

“Alphabet” Selenoproteins: Their Characteristics and Physiological Roles


Abstract
:1. Introduction
2. “Alphabet” Selenoproteins Structures
2.1. Selenoprotein I (SELENOI)
2.2. Selenoprotein O (SELENOO)
2.3. Selenoprotein N (SELENON)
2.4. Selenoproteins H, T, V, and W (SELENOH, SELENOT, SELENOV, and SELENOW)
2.5. Selenoprotein M (SELENOM) and the 15-kDa Selenoprotein of the Selenoprotein M Family (Sep15, Selenoprotein F, and SELENOF)
2.6. Selenoproteins K (SELENOK) and S (SELENOS)
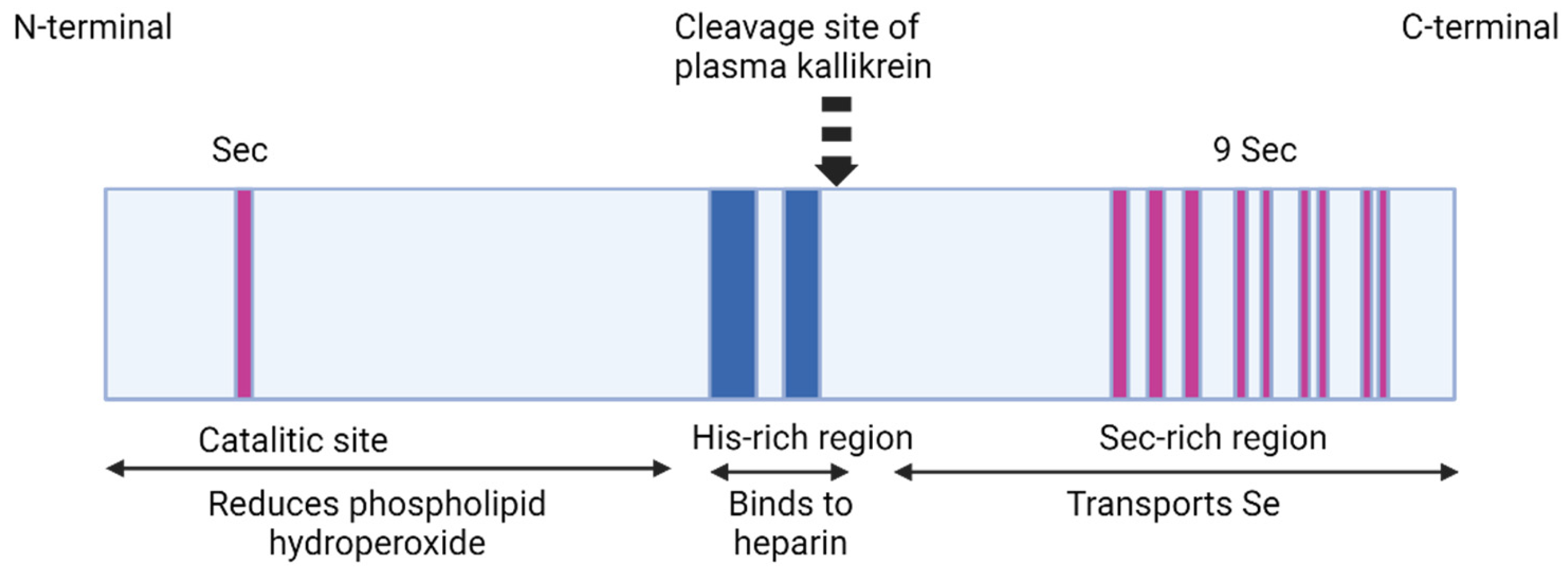
2.7. Selenoprotein P (SELENOP, Sepp1)
3. Signaling Pathways Mediated by “Alphabet” Selenoproteins
3.1. Signaling Pathways That Are Mediated by the Endoplasmic Reticulum (ER) Membrane Selenoproteins (SELENON, SELENOK, SELENOT, SELENOI)

{kind=link}
{kind=link}
{kind=link}
| Selenoprotein Name | Cellular Localization | Function |
|---|---|---|
| Selenoprotein F (Sep15, selenoprotein M family) | ER lumen | Protein folding [91] |
| Selenoprotein H | Nucleus | Nucleolar oxidoreductase [92] Redox homeostasis [92] Cell cycle regulator [93] |
| Selenoprotein I | ER membrane/Golgi apparatus | Phospholipid biosynthesis [9,10] |
| Selenoprotein K | ER membrane | ER responses to stress [94] Ca2+-dependent signal transmissions [94] Immune response process [95] |
| Selenoprotein M | ER lumen | Thiol-disulfide oxidoreductase [96] Calcium homeostasis [97] Hypothalamic signaling via leptin [98] |
| Selenoprotein N | ER membrane | Calcium signaling [82,83,84,85,86,87] Protects ER from oxidative stress [88] Role in early muscle protein folding [88] |
| Selenoprotein O | Mitochondria (in mammals) | Probable redox function [11,12,13] |
| Selenoprotein P | Bound to endothelial cell wall | Selenium transport [59,99] Oxidative stress [65] |
| Selenoprotein R (MsrB1) | Cytoplasm Nucleus | Methionine sulfoxide reductase [100] |
| Selenoprotein S | ER membrane and lumen | Anti ER stress effects [101] Antioxidant protection [102] Removal of misfolded proteins [43] Inflammatory responses [103] |
| Selenoprotein T | ER membrane | Hormone synthesis [104] Calcium mobilization [105] Redox regulation [25] |
| Selenoprotein V | Cytoplasm of testes and mammal placenta | Testes specific expression [10] |
| Selenoprotein W | Cytoplasm | Antioxidant role in oxidative stress [19] |
3.2. Signaling Pathways Mediated by the Endoplasmic Reticulum (ER) Lumen Selenoproteins (SELENOF, SELENOS, SELENOM)
3.3. Signaling Functions Mediated by Selenoproteins of the Cell Nucleus (SELENOH, SELENOR)
3.4. Signaling Pathways Mediated by Cytoplasm Selenoproteins (SELENOR)
3.5. Signaling Pathways Mediated by SELENOP
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stadtman, T.C. Selenium Biochemistry. Science 1974, 183, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular Pathways and Physiological Roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, D.L.; Choi, I.S.; Ohama, T.; Jung, J.-E.; Diamond, A.M. Selenocysteine TRNA[Ser]Sec Isoacceptors as Central Components in Selenoprotein Biosynthesis in Eukaryotes. In Selenium in Biology and Human Health; Raymond, F.B., Ed.; Springer: New York, NY, USA, 2011; pp. 25–44. [Google Scholar]
- Lee, B.J.; Worland, P.J.; Davis, J.N.; Stadtman, T.C.; Hatfield, D.L. Identification of a Selenocysteyl-TRNA(Ser) in Mammalian Cells That Recognizes the Nonsense Codon, UGA. J. Biol. Chem. 1989, 264, 9724–9727. [Google Scholar] [CrossRef] [PubMed]
- Böck, A.; Forchhammer, K.; Heider, J.; Baron, C. Selenoprotein Synthesis: An Expansion of the Genetic Code. Trends Biochem. Sci. 1991, 16, 463–467. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of Mammalian Selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Dikiy, A.; Novoselov, S.V.; Fomenko, D.E.; Sengupta, A.; Carlson, B.A.; Cerny, R.L.; Ginalski, K.; Grishin, N.V.; Hatfield, D.L.; Gladyshev, V.N. SelT, SelW, SelH, and Rdx 12: Genomics and Molecular Insights into the Functions of Selenoproteins of a Novel Thioredoxin-like Family. Biochemistry 2007, 46, 6871–6882. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Labunskyy, V.M.; Fomenko, D.E.; Araç, D.; Chelliah, Y.; Amezcua, C.A.; Rizo, J.; Gladyshev, V.N.; Deisenhofer, J. NMR Structures of the Selenoproteins Sep15 and SelM Reveal Redox Activity of a New Thioredoxin-like Family. J. Biol. Chem. 2006, 281, 3536–3543. [Google Scholar] [CrossRef] [PubMed]
- Henneberry, A.L.; Mcmaster, C.R. Cloning and Expression of a Human Choline/Ethanolaminephosphotransferase: Synthesis of Phosphatidylcholine and Phosphatidylethanolamine. Biochem. J. 1999, 339 Pt 2, 291–298. [Google Scholar] [CrossRef]
