarini, A. Multi-target Design Strategies in the Context of Alzheimer’s Disease: Acetylcholinesterase Inhibition and NMDA Receptor Antagonism as the Driving Forces. Neurochem. Res. 2014, 39, 1914–1923. [Google Scholar] [CrossRef]
- Lau, L.F.; Huganir, R.L. Differential tyrosine phosphorylation of N methyl-D-aspartate receptor subunits. J. Biol. Chem. 1995, 270, 20036–20041. [Google Scholar] [CrossRef]
- Pan, L.; Li, T.; Wang, R. Roles of Phosphorylation of N-Methyl-D-Aspartate Receptor in Chronic Pain. Cell Mol. Neurobiol. 2023, 43, 155–175. [Google Scholar] [CrossRef]
- Wang, M.; Wong, A.H.; Liu, F. Interactions between NMDA and dopamine receptors: A potential therapeutic target. Brain Res. 2012, 1476, 154–163. [Google Scholar] [CrossRef]
- Linda, N.; Kelan, L.; Thomas, B.P.; Lucke-Wold, J.Z.; Cavendish, M.S.; Crowe, R.; Matsumoto, R. Dextromethorphan: An update on its utility for neurological and neuropsychiatric disorders. Pharmacol. Ther. 2016, 159, 1–22. [Google Scholar] [CrossRef]
- Yi, B.; Sahn, J.J.; Ardestani, P.M.; Evans, A.K.; Scott, L.L.; Chan, J.Z.; Iyer, S.; Crisp, A.; Zuniga, G.; Pierce, J.T.; et al. Small molecule modulator of sigma 2 receptor is neuroprotective and reduces cognitive deficits and neuroinflammation in experimental models of Alzheimer’s disease. J. Neurochem. 2017, 140, 561–575. [Google Scholar] [CrossRef]
- Lin, C.; Huang, Y.; Lin, C.; Lane, H.; Tsai, G. NMDA Neurotransmission Dysfunction in Mild Cognitive Impairment and Alzheimers Disease. Curr. Pharm. Des. 2014, 20, 5169–5179. [Google Scholar] [CrossRef]
- Yang, K.; Wang, C.; Sun, T. The Roles of Intracellular Chaperone Proteins, Sigma Receptors, in Parkinson’s Disease (PD) and Major Depressive Disorder (MDD). Front. Pharmacol. 2019, 10, 528. [Google Scholar] [CrossRef]
- Garzón, J.; Rodríguez-Muñoz, M.; Sánchez-Blázquez, P. Direct association of Mu-opioid and NMDA glutamate receptors supports their cross-regulation: Molecular implications for opioid tolerance. Curr. Drug Abuse Rev. 2012, 5, 199–226. [Google Scholar] [CrossRef]
- Paul, L.C. The NMDA Receptor NR2B Subunit: A Valid Therapeutic Target for Multiple CNS Pathologies. Curr. Med. Chem. 2004, 11, 389–396. [Google Scholar]
- Collingridge, G.L.; Volianskis, A.; Bannister, N. The NMDA receptor as a target for cognitive enhancement. Neuropharmacology 2013, 64, 13–26. [Google Scholar] [CrossRef]
- Hansen, K.B.; Ogden, K.K.; Yuan, H.; Traynelis, S.F. Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 2014, 81, 1084–1096. [Google Scholar] [CrossRef]
- Patel, L.; Grossberg, G.T. Combination therapy for Alzheimer’s disease. Drugs Aging 2011, 28, 539–546. [Google Scholar] [CrossRef]
- Bittner, E.A.; Martyn, J.A. Neuromuscular physiology and pharmacology. In Pharmacology and Physiology for Anaesthesia; Hemmings, H.C., Jr., Egan, T.D., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 309–324. [Google Scholar]
- Reddy, V.P. Fluorinated Compounds in Enzyme-Catalysed Reactions in Organofluorine Compounds in Biology and Medicine; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 29–57. [Google Scholar]
- Giacobini, E. Cholinergic function and Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, S1–S5. [Google Scholar] [CrossRef]
