
Novel COX11 Mutations Associated with Mitochondrial Disorder: Functional Characterization in Patient Fibroblasts and Saccharomyces cerevisiae

 , ,
, ,  , ,
, ,  , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. Clinical Description
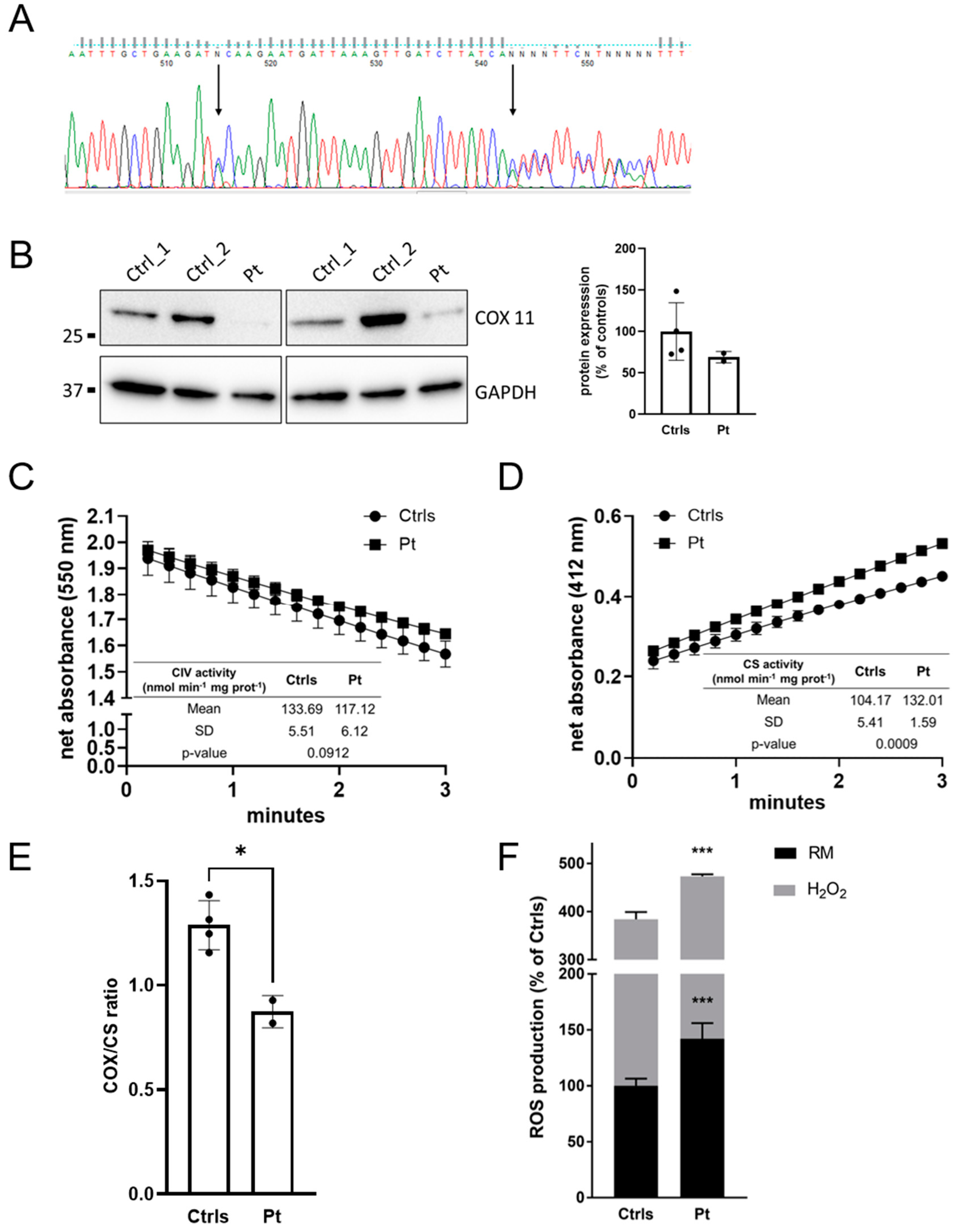
2.2. Genetic Analysis
2.3. Muscle and Cellular Studies
2.4. Mutation Analysis and Characterization in Yeast
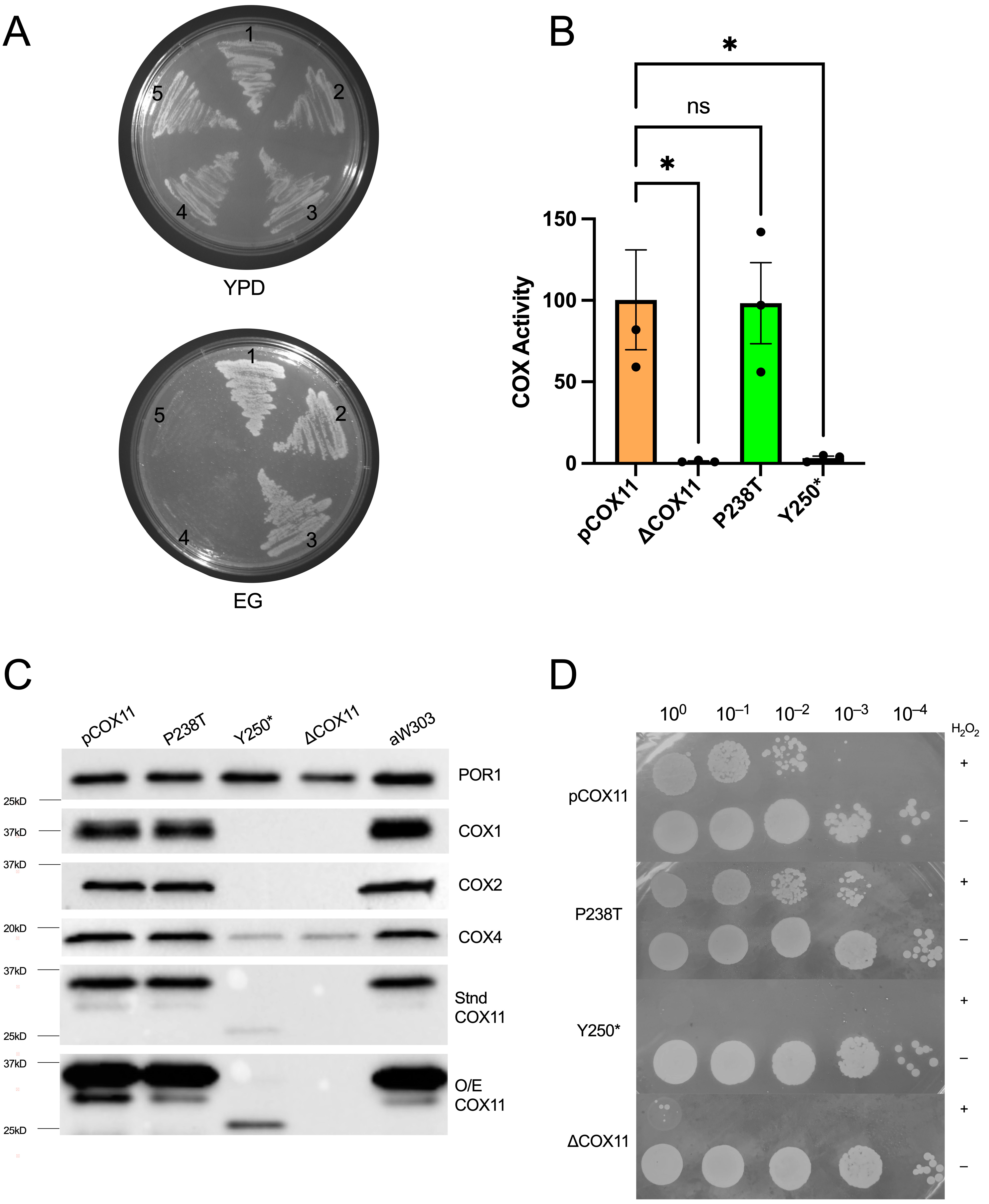
2.4.1. Respiratory Competence Is Maintained in the P238T Mutant
2.4.2. Stable COX11 Expression in Both Yeast Mutants
2.4.3. Peroxide Sensitivity Phenotypes Correlate with Respiratory Competence
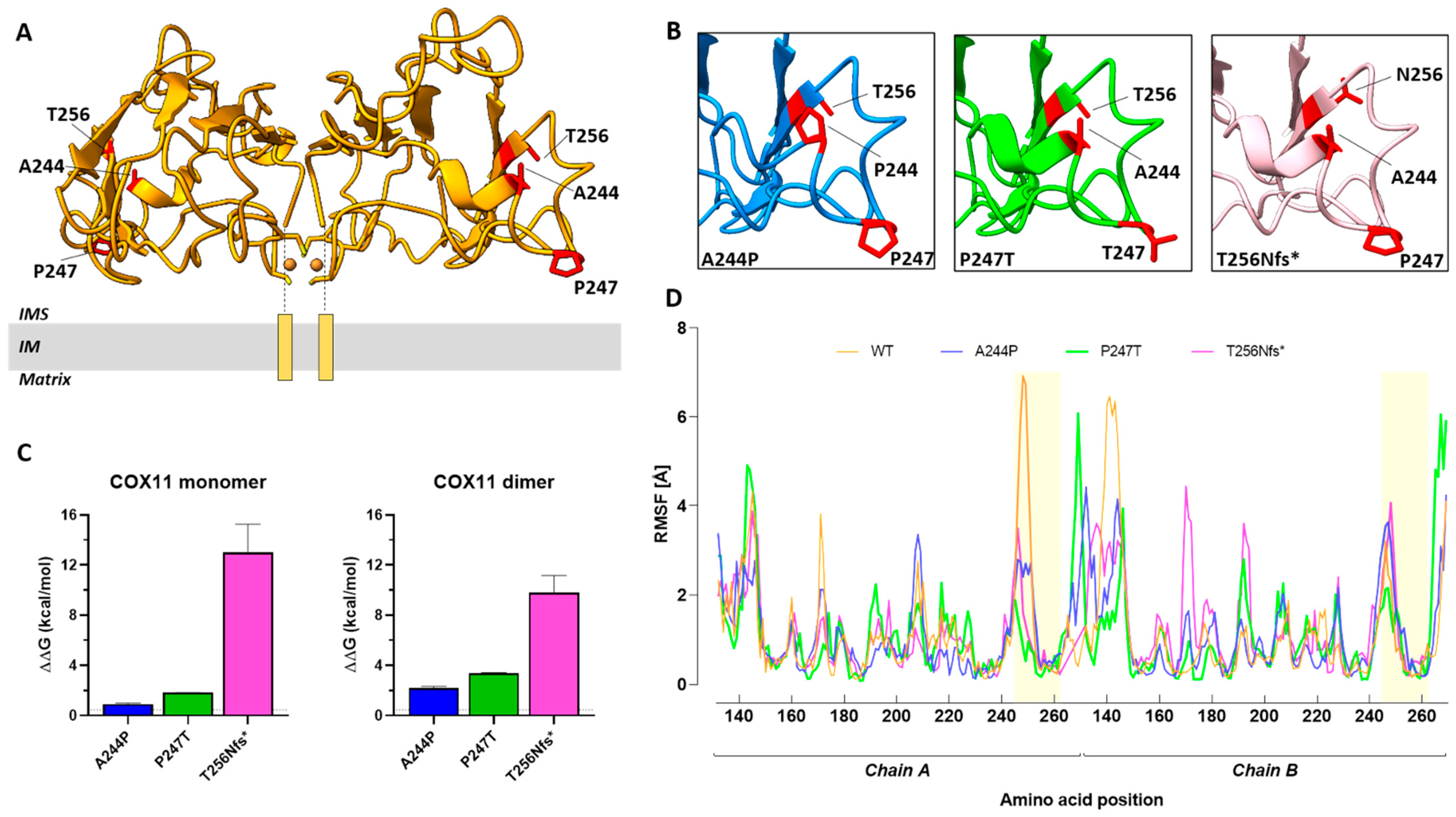
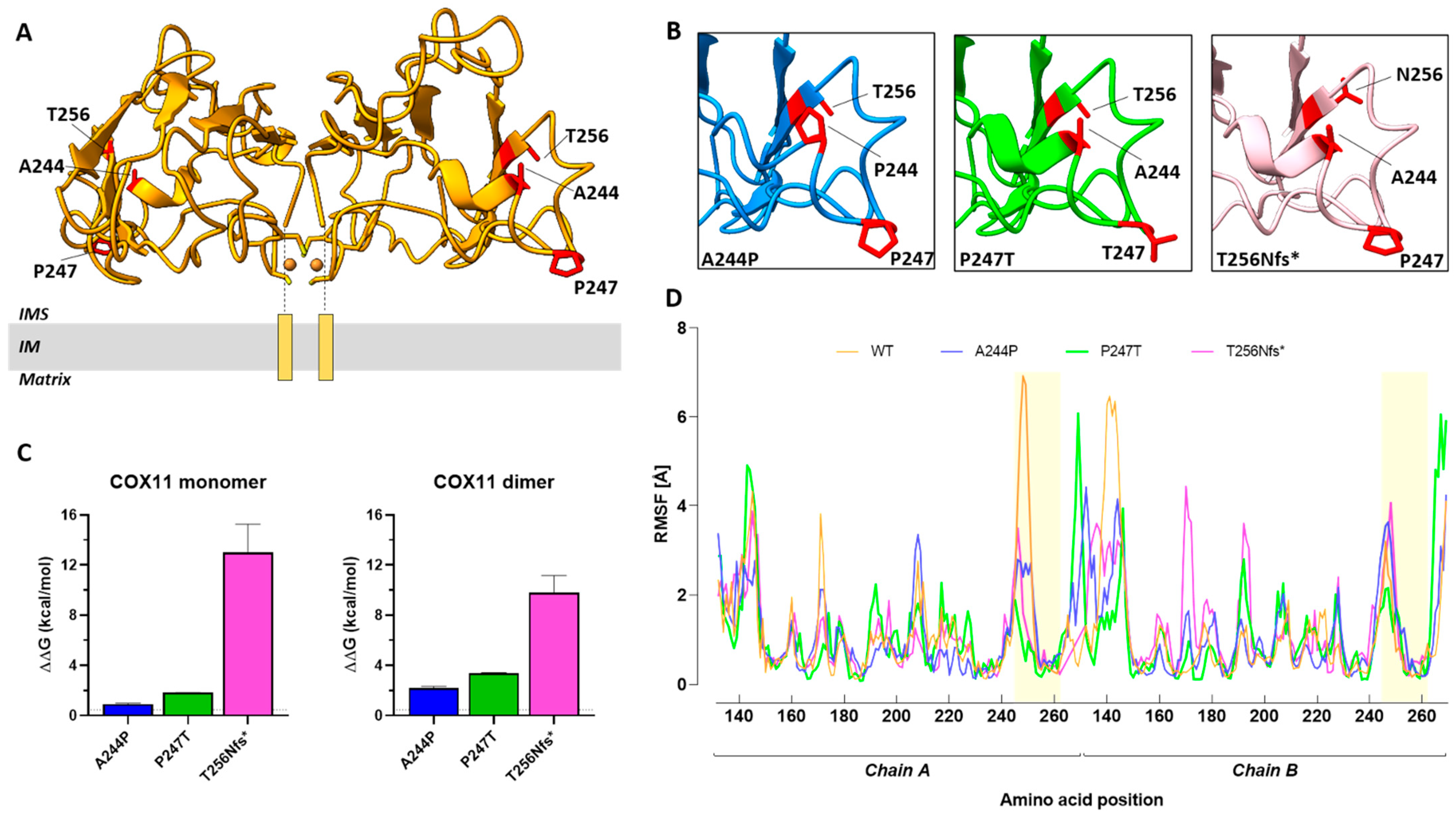
2.5. Structure-Based Analysis of Human COX11 Mutations
3. Discussion
4. Materials and Methods
4.1. Analyses on Patient’s Tissues
4.2. Generation of Yeast Mutants
4.3. Characterization of Yeast Mutants
4.4. Human COX11 Protein Modelling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghezzi, D.; Zeviani, M. Assembly Factors of Human Mitochondrial Respiratory Chain Complexes: Physiology and Pathophysiology. In Mitochondrial Oxidative Phosphorylation: Nuclear-Encoded Genes, Enzyme Regulation, and Pathophysiology; Springer: New York, NY, USA, 2012; pp. 65–106. [Google Scholar]
- Fernández-Vizarra, E.; Tiranti, V.; Zeviani, M. Assembly of the Oxidative Phosphorylation System in Humans: What We Have Learned by Studying Its Defects. Biochim. Biophys. Acta-Mol. Cell Res. 2009, 1793, 200–211. [Google Scholar] [CrossRef]