- Mariotti, M.; Ridge, P.G.; Zhang, Y.; Lobanov, A.V.; Pringle, T.H.; Guigo, R.; Hatfield, D.L.; Gladyshev, V.N. Composition and Evolution of the Vertebrate and Mammalian Selenoproteomes. PLoS ONE 2012, 7, e33066. [Google Scholar] [CrossRef]
- Han, S.J.; Lee, B.C.; Yim, S.H.; Gladyshev, V.N.; Lee, S.R. Characterization of Mammalian Selenoprotein O: A Redox-Active Mitochondrial Protein. PLoS ONE 2014, 9, e95518. [Google Scholar] [CrossRef]
- Dudkiewicz, M.; Szczepińska, T.; Grynberg, M.; Pawłowski, K. A Novel Protein Kinase-like Domain in a Selenoprotein, Widespread in the Tree of Life. PLoS ONE 2012, 7, e32138. [Google Scholar] [CrossRef] [PubMed]
- Lenart, A.; Pawłowski, K. Intersection of Selenoproteins and Kinase Signalling. Biochim. Biophys. Acta 2013, 1834, 1279–1284. [Google Scholar] [CrossRef]
- Petit, N.; Lescure, A.; Rederstorff, M.; Krol, A.; Moghadaszadeh, B.; Wewer, U.M.; Guicheney, P. Selenoprotein N: An Endoplasmic Reticulum Glycoprotein with an Early Developmental Expression Pattern. Hum. Mol. Genet. 2003, 12, 1045–1053. [Google Scholar] [CrossRef]
- Strynadka, N.C.; James, M.N. Crystal Structures of the Helix-Loop-Helix Calcium-Binding Proteins. Annu. Rev. Biochem. 1989, 58, 951–958. [Google Scholar] [CrossRef]
- Chernorudskiy, A.; Varone, E.; Francesca Colombo, S.; Fumagalli, S.; Cagnotto, A.; Cattaneo, A.; Briens, M.; Baltzinger, M.; Kuhn, L.; Bachi, A.; et al. Selenoprotein N Is an Endoplasmic Reticulum Calcium Sensor That Links Luminal Calcium Levels to a Redox Activity. Proc. Natl. Acad. Sci. USA 2020, 117, 21288–21298. [Google Scholar] [CrossRef]
- Gu, Q.P.; Sun, Y.; Ream, L.W.; Whanger, P.D. Selenoprotein W Accumulates Primarily in Primate Skeletal Muscle, Heart, Brain and Tongue. Mol. Cell Biochem. 2000, 204, 49–56. [Google Scholar] [CrossRef]
- Vendeland, S.C.; Beilstein, M.A.; Yeh, J.-Y.; Ream, W.; Whanger, P.D. Rat Skeletal Muscle Selenoprotein W: cDNA Clone and mRNA Modulation by Dietary Selenium. Proc. Natl. Acad. Sci. USA 1995, 92, 8749–8753. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.T.; Carlson, B.A.; Anderson, C.B.; Hatfield, D.L. Translational Redefinition of UGA Codons Is Regulated by Selenium Availability. J. Biol. Chem. 2013, 288, 19401–19413. [Google Scholar] [CrossRef]
- Beilstein, M.A.; Vendeland, S.C.; Barofsky, E.; Jensen, O.N.; Whanger, P.D. Selenoprotein W of Rat Muscle Binds Glutathione and an Unknown Small Molecular Weight Moiety. J. Inorg. Biochem. 1996, 61, 117–124. [Google Scholar] [CrossRef]
- Musiani, F.; Ciurli, S.; Dikiy, A. Interaction of Selenoprotein W with 14-3-3 Proteins: A Computational Approach. J. Proteome Res. 2011, 10, 968–976. [Google Scholar] [CrossRef]
- Park, Y.H.; Jeon, Y.H.; Kim, I.Y. Selenoprotein W Promotes Cell Cycle Recovery from G2 Arrest through the Activation of CDC25B. Biochim. Biophys. Acta 2012, 1823, 2217–2226. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.H.; Park, Y.H.; Kwon, J.H.; Lee, J.H.; Kim, I.Y. Inhibition of 14-3-3 Binding to Rictor of MTORC2 for Akt Phosphorylation at Ser473 Is Regulated by Selenoprotein W. Biochim. Biophys. Acta 2013, 1833, 2135–2142. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Kryukov, V.M.; Gladyshev, V.N. New Mammalian Selenocysteine-Containing Proteins Identified with an Algorithm That Searches for Selenocysteine Insertion Sequence Elements. J. Biol. Chem. 1999, 274, 33888–33897. [Google Scholar] [CrossRef] [PubMed]
- Hamieh, A.; Cartier, D.; Abid, H.; Calas, A.; Burel, C.; Bucharles, C.; Jehan, C.; Grumolato, L.; Landry, M.; Lerouge, P.; et al. Selenoprotein T Is a Novel OST Subunit That Regulates UPR Signaling and Hormone Secretion. EMBO Rep. 2017, 18, 1935–1946. [Google Scholar] [CrossRef]
- Novoselov, S.V.; Kryukov, G.V.; Xu, X.M.; Carlson, B.A.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein H Is a Nucleolar Thioredoxin-like Protein with a Unique Expression Pattern. J. Biol. Chem. 2007, 282, 11960–11968. [Google Scholar] [CrossRef] [PubMed]
- Panee, J.; Stoytcheva, Z.R.; Liu, W.; Berry, M.J. Selenoprotein H Is a Redox-Sensing High Mobility Group Family DNA-Binding Protein That Up-Regulates Genes Involved in Glutathione Synthesis and Phase II Detoxification. J. Biol. Chem. 2007, 282, 23759–23765. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; Huang, J.-Q.; Xiao, Y.; Wu, Y.-Y.; Ren, F.-Z.; Lei, X.G. Knockout of Selenoprotein V Affects Regulation of Selenoprotein Expression by Dietary Selenium and Fat Intakes in Mice. J. Nutr. 2020, 150, 483–491. [Google Scholar] [CrossRef]