- Terry, A.V.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Parsons, C.G.; Danysz, W.; Dekundy, A.; Pulte, I. Memantine and cholinesterase inhibitors: Complementary+ mechanisms in the treatment of Alzheimer’s disease. Neurotox Res. 2013, 24, 358–369. [Google Scholar] [CrossRef]
- Parsons, C.G.; Stöffler, A.; Danysz, W. Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system—Too little activation is bad, too much is even worse. Neuropharmacology 2007, 53, 699–723. [Google Scholar] [CrossRef]
- Schmidtke, K.; Holthoff, V.; Kressig, R.W.; Molinuevo, J.L. Combination of Memantine and cholinesterase inhibitors in the treatment of AD. Neurol. News 2011, 1, 1–8. [Google Scholar]
- Marotta, G.; Basagni, F.; Rosini, M.; Minarini, A. Memantine Derivatives as Multitarget Agents in Alzheimer’s Disease. Molecules 2020, 25, 4005. [Google Scholar] [CrossRef]
- Roceri, M.; Hendriks, W.; Racagni, G.; Ellenbroek, B.A.; Riva, M.A. Early maternal deprivation reduces the expression of BDNF and NMDA receptor subunits in rat hippocampus. Mol. Psychiatry 2002, 7, 609–616. [Google Scholar] [CrossRef]
- Pickering, C.; Gustafsson, L.; Cebere, A.; Nylander, I.; Liljequist, S. Repeated maternal separation of male Wistar rats alters glutamate receptor expression in the hippocampus but not the prefrontal cortex. Brain Res. 2006, 1099, 101–108. [Google Scholar] [CrossRef]
- Oreland, S.; Nylander, I.; Pickering, C. Prolonged maternal separation decreases granule cell number in the dentate gyrus of 3-week-old male rats. Int. J. Dev. Neurosci. 2010, 28, 139–144. [Google Scholar] [CrossRef]
- Folarin, O.R.; Snyder, A.M.; Peters, D.G.; Olopade, F.; Connor, J.R.; Olopade, J.O. Brain metal distribution and neuro-inflammatory profiles after chronic vanadium administration and withdrawal in mice. Front. Neuroanat. 2017, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Gustav, M.; Hans, F.; Magnus, H.; Eskil, E.; Tadeusz, W. Flow cytometric analysis of mitochondria from CA1 and CA3 regions of rat hippocampus reveals differences in permeability transition pore activation. J. Neurochem. 2003, 87, 532–544. [Google Scholar]
- Majlath, Z.; Vecsei, L. NMDA antagonists as Parkinson’s disease therapy: Disseminating the evidence. Neurodegener. Dis. Manag. 2014, 4, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Zhao, C.; Toni, N.; Yao, J.; Gage, F.H. Distinct roles of NMDA receptors at different stages of granule cell development in the adult brain. eLife 2015, 4, e07871. [Google Scholar] [CrossRef]
- Olopade, F.E.; Femi-Akinlosotu, O.M.; Adekanmbi, A.J.; Ighogboja, O.O.; Shokunbi, M.T. Chronic caffeine ingestion improves motor function and increases dendritic length and arborization in the motor cortex, striatum, and cerebellum. J. Caffeine Adenosine Res. 2021, 11, 3–14. [Google Scholar] [CrossRef]
- Maiolo, L.; Guarino, V.; Saracino, E. Glial interfaces: Advanced materials and devices to uncover the role of astro-glial cells in brain function and dysfunction. Adv. Healthc. Mater. 2021, 10, 2001268. [Google Scholar] [CrossRef]
- Igado, O.O.; Andrioli, A.; Azeez, I.A.; Girolamo, F.; Errede, M.; Aina, O.O.; Glaser, K.; Holzgrabe, U.; Bentivoglio, M.; Olopade, J.O. The ameliorative effects of a phenolic derivative of Moringa oleifera leave against vanadium-induced neurotoxicity in mice. IBRO Rep. 2020, 9, 164–182. [Google Scholar] [CrossRef]