- Nývltová, E.; Dietz, J.V.; Seravalli, J.; Khalimonchuk, O.; Barrientos, A. Coordination of Metal Center Biogenesis in Human Cytochrome c Oxidase. Nat. Commun. 2022, 13, 3615. [Google Scholar] [CrossRef]
- Tzagoloff, A.; Capitanio, N.; Nobrega, M.P.; Gatti, D. Cytochrome Oxidase Assembly in Yeast Requires the Product of COX11, a Homolog of the P. Denitrificans Protein Encoded by ORF3. EMBO J. 1990, 9, 2759–2764. [Google Scholar] [CrossRef] [PubMed]
- Hederstedt, L. Diversity of Cytochrome c Oxidase Assembly Proteins in Bacteria. Microorganisms 2022, 10, 926. [Google Scholar] [CrossRef]
- Banting, G.S.; Glerum, D.M. Mutational Analysis of the Saccharomyces Cerevisiae Cytochrome c Oxidase Assembly Protein Cox11p. Eukaryot. Cell 2006, 5, 568–578. [Google Scholar] [CrossRef]
- Carr, H.S.; George, G.N.; Winge, D.R. Yeast Cox11, a Protein Essential for Cytochrome COxidase Assembly, Is a Cu(I)-Binding Protein. J. Biol. Chem. 2002, 277, 31237–31242. [Google Scholar] [CrossRef]
- Veniamin, S.; Sawatzky, L.G.; Banting, G.S.; Glerum, D.M. Characterization of the Peroxide Sensitivity of COX-Deficient Yeast Strains Reveals Unexpected Relationships between COX Assembly Proteins. Free Radic. Biol. Med. 2011, 51, 1589–1600. [Google Scholar] [CrossRef]
- Bode, M.; Woellhaf, M.W.; Bohnert, M.; van der Laan, M.; Sommer, F.; Jung, M.; Zimmermann, R.; Schroda, M.; Herrmann, J.M. Redox-Regulated Dynamic Interplay between Cox19 and the Copper-Binding Protein Cox11 in the Intermembrane Space of Mitochondria Facilitates Biogenesis of Cytochrome c Oxidase. Mol. Biol. Cell 2015, 26, 2385–2401. [Google Scholar] [CrossRef]