- Korotkov, K.V.; Novoselov, S.V.; Hatfield, D.L.; Gladyshev, V.N. Mammalian Selenoprotein in Which Selenocysteine (Sec) Incorporation Is Supported by a New Form of Sec Insertion Sequence Element. Mol. Cell. Biol. 2002, 22, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Ferguson, A.D.; Fomenko, D.E.; Chelliah, Y.; Hatfield, D.L.; Gladyshev, V.N. A Novel Cysteine-Rich Domain of Sep15 Mediates the Interaction with UDP-Glucose:Glycoprotein Glucosyltransferase. J. Biol. Chem. 2005, 280, 37839–37845. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. The Sep15 Protein Family: Roles in Disulfide Bond Formation and Quality Control in the Endoplasmic Reticulum. IUBMB Life 2007, 59, 1–5. [Google Scholar] [CrossRef]
- Heider, J.; Bock, A. Selenium Metabolism in Micro-Organisms. Adv. Microb. Physiol. 1993, 35, 71–109. [Google Scholar] [CrossRef]
- Molinari, M.; Helenius, A. Glycoproteins Form Mixed Disulphides with Oxidoreductases during Folding in Living Cells. Nature 1999, 402, 90–93. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Yoo, M.H.; Hatfield, D.L.; Gladyshev, V.N. Sep15, a Thioredoxin-like Selenoprotein, Is Involved in the Unfolded Protein Response and Differentially Regulated by Adaptive and Acute ER Stresses. Biochemistry 2009, 48, 8458–8465. [Google Scholar] [CrossRef]
- Novoselov, S.V.; Calvisi, D.F.; Labunskyy, V.M.; Factor, V.M.; Carlson, B.A.; Fomenko, D.E.; Moustafa, M.E.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein Deficiency and High Levels of Selenium Compounds Can Effectively Inhibit Hepatocarcinogenesis in Transgenic Mice. Oncogene 2005, 24, 8003–8011. [Google Scholar] [CrossRef]
- Kumaraswamy, E.; Malykh, A.; Korotkov, K.V.; Kozyavkin, S.; Hu, Y.; Kwon, S.Y.; Moustafa, M.E.; Carlson, B.A.; Berry, M.J.; Lee, B.J.; et al. Structure-Expression Relationships of the 15-KDa Selenoprotein Gene: Possible Role of the Protein in Cancer Etiology. J. Biol. Chem. 2000, 275, 35540–35547. [Google Scholar] [CrossRef]
- Kumaraswamy, E.; Korotkov, K.V.; Diamond, A.M.; Gladyshev, V.N.; Hatfield, D.L. Genetic and Functional Analysis of Mammalian Sep15 Selenoprotein. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2002; Volume 347, pp. 187–197. [Google Scholar] [CrossRef]
- Hu, Y.J.; Korotkov, K.V.; Mehta, R.; Hatfield, D.L.; Rotimi, C.N.; Luke, A.; Prewitt, T.E.; Cooper, R.S.; Stock, W.; Vokes, E.E.; et al. Distribution and Functional Consequences of Nucleotide Polymorphisms in the 3-Untranslated Region of the Human Sep15 Gene. Cancer Res. 2001, 61, 2307–2310. [Google Scholar]
- Apostolou, S.; Klein, J.O.; Mitsuuchi, Y.; Shetler, J.N.; Poulikakos, P.I.; Jhanwar, S.C.; Kruger, W.D.; Testa, J.R. Growth Inhibition and Induction of Apoptosis in Mesothelioma Cells by Selenium and Dependence on Selenoprotein SEP15 Genotype. Oncogene 2004, 23, 5032–5040. [Google Scholar] [CrossRef]
- Irons, R.; Tsuji, P.A.; Carlson, B.A.; Ouyang, P.; Yoo, M.H.; Xu, E.M.; Hatfield, D.L.; Gladyshev, V.N.; Davis, C.D. Deficiency in the 15-KDa Selenoprotein Inhibits Tumorigenicity and Metastasis of Colon Cancer Cells. Cancer Prev. Res. 2010, 3, 630–639. [Google Scholar] [CrossRef]
- Tsuji, P.A.; Carlson, B.A.; Naranjo-Suarez, S.; Yoo, M.H.; Xu, X.M.; Fomenko, D.E.; Gladyshev, V.N.; Hatfield, D.L.; Davis, C.D. Knockout of the 15 KDa Selenoprotein Protects against Chemically-Induced Aberrant Crypt Formation in Mice. PLoS ONE 2012, 7, e50574. [Google Scholar] [CrossRef]
- Shchedrina, V.A.; Everley, R.A.; Zhang, Y.; Gygi, S.P.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein K Binds Multiprotein Complexes and Is Involved in the Regulation of Endoplasmic Reticulum Homeostasis. J. Biol. Chem. 2011, 286, 42937–42948. [Google Scholar] [CrossRef]
- Christensen, L.C.; Jensen, N.W.; Vala, A.; Kamarauskaite, J.; Johansson, L.; Winther, J.R.; Hofmann, K.; Teilum, K.; Ellgaard, L. The Human Selenoprotein VCP-Interacting Membrane Protein (VIMP) Is Non-Globular and Harbors a Reductase Function in an Intrinsically Disordered Region. J. Biol. Chem. 2012, 287, 26388–26399. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, F.; Rozovsky, S. The Intrinsically Disordered Membrane Protein Selenoprotein S Is a Reductase in Vitro. Biochemistry 2013, 52, 3051–3061. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rozovsky, S. Contribution of Selenocysteine to the Peroxidase Activity of Selenoprotein s. Biochemistry 2013, 52, 5514–5516. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.E.; Jowett, J.B.M.; Elliott, K.S.; Gao, Y.; Gluschenko, K.; Wang, J.; Azim, D.M.A.; Cai, G.; Mahaney, M.C.; Comuzzie, A.G.; et al. Genetic Variation in Selenoprotein S Influences Inflammatory Response. Nat. Genet. 2005, 37, 1234–1241. [Google Scholar] [CrossRef]