- Stojanovic, I.R.; Kostic, M.; Ljubisavljevic, S. The role of glutamate and its receptors in multiple sclerosis. J. Neural. Transm. 2014, 121, 945–955. [Google Scholar] [CrossRef]
- Maurer, L.L.; Philbert, M.A. The mechanisms of neurotoxicity and the selective vulnerability of nervous system sites. Handb. Clin. Neurol. 2015, 131, 61–70. [Google Scholar] [CrossRef]
- Paluch-Oles, J.; Magrys, A.; Koziol-Montewka, M.; Koszar-ny, A.; Majdan, M. Identification of latent tuberculosis infection in rheumatic patients under consideration for treatment with anti-TNF-alpha agents. Arch. Med. Sci. 2013, 9, 112–117. [Google Scholar] [CrossRef]
- Zheng, X.; Zhou, J.; Xia, Y. The role of TNF-α in regulating ketamne-induced hippocampal neurotoxicity. Arch. Med. Sci. 2015, 11, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Dolga, A.M.; Granic, I.; Blank, T.; Knaus, H.G.; Spiess, J.; Luiten, P.G.; Eisel, U.L.; Nijholt, I.M. TNF-alpha-mediates neuroprotection against glutamate-induced excitotoxicity via NF-kappaB-dependent up-regulation of K2.2 channels. J. Neurochem. 2008, 107, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Isaias, G.; Carolina, D.M.; Elisa, M.K.; Tania, M.; Maria, C.W.; Avellar, C.S. MK-801 and 7-Ni attenuate the activation of brain NF-κB induced by LPS. Neuropharmacology 2003, 45, 1120–1129. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, Y.; Xu, Y.; Qu, P.; Wang, M. Memantine protects against ischemia/reperfusion-induced brain endothelial permeability. IUBMB Life 2018, 70, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Bwala, D.A.; Ladagu, A.D.; Olopade, F.E.; Siren, A.L.; Kwari, H.; Yahaya, A.; Olopade, J.O. Neurotoxic profiles of vanadium when administered at the onset of myelination in rats: The protective role of vitamin E. Trop. Vet. 2014, 32, 36–46. [Google Scholar]
- Kennedy, D.; Okello, E.; Chazot, P.; Howes, M.J.; Ohiomokhare, S.; Jackson, P.; Haskell-Ramsay, C.; Khan, J.; Forster, J.; Wightman, E. Volatile Terpenes and Brain Function: Investigation of the Cognitive and Mood Effects of Mentha × Piperita, L. Essential Oil with In Vitro Properties Relevant to Central Nervous System Function. Nutrients 2018, 10, 1029. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Sharma, T.A. NMDA Receptor Protocols; Springer: Berlin/Heidelberg, Germany, 1999; pp. 93–102. [Google Scholar]
- Reynolds, I.J. [3H](+) MK801 Radioligand Binding Assay at the N-Methyl-D-Aspartate Receptor. Curr. Protoc. Pharmacol. 2001, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chazot, P.L.; Cik, M.; Stephenson, F.A. Transient expression of functional NMDA receptors in mammalian cells. Methods Mol. Biol. 1999, 128, 33–42. [Google Scholar] [CrossRef]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.K.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal. Biochem. J. 2001, 126, 131–136. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Bio. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Jacques-Silva, M.C.; Nogueira, C.W.; Broch, L.C.; Flores, E.M.; Rocha Note, J.B. Diphenyl diselenide and ascorbic acid changes deposition of selenium and ascorbic acid in liver and brain of mice. Pharmacol. Toxicol. 2001, 88, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.F. Ferrous ion oxidation in the presence of ferric ion indicator xylenol orange for measurement of hydrogen peroxides. Methods Enzymol. 1994, 233, 182–189. [Google Scholar]