- Radin, I.; Kost, L.; Gey, U.; Steinebrunner, I.; Rödel, G. The Mitochondrial Copper Chaperone COX11 Has an Additional Role in Cellular Redox Homeostasis. PLoS ONE 2021, 16, e0261465. [Google Scholar] [CrossRef]
- Brischigliaro, M.; Zeviani, M. Cytochrome c Oxidase Deficiency. Biochim. Biophys. Acta-Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef]
- Lee, I.-C.; Chiang, K.-L. Clinical Diagnosis and Treatment of Leigh Syndrome Based on SURF1: Genotype and Phenotype. Antioxidants 2021, 10, 1950. [Google Scholar] [CrossRef] [PubMed]
- Čunátová, K.; Reguera, D.P.; Houštěk, J.; Mráček, T.; Pecina, P. Role of Cytochrome c Oxidase Nuclear-Encoded Subunits in Health and Disease. Physiol. Res. 2020, 69, 947–965. [Google Scholar] [CrossRef] [PubMed]
- Brix, N.; Jensen, J.M.; Pedersen, I.S.; Ernst, A.; Frost, S.; Bogaard, P.; Petersen, M.B.; Bender, L. Mitochondrial Disease Caused by a Novel Homozygous Mutation (Gly106del) in the SCO1 Gene. Neonatology 2019, 116, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Gurgel-Giannetti, J.; Oliveira, G.; Brasileiro Filho, G.; Martins, P.; Vainzof, M.; Hirano, M. Mitochondrial Cardioencephalomyopathy Due to a Novel SCO2 Mutation in a Brazilian Patient. JAMA Neurol. 2013, 70, 258. [Google Scholar] [CrossRef] [PubMed]
- Rius, R.; Bennett, N.K.; Bhattacharya, K.; Riley, L.G.; Yüksel, Z.; Formosa, L.E.; Compton, A.G.; Dale, R.C.; Cowley, M.J.; Gayevskiy, V.; et al. Biallelic Pathogenic Variants in COX11 Are Associated with an Infantile-onset Mitochondrial Encephalopathy. Hum. Mutat. 2022, 43, 1970–1978. [Google Scholar] [CrossRef]
- Shibata, A.; Kasai, M.; Hoshino, A.; Tanaka, T.; Mizuguchi, M. RANBP2 Mutation Causing Autosomal Dominant Acute Necrotizing Encephalopathy Attenuates Its Interaction with COX11. Neurosci. Lett. 2021, 763, 136173. [Google Scholar] [CrossRef]