- Du, X.A.; Wang, H.M.; Dai, X.X.; Kou, Y.; Wu, R.P.; Chen, Q.; Cao, J.L.; Mo, X.Y.; Xiong, Y.M. Role of Selenoprotein S (SEPS1)-105G>A polymorphisms and PI3K/Akt Signaling Pathway in Kashin-Beck Disease. Osteoarthr. Cartil. 2015, 23, 210–216. [Google Scholar] [CrossRef]
- Santos, L.R.; Durães, C.; Mendes, A.; Prazeres, H.; Alvelos, M.I.; Moreira, C.S.; Canedo, P.; Esteves, C.; Neves, C.; Carvalho, D.; et al. A Polymorphism in the Promoter Region of the Selenoprotein S Gene (SEPS1) Contributes to Hashimoto’s Thyroiditis Susceptibility. J. Clin. Endocrinol. Metab. 2014, 99, E719–E723. [Google Scholar] [CrossRef]
- Sutherland, A.; Kim, D.H.; Relton, C.; Ahn, Y.O.; Hesketh, J. Polymorphisms in the Selenoprotein S and 15-kDa Selenoprotein Genes are Associated with Altered Susceptibility to Colorectal Cancer. Genes Nutr. 2010, 5, 215–223. [Google Scholar] [CrossRef]
- Meplan, C.; Hughes, D.J.; Pardini, B.; Naccarati, A.; Soucek, P.; Vodickova, L.; Hlavata, I.; Vrana, D.; Vodicka, P.; Hesketh, J.E. Genetic Variants in Selenoprotein Genes Increase Risk of Colorectal Cancer. Carcinogenesis 2010, 31, 1074–1079. [Google Scholar] [CrossRef]
- Mukhtar, M.; Ashfield, N.; Vodickova, L.; Vymetalkova, V.; Levy, M.; Liska, V.; Bruha, J.; Bendova, P.; O’Sullivan, J.; Doherty, G.; et al. The Associations of Selenoprotein Genetic Variants With the Risks of Colorectal Adenoma and Colorectal Cancer: Case–Control Studies in Irish and Czech Populations. Nutrients 2022, 14, 2718. [Google Scholar] [CrossRef]
- Li, J.; Zhu, Y.; Zhou, Y.; Jiang, H.; Chen, Z.; Lu, B.; Shen, X. The Sels rs34713741 Polymorphism is Associated with Susceptibility to Colorectal Cancer and Gastric Cancer: A Meta-Analysis. Genet. Test. Mol. Biomark. 2020, 24, 835–844. [Google Scholar] [CrossRef]
- Speckmann, B.; Gerloff, K.; Simms, L.; Oancea, I.; Shi, W.; McGuckin, M.A.; Radford-Smith, G.; Khanna, K.K. Selenoprotein S is a Marker but not a Regulator of Endoplasmic Reticulum Stress in Intestinal Epithelial Cells. Free Radic. Biol. Med. 2014, 67, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Zhou, X.Y.; Li, C.S.; Liu, L.Q.; Huang, S.Y.; Zhou, S.N. Selenoprotein S Expression in the Rat Brain Following Focal Cerebral Ischemia. Neurol. Sci. 2013, 34, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Rueli, R.H.L.H.; Torres, D.J.; Dewing, A.S.T.; Kiyohara, A.C.; Barayuga, S.M.; Bellinger, M.T.; Uyehara-Lock, J.H.; White, L.R.; Moreira, P.I.; Berry, M.J.; et al. Selenoprotein S Reduces Endoplasmic Reticulum Stress-Induced Phosphorylation of Tau: Potential Role in Selenate Mitigation of Tau Pathology. J. Alzheimers Dis. 2016, 55, 749–762. [Google Scholar] [CrossRef]
- Ghelichkhani, F.; Gonzales, F.A.; Kapitonova, M.A.; Schaefer-Ramadan, S.; Liu, J.; Cheng, R.; Rozovsky, S. Selenoprotein S: A Versatile Disordered Protein. Arch. Biochem. Biophys. 2022, 731, 109427. [Google Scholar] [CrossRef] [PubMed]
- Raymond, F. Burk. Effect of Dietary Selenium Level on 75Se Binding to Rat Plasma Proteins. Proc. Soc. Exp. Biol. Med. 1973, 143, 719–722. [Google Scholar] [CrossRef]
- Rotruck, J.T.; Pope, A.L.; Ganther, H.E.; Swanson, A.B.; Hafeman, D.G.; Hoekstra, W.G. Selenium: Biochemical Role as a Component of Glatathione Peroxidase. Science 1973, 179, 588–590. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Saito, Y. Selenoprotein P; P for Plasma, Prognosis, Prophylaxis, and More. Biol. Pharm. Bull. 2020, 43, 366–374. [Google Scholar] [CrossRef]
- Burk, R.F.; Kristina, E.H. Selenoprotein P: An Extracellular Protein with Unique Physical Characteristics and a Role in Selenium Homeostasis. Annu. Rev. Nutr. 2005, 25, 215–235. [Google Scholar] [CrossRef]
- Lobanov, A.V.; Hatfield, D.L.; Gladyshev, V.N. Reduced Reliance on the Trace Element Selenium during Evolution of Mammals. Genome Biol. 2008, 9, R62. [Google Scholar] [CrossRef]
- Saito, Y.; Takahashi, K. Characterization of Selenoprotein P as a Selenium Supply Protein. Eur. J. Biochem. 2002, 269, 5746–5751. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Gladyshev, V.N. Selenium Metabolism in Zebrafish: Multiplicity of Selenoprotein Genes and Expression of a Protein Containing 17 Selenocysteine Residues. Genes Cells 2000, 5, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Hayashi, T.; Tanaka, A.; Watanabe, Y.; Suzuki, M.; Saito, E.; Takahashi, K. Selenoprotein P in Human Plasma as an Extracellular Phospholipid Hydroperoxide Glutathione Peroxidase: Isolation and Enzymatic Characterization of Human Selenoprotein P. J. Biol. Chem. 1999, 274, 2866–2871. [Google Scholar] [CrossRef] [PubMed]