- Ellman, G.L. Tissue sulfhydryl groups. Arc. Biochem. Biophy. 1959, 82, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Noraberg, J.; Kristensen, B.W.; Zimmer, J. Markers for neuronal degeneration in organotypic slice cultures. Brain Res. Protoc. 1999, 3, 278–290. [Google Scholar] [CrossRef]
- Adebiyi, O.E.; Olayemi, F.O.; Olopade, J.O.; Tan, N.H. Beta-sitosterol enhances motor coordination, attenuates memory loss and demyelination in a vanadium-induced model of experimental neurotoxicity. Pathophysiology 2019, 26, 21–29. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | [3H] Ligand | Species | Source |
|---|---|---|---|
| 5-HT2A | Ketanserin | Human | Cloned |
| Dopamine 1 (D1) | SCH23390 | Human | Cloned |
| Dopamine 1(D2) | N-Methylspiperone | Human | Cloned |
| Dopamine Transporter (DAT) | WIN35428 | Human | Cloned |
| κ-opioid receptors (KOR) | U69593 (2007-07-27) | Rat | Cloned |
| µ-opioid receptors (MOR) | DAMGO (2007-07-27) | Human | Cloned |
| Norepinephrine Transporter (NET) | Nisoxetine | Human | Cloned |
| Serotonin Transporter (SERT) | Citalopram | Human | Cloned |
| Sigma1 | Pentazocine (+) | Rat | Brain |
| Sigma2 | DTG | Rat | PC12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ladagu, A.D.; Olopade, F.E.; Chazot, P.; Oyagbemi, A.A.; Ohiomokhare, S.; Folarin, O.R.; Gilbert, T.T.; Fuller, M.; Luong, T.; Adejare, A.; et al. Attenuation of Vanadium-Induced Neurotoxicity in Rat Hippocampal Slices (In Vitro) and Mice (In Vivo) by ZA-II-05, a Novel NMDA-Receptor Antagonist. Int. J. Mol. Sci. 2023, 24, 16710. https://doi.org/10.3390/ijms242316710
Ladagu AD, Olopade FE, Chazot P, Oyagbemi AA, Ohiomokhare S, Folarin OR, Gilbert TT, Fuller M, Luong T, Adejare A, et al. Attenuation of Vanadium-Induced Neurotoxicity in Rat Hippocampal Slices (In Vitro) and Mice (In Vivo) by ZA-II-05, a Novel NMDA-Receptor Antagonist. International Journal of Molecular Sciences. 2023; 24(23):16710. https://doi.org/10.3390/ijms242316710
Chicago/Turabian StyleLadagu, Amany Digal, Funmilayo Eniola Olopade, Paul Chazot, Ademola A. Oyagbemi, Samuel Ohiomokhare, Oluwabusayo Racheal Folarin, Taidinda Tashara Gilbert, Madison Fuller, Toan Luong, Adeboye Adejare, and et al. 2023. "Attenuation of Vanadium-Induced Neurotoxicity in Rat Hippocampal Slices (In Vitro) and Mice (In Vivo) by ZA-II-05, a Novel NMDA-Receptor Antagonist" International Journal of Molecular Sciences 24, no. 23: 16710. https://doi.org/10.3390/ijms242316710
APA StyleLadagu, A. D., Olopade, F. E., Chazot, P., Oyagbemi, A. A., Ohiomokhare, S., Folarin, O. R., Gilbert, T. T., Fuller, M., Luong, T., Adejare, A., & Olopade, J. O. (2023). Attenuation of Vanadium-Induced Neurotoxicity in Rat Hippocampal Slices (In Vitro) and Mice (In Vivo) by ZA-II-05, a Novel NMDA-Receptor Antagonist. International Journal of Molecular Sciences, 24(23), 16710. https://doi.org/10.3390/ijms242316710