- Battini, R.; Sgandurra, G.; Menici, V.; Scalise, R.; Olivieri, I.; Di Pietro, R.; Lucibello, S.; Giannini, M.T.; Cioni, G. Movement Disorders-Childhood Rating Scale 4-18 Revised in Children with Dyskinetic Cerebral Palsy. Eur. J. Phys. Rehabil. Med. 2020, 56, 272–278. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Sue, C.M.; Davidson, M.M.; Tanji, K.; Nishino, I.; Sadlock, J.E.; Krishna, S.; Walker, W.; Selby, J.; Glerum, D.M.; et al. Fatal Infantile Cardioencephalomyopathy with COX Deficiency and Mutations in SCO2, a COX Assembly Gene. Nat. Genet. 1999, 23, 333–337. [Google Scholar] [CrossRef]
- Dickinson, E.K.; Adams, D.L.; Schon, E.A.; Glerum, D.M. A Human SCO2 Mutation Helps Define the Role of Sco1p in the Cytochrome Oxidase Assembly Pathway. J. Biol. Chem. 2000, 275, 26780–26785. [Google Scholar] [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of Mitochondrial Respiratory Chain Enzymatic Activities on Tissues and Cultured Cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef]
- Bucher, S.; Le Guillou, D.; Allard, J.; Pinon, G.; Begriche, K.; Tête, A.; Sergent, O.; Lagadic-Gossmann, D.; Fromenty, B. Possible Involvement of Mitochondrial Dysfunction and Oxidative Stress in a Cellular Model of NAFLD Progression Induced by Benzo[a]Pyrene/Ethanol CoExposure. Oxid. Med. Cell. Longev. 