- Takebe, G.; Yarimizu, J.; Saito, Y.; Hayashi, T.; Nakamura, H.; Yodoi, J.; Nagasawa, S.; Takahashi, K. A Comparative Study on the Hydroperoxide and Thiol Specificity of the Glutathione Peroxidase Family and Selenoprotein P. J. Biol. Chem. 2002, 277, 41254–41258. [Google Scholar] [CrossRef] [PubMed]
- Akesson, B.; Mårtensson, B. Heparin Interacts with a Selenoprotein in Human Plasma. J. Inorg. Biochem. 1998, 33, 257–261. [Google Scholar] [CrossRef] [PubMed]
- John, L.H. The Properties of a Rat Serum Protein Labelled by the Injection of Sodium Selenite. Biochim. Biophys. Acta 1977, 500, 61–70. [Google Scholar] [CrossRef]
- Chittum, H.S.; Himeno, S.; Hill, K.E.; Burk, R.F.; Zinn, K.R. Multiple Forms of Selenoprotein P in Rat Plasma. Arch. Biochem. Biophys. 1996, 325, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Hondal, R.J.; Ma, S.; Caprioli, R.M.; Hill, K.E.; Burk, R.F. Heparin-Binding Histidine and Lysine Residues of Rat Selenoprotein P. J. Biol. Chem. 2001, 276, 15823–15831. [Google Scholar] [CrossRef]
- Arteel, G.E.; Franken, S. Binding of Selenoprotein P to Heparin: Characterization with Surface Plasmon Resonance. Biol. Chem. 2000, 381, 265–268. [Google Scholar] [CrossRef]
- Ma, S.; Hill, K.E.; Burk, R.F.; Caprioli, R.M. Mass Spectrometric Identification of N- and O-Glycosylation Sites of Full-Length Rat Selenoprotein P and Determination of Selenide−Sulfide and Disulfide Linkages in the Shortest Isoform. Biochemistry 2003, 42, 9703–9711. [Google Scholar] [CrossRef]
- Himeno, S.; Chittum, H.S.; Burk, R.F. Isoforms of Selenoprotein P in Rat Plasma: Evidence for a Full-Length Form and Another Form that Terminates at the Second UGA in the Open Reading Frame. J. Biol. Chem. 1996, 271, 15769–15775. [Google Scholar] [CrossRef]
- Ma, S.; Hill, K.E.; Caprioli, R.M.; Burk, R.F. Mass Spectrometric Characterization of Full-Length Rat Selenoprotein P and Three Isoforms Shortened at the C Terminus. Evidence That Three UGA Codons in the MRNA Open Reading Frame Have Alternative Functions of Specifying Selenocysteine Insertion or Translation Termination. J. Biol. Chem. 2002, 277, 12749–12754. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, J.; Ronga, L.; Szpunar, J.; Lobinski, R. Characterization and Quantification of Selenoprotein P: Challenges to Mass Spectrometry. Int. J. Mol. Sci. 2021, 22, 6283. [Google Scholar] [CrossRef] [PubMed]
- Kristina, E.; Lloyd, R.S.; Burk, R.F. Conserved Nucleotide Sequences in the Open Reading Frame and 3′ Untranslated Region of Selenoprotein P mRNA. Proc. Natl. Acad. Sci. USA 1993, 90, 537–541. [Google Scholar] [CrossRef]
- Saito, Y.; Sato, N.; Hirashima, M.; Takebe, G.; Nagasawa, S.; Takahashi, K. Domain Structure of Bi-Functional Selenoprotein P. Biochem. J. 2004, 381 Pt 3, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Selenoprotein P-Expression, Functions, and Roles in Mammals. Biochim. Biophys. Acta 2009, 1790, 1441–1447. [Google Scholar] [CrossRef]
- Kurokawa, S.; Bellinger, F.P.; Hill, K.E.; Burk, R.F.; Berry, M.J. Isoform-Specific Binding of Selenoprotein P to the β-Propeller Domain of Apolipoprotein e Receptor 2 Mediates Selenium Supply. J. Biol. Chem. 2014, 289, 9195–9207. [Google Scholar] [CrossRef]
- Burk, R.F.; Olson, G.E.; Hill, K.E.; Winfrey, V.P.; Motley, A.K.; Kurokawa, S. Maternal-Fetal Transfer of Selenium in the Mouse. FASEB J. 2013, 27, 3249–3256. [Google Scholar] [CrossRef]
- Hill, K.E.; Zhou, J.; Austin, L.M.; Motley, A.K.; Ham, A.J.L.; Olson, G.E.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. The Selenium-Rich C-Terminal Domain of Mouse Selenoprotein P Is Necessary for the Supply of Selenium to Brain and Testis but Not for the Maintenance of Whole Body Selenium. J. Biol. Chem. 2007, 282, 10972–10980. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Regulation of Selenium Metabolism and Transport. Annu. Rev. Nutr. 2015, 35, 109–134. [Google Scholar] [CrossRef]
- Ushioda, R.; Miyamoto, A.; Inoue, M.; Watanabe, S.; Okumura, M.; Maegawa, K.I.; Uegaki, K.; Fujii, S.; Fukuda, Y.; Umitsu, M.; et al. Redox-Assisted Regulation of Ca2+ Homeostasis in the Endoplasmic Reticulum by Disulfide Reductase ERdj5. Proc. Natl. Acad. Sci. USA 2016, 113, E6055–E6063. [Google Scholar] [CrossRef]