2018, 2018, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, A.D.J.; Ciofi-Baffoni, S.; Banci, L.; Bertini, I.; Boelens, R.; Bonvin, A.M.J.J. Modeling Protein−Protein Complexes Involved in the Cytochrome c Oxidase Copper-Delivery Pathway. J. Proteome Res. 2007, 6, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Cantini, F.; Ciofi-Baffoni, S.; Gonnelli, L.; Mangani, S. Solution Structure of Cox11, a Novel Type of β-Immunoglobulin-like Fold Involved in CuB Site Formation of Cytochrome c Oxidase. J. Biol. Chem. 2004, 279, 34833–34839. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX Web Server: An Online Force Field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef]
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-Flex 2.0: A Web Server for Fast Simulations of Flexibility of Protein Structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt1 | Pt2 | Our Case | |
|---|---|---|---|
| Type of mutations | Homozygous c.730G>C (p.A244P) | Homozygous c.35_36delinsG (p.V12Gfs*21) | Compound heterozygous c.739C>A (p.P247T), c.766dupA (p.T256Nfs*8) |
| Last evaluation | 9 months | 3 years | 11 years |
| Age of onset | Congenital | Congenital | Early infancy |
| First manifestation | Sensorineural deafness (neonatal period) | Severe intrauterine growth retardation | Developmental delay, hypotonia, cerebellar and pyramidal signs |
| Developmental delay | Y | Y | Y |
| Microcephaly | NA | Y | Y |
| Hypotonia | Y | NA | Y |
| Feeding and gastrointestinal disorders | Poor feeding, nasogastric feeding | Gastrointestinal dysmotility and gastroesophageal reflux | N |
| Respiratory distress | Y (from 3 months) | N | N |
| Visual disturbances | Y | Y | Pale optic papilla and exotropia |
| Dyskinetic movements | Y (after 3 months) | N | Y (from 5 years) |