- Li, Y.; Camacho, P. Ca2+-Dependent Redox Modulation of SERCA 2b by ERp57. J. Cell Biol. 2004, 164, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α-Mediated Stimulation of Inositol 1,4,5-Triphosphate Receptor Activity in Endoplasmic Reticulum Stress-Induced Apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Higo, T.; Hattori, M.; Nakamura, T.; Natsume, T.; Michikawa, T.; Mikoshiba, K. Subtype-Specific and ER Lumenal Environment-Dependent Regulation of Inositol 1,4,5-Trisphosphate Receptor Type 1 by ERp44. Cell 2005, 120, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.A.; Hess, D.T.; Nogueira, L.; Yong, S.; Bowles, D.E.; Eu, J.; Laurita, K.R.; Meissner, G.; Stamler, J.S. Oxygen-Coupled Redox Regulation of the Skeletal Muscle Ryanodine Receptor-Ca 2+ Release Channel by NADPH Oxidase 4. Proc. Natl. Acad. Sci. USA 2011, 108, 16098–16103. [Google Scholar] [CrossRef]
- Chernorudskiy, A.L.; Zito, E. Regulation of Calcium Homeostasis by ER Redox: A Close-Up of the ER/Mitochondria Connection. J. Mol. Biol. 2017, 429, 620–632. [Google Scholar] [CrossRef]
- Marino, M.; Stoilova, T.; Giorgi, C.; Bachi, A.; Cattaneo, A.; Auricchio, A.; Pinton, P.; Zito, E. SEPN1, an Endoplasmic Reticulum-Localized Selenoprotein Linked to Skeletal Muscle Pathology, Counteracts Hyperoxidation by Means of Redox-Regulating SERCA2 Pump Activity. Hum. Mol. Genet. 2014, 24, 1843–1855. [Google Scholar] [CrossRef]
- Pozzer, D.; Varone, E.; Chernorudskiy, A.; Schiarea, S.; Missiroli, S.; Giorgi, C.; Pinton, P.; Canato, M.; Germinario, E.; Nogara, L.; et al. A Maladaptive ER Stress Response Triggers Dysfunction in Highly Active Muscles of Mice with SELENON Loss. Redox Biol. 2019, 20, 354–366. [Google Scholar] [CrossRef]
- Stanishevska, N.V. Selenoproteins and Their Emerging Roles in Signaling Pathways. Regul. Mech. Biosyst. 2020, 11, 186–189. [Google Scholar] [CrossRef]
- Bang, J.; Huh, J.H.; Na, J.W.; Lu, Q.; Carlson, B.A.; Tobe, R.; Tsuji, P.A.; Gladyshev, V.N.; Hatfield, D.L.; Lee, B.J. Cell Proliferation and Motility Are Inhibited by G1 Phase Arrest in 15-KDa Selenoprotein-Deficient Chang Liver Cells. Mol. Cells 2015, 38, 457–465. [Google Scholar] [CrossRef]
- Cox, A.G.; Tsomides, A.; Kim, A.J.; Saunders, D.; Hwang, K.L.; Evason, K.J.; Heidel, J.; Brown, K.K.; Yuan, M.; Lien, E.C.; et al. Selenoprotein H Is an Essential Regulator of Redox Homeostasis That Cooperates with P53 in Development and Tumorigenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E5562–E5571. [Google Scholar] [CrossRef]
- Bertz, M.; Kühn, K.; Koeberle, S.C.; Müller, M.F.; Hoelzer, D.; Thies, K.; Deubel, S.; Thierbach, R.; Kipp, A.P. Selenoprotein H Controls Cell Cycle Progression and Proliferation of Human Colorectal Cancer Cells. Free Radic. Biol. Med. 2018, 127, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Fredericks, G.J.; Hoffmann, F.K.W.; Rose, A.H.; Osterheld, H.J.; Hess, F.M.; Mercier, F.; Hoffmann, P.R. Stable Expression and Function of the Inositol 1,4,5-Triphosphate Receptor Requires Palmitoylation by a DHHC6/Selenoprotein K Complex. Proc. Natl. Acad. Sci. USA 2014, 111, 16478–16483. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, R.; Huang, Y.; Wang, M.; Yang, F.; Huang, D.; Wu, C.; Li, Y.; Tang, Y.; Zhang, R.; et al. Selenoprotein K Modulate Intracellular Free Ca2+ by Regulating Expression of Calcium Homoeostasis Endoplasmic Reticulum Protein. Biochem. Biophys. Res. Commun. 2017, 484, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, E.; Accardo, M.; Capone, F.; Colonna, G.; Castello, G.; Costantini, S. Assessment of the Selenoprotein M (SELM) over-Expression on Human Hepatocellular Carcinoma Tissues by Immunohistochemistry. Eur. J. Histochem. 2014, 58, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.A.; Bellinger, F.P.; Berry, M.J. The Neuroprotective Functions of Selenoprotein M and Its Role in Cytosolic Calcium Regulation. Antioxid. Redox Signal. 2010, 12, 809–818. [Google Scholar] [CrossRef]
- Gong, T.; Hashimoto, A.C.; Sasuclark, A.R.; Khadka, V.S.; Gurary, A.; Pitts, M.W. Selenoprotein M Promotes Hypothalamic Leptin Signaling and Thioredoxin Antioxidant Activity. Antioxid. Redox Signal. 2021, 35, 775–787. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E.; Motley, A.K.; Winfrey, V.P.; Kurokawa, S.; Mitchell, S.L.; Zhang, W. Selenoprotein P and Apolipoprotein e Receptor-2 Interact at the Blood-Brain Barrier and also within the Brain to Maintain an Essential Selenium Pool That Protects against Neurodegeneration. FASEB J. 2014, 28, 3579–3588. [Google Scholar] [CrossRef]