| Cerebellar signs | N | N | Y (from the first months of life) |
| Pyramidal signs | N | Y (increased tone and brisk deep tendon reflexes at 3 years) | Y (from the first months of life with progressive spasticity) |
| Epileptic seizures | Y (clonic seizures from the first months of life) | Y (focal seizures for 3 years) | N |
| Sleep disorders | NA | NA | Y (from 7.6 years) |
| EEG | NA | Generalized epileptiform discharges at 7 months | Posterior slow activity triggered by eye closure, slow wave sequences and paroxysmal abnormalities (activated by sleep) in the middle vertex and centro-parietal areas. During sleep, diffuse, unusual, rapid activity. |
| CSF lactate | ↑ | ↑ | NA |
| Plasma lactate | ↑ intermittently | NA | NA (↑ at MRS and UOA analysis) |
| PAA and UOA | NA | Tandem mass spectrometry and urine gas chromatography were normal | Altered * |
| Brain MRI | At the beginning, it was normal. At 3 months, new lesions in the cerebellar peduncles. At 7 months, brain atrophy, dentate nucleus lesions, and diffuse WM disease. | At 1 year, bilateral basal ganglia hyperintensity, mild brain atrophy. MRS showed lactate peak in basal ganglia. | Relapsing and remitting lesions hyperintense in T2, hypointense in T1 and with restricted diffusion in the acute phase in the peridentate WM, mesial nuclei of the thalami, CC splenium, lenticular nuclei and mesencephalic tegmentum. ↓ of grey/white matter contrast in the cerebellar hemispheres. ↑ lactate peak in the cerebellum and lenticular nuclei. |