- Martinez, Y.; Li, X.; Liu, G.; Bin, P.; Yan, W.; Mas, D.; Valdivie, M.; Hu, C.A.; Ren, W.; Yin, Y. The role of methionine on metabolism, oxidative stress, and diseases. Amino. Acids. 2017, 49, 2091–2098. [Google Scholar] [CrossRef]
- Men, L.; Yu, S.; Yao, J.; Li, Y.; Ren, D.; Du, J. Selenoprotein S Protects against Adipocyte Death through Mediation of the IRE1α-SXBP1 Pathway. Biochem. Biophys. Res. Commun. 2018, 503, 2866–2871. [Google Scholar] [CrossRef]
- Li, X.; Chen, M.; Yang, Z.; Wang, W.; Lin, H.; Xu, S. Selenoprotein S Silencing Triggers Mouse Hepatoma Cells Apoptosis and Necrosis Involving in Intracellular Calcium Imbalance and ROS-mPTP-ATP. Biochim. Biophys. Acta Gene Subj. 2018, 1862, 2113–2123. [Google Scholar] [CrossRef]
- Ye, Y.; Bian, W.; Fu, F.; Hu, J.; Liu, H. Selenoprotein S Inhibits Inflammation-Induced Vascular Smooth Muscle Cell Calcification. J. Biol. Inorg. Chem. 2018, 23, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Anouar, Y.; Lihrmann, I.; Falluel-Morel, A.; Boukhzar, L. Selenoprotein T Is a Key Player in ER Proteostasis, Endocrine Homeostasis and Neuroprotection. Free Radic. Biol. Med. 2018, 127, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Grumolato, L.; Ghzili, H.; Montero-Hadjadje, M.; Gasman, S.; Lesage, J.; Tanguy, Y.; Galas, L.; Ait-Ali, D.; Leprince, J.; Guérineau, N.C.; et al. Selenoprotein T Is a PACAP-regulated Gene Involved in Intracellular Ca2+ Mobilization and Neuroendocrine Secretion. FASEB J. 2008, 22, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, C.; Gu, G.; Wang, Q.; Guo, M. Selenoprotein N Was Required for the Regulation of Selenium on the Uterine Smooth Muscle Contraction in Mice. Biol. Trace Elem. Res. 2018, 183, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Rederstorff, M.; Castets, P.; Arbogast, S.; Lainé, J.; Vassilopoulos, S.; Beuvin, M.; Dubourg, O.; Vignaud, A.; Ferry, A.; Krol, A.; et al. Increased Muscle Stress-Sensitivity Induced by Selenoprotein N Inactivation in Mouse: A Mammalian Model for SEPN1-Related Myopathy. PLoS ONE 2011, 6, e23094. [Google Scholar] [CrossRef] [PubMed]
- Scoto, M.; Cirak, S.; Mein, R.; Feng, L.; Manzur, A.Y.; Robb, S.; Childs, A.-M.; Quinlivan, R.M.; Roper, H.; Jones, D.H.; et al. SEPN1-Related Myopathies: Clinical Course in a Large Cohort of Patients. Neurology 2011, 76, 2073–2078. [Google Scholar] [CrossRef]
- Bosl, M.R.; Takaku, K.; Oshima, M.; Nishimura, S.; Taketo, M.M. Early Embryonic Lethality Caused by Targeted Disruption of the Mouse Selenocysteine tRNA Gene (Trsp). Proc. Natl. Acad. Sci. USA 1997, 94, 5531–5534. [Google Scholar] [CrossRef]
- Kim, L.K.; Matsufuji, T.; Matsufuji, S.; Carlson, B.A.; Kim, S.S.; Hatfield, D.L.; Li, B.J. Methylation of the Ribosyl Moiety at Position 34 of Selenocysteine tRNA[Ser]Sec is Governed by Both Primary and Tertiary Structure. RNA 2000, 6, 1306–1315. [Google Scholar] [CrossRef]
- Warner, G.J.; Berry, M.J.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Faust, J.R. Inhibition of Selenoprotein Synthesis by Selenocysteine tRNA[Ser]Sec Lacking Isopentenyladenosine. J. Biol. Chem. 2000, 275, 28110–28119. [Google Scholar] [CrossRef]
- Moustafa, M.E.; Carlson, B.A.; El-Saadani, M.A.; Kryukov, G.V.; Sun, Q.A.; Harney, J.W.; Hill, K.E.; Combs, G.F.; Feigenbaum, L.; Mansur, D.B.; et al. Selective Inhibition of Selenocysteine tRNA Maturation and Selenoprotein Synthesis in Transgenic Mice Expressing Isopentenyladenosine-Deficient Selenocysteine tRNA. Mol. Cell. Biol. 2001, 21, 3840–3852. [Google Scholar] [CrossRef]
- Hornberger, T.A.; McLoughlin, T.J.; Leszczynski, J.K.; Armstrong, D.D.; Jameson, R.R.; Bowen, P.E.; Hwang, E.-S.; Hou, H.; Moustafa, M.E.; Carlson, B.A.; et al. Selenoprotein-Deficient Transgenic Mice Exhibit Enhanced Exercise-Induced Muscle Growth. J. Nutr. 2003, 133, 3091–3097. [Google Scholar] [CrossRef] [PubMed]
- Marciel, M.P.; Khadka, V.S.; Deng, Y.; Kilicaslan, P.; Pham, A.; Bertino, P.; Lee, K.; Chen, S.; Glibetic, N.; Hoffmann, F.W.; et al. Selenoprotein K Deficiency Inhibits Melanoma by Reducing Calcium Flux Required for Tumor Growth and Metastasis. Oncotarget 2018, 9, 30937. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Hoffmann, F.W.; Kumar, M.; Huang, Z.; Roe, K.; Nguyen-Wu, E.; Hashimoto, A.S.; Hoffman, P.R. Selenoprotein K Knockout Mice Exhibit Deficient Calcium Flux in Immune Cells and Impaired Immune Responses. J. Immunol. 2011, 186, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Horibata, Y.; Ando, H.; Sugimoto, H. Locations and Contributions of the Phosphotransferases EPT1 and CEPT1 to the Biosynthesis of Ethanolamine Phospholipids. J. Lipid Res. 2020, 61, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, F.; Smith, T.K. The Kennedy Pathway-De Novo Synthesis of Phosphatidylethanolamine and Phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Martinez-Rodriguez, V.; Hoffman, P.R. Roles for Selenoprotein I and Ethanolamine Phospholipid Synthesis in T Cell Activation. Int. J. Mol. Sci. 2021, 22, 11174. [Google Scholar] [CrossRef] [PubMed]
- Nunes, L.G.A.; Pitts, M.W.; Hoffman, P.R. Selenoprotein I (Selenoi) as A Critical Enzyme in The Central Nervous System. Arch. Biochem. Biophys. 2022, 729, 109376. [Google Scholar] [CrossRef]
- Avery, J.C.; Yamazaki, Y.; Hoffman, F.W.; Folgelgren, B.; Hoffman, P.R. Selenoprotein I is Essential for Murine Embryogenesis. Arch. Biochem. Biophys. 2020, 689, 108444. [Google Scholar] [CrossRef]
- Meyyazhagan, A.; Orlacchio, A. Hereditary Spastic Paraplegia: An Update. Int. J. Mol. Sci. 2022, 23, 1697. [Google Scholar] [CrossRef]
- Rickman, O.J.; Baple, E.L.; Crosby, A.H. Lipid Metabolic Pathways Converge in Motor Neuron Degenerative Diseases. Brain 2020, 143, 1073–1087. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, K.; Xing, G.; Li, L.; Ma, B.; Hu, Z.; Duan, L.; Liu, X. Phospholipid Remodeling is Critical for Stem Cell Pluripotency by Facilitating Mesenchymal-to-Epithelial Transition. Sci. Adv. 2019, 5, eaax7525. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Rong, X.; Palladino, E.N.; Wang, J.; Fogelman, A.M.; Martín, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kwon, J.H.; Jeon, Y.H.; Ko, K.Y.; Lee, S.R.; Kim, I.Y. Pro178 and Pro183 of Selenoprotein S Are Essential Residues for Interaction with P97(VCP) during Endoplasmic Reticulum-Associated Degradation. J. Biol. Chem. 2014, 289, 13758–13768. [Google Scholar] [CrossRef]
- Cui, S.; Men, L.; Li, Y.; Zhong, Y.; Yu, S.; Li, F.; Du, J. Selenoprotein S Attenuates Tumor Necrosis Factor-a-Induced Dysfunction in Endothelial Cells. Mediat. Inflamm. 2018, 2018, 1625414. [Google Scholar] [CrossRef]
- Gan, F.; Hu, Z.; Zhou, Y.; Huang, K. Overexpression and Low Expression of Selenoprotein S Impact Ochratoxin A-Induced Porcine Cytotoxicity and Apoptosis in Vitro. J. Agric. Food Chem. 2017, 65, 6972–6981. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Xie, S.; Xu, F.; Liu, A.; Wang, Y.; Chen, D.; Pan, Y.; Huang, L.; Peng, D.; Wang, X.; et al. Toxicity, Oxidative stress and Metabolism. Food Chem. Toxicol. 2018, 112, 320–331. [Google Scholar] [CrossRef]
- Darbuka, E.; Gürkaşlar, C.; Yaman, I. Ochratoxin A Induces ERK1/2 Phosphorylation-Dependent Apoptosis Through NF-κB/ERK Axis In Human Proximal Tubule HK-2 Cell Line. Toxicon 2021, 199, 79–86. [Google Scholar] [CrossRef]
- Pitts, M.W.; Reeves, M.A.; Hashimoto, A.C.; Ogawa, A.; Kremer, P.; Seale, L.A.; Berry, M.J. Deletion of Selenoprotein M Leads to Obesity Without Cognitive Deficits. JBC 2013, 288, 26121–26134. [Google Scholar] [CrossRef]
- Du, X.; Li, H.; Wang, Z.; Qiu, S.; Liu, Q.; Ni, J. Selenoprotein P and Selenoprotein M Block Zn2+-Mediated Aβ42 Aggregation and Toxicity. Metallomics 2013, 5, 861–870. [Google Scholar] [CrossRef]
- Jiang, H.; Shi, Q.Q.; Ge, L.Y.; Zhuang, Q.F.; Xue, D.; Xu, H.Y.; He, X.Z. Selenoprotein M Stimulates the Proliferative and Metastatic Capacities of Renal Cell Carcinoma through Activating the PI3K/AKT/MTOR Pathway. Cancer Med. 2019, 8, 4836–4844. [Google Scholar] [CrossRef]
- Bang, J.; Jang, M.; Huh, J.H.; Na, J.W.; Shim, M.; Carlson, B.A.; Tobe, R.; Tsuji, P.A.; Gladyshev, V.N.; Hatfield, D.L.; et al. Deficiency of the 15-KDa Selenoprotein Led to Cytoskeleton Remodeling and Non-Apoptotic Membrane Blebbing through a RhoA/ROCK Pathway. Biochem. Biophys. Res. Commun. 2015, 456, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Huang, Y.; Zou, C.; Wu, Y.; Huang, Y.; Ni, J.; Tian, J. Transcriptional Regulation of Selenoprotein F by Heat Shock Factor 1 during Selenium Supplementation and Stress Response. Cells 2019, 8, 479. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Kim, M.; Youn, B.S.; Lee, N.S.; Park, J.W.; Lee, I.K.; Lee, Y.S.; Kim, J.B.; Cho, Y.M.; Lee, H.K.; et al. Glutathione peroxidase 3 mediates the antioxidant effect of peroxisome proliferator-activated receptor gamma in human skeletal muscle cells. Mol. Cell. Biol. 2009, 29, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Hansel, A.; Heinemann, S.H.; Hoshi, T. Heterogenity and