| Death | 9 months | Still alive | Still alive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caron-Godon, C.A.; Della Vecchia, S.; Romano, A.; Doccini, S.; Dal Canto, F.; Pasquariello, R.; Rubegni, A.; Battini, R.; Santorelli, F.M.; Glerum, D.M.; et al. Novel COX11 Mutations Associated with Mitochondrial Disorder: Functional Characterization in Patient Fibroblasts and Saccharomyces cerevisiae. Int. J. Mol. Sci. 2023, 24, 16636. https://doi.org/10.3390/ijms242316636
Caron-Godon CA, Della Vecchia S, Romano A, Doccini S, Dal Canto F, Pasquariello R, Rubegni A, Battini R, Santorelli FM, Glerum DM, et al. Novel COX11 Mutations Associated with Mitochondrial Disorder: Functional Characterization in Patient Fibroblasts and Saccharomyces cerevisiae. International Journal of Molecular Sciences. 2023; 24(23):16636. https://doi.org/10.3390/ijms242316636
Chicago/Turabian StyleCaron-Godon, Chenelle A., Stefania Della Vecchia, Alessandro Romano, Stefano Doccini, Flavio Dal Canto, Rosa Pasquariello, Anna Rubegni, Roberta Battini, Filippo Maria Santorelli, D. Moira Glerum, and et al. 2023. "Novel COX11 Mutations Associated with Mitochondrial Disorder: Functional Characterization in Patient Fibroblasts and Saccharomyces cerevisiae" International Journal of Molecular Sciences 24, no. 23: 16636. https://doi.org/10.3390/ijms242316636
APA StyleCaron-Godon, C. A., Della Vecchia, S., Romano, A., Doccini, S., Dal Canto, F., Pasquariello, R., Rubegni, A., Battini, R., Santorelli, F. M., Glerum, D. M., & Nesti, C. (2023). Novel COX11 Mutations Associated with Mitochondrial Disorder: Functional Characterization in Patient Fibroblasts and Saccharomyces cerevisiae. International Journal of Molecular Sciences, 24(23), 16636. https://doi.org/10.3390/ijms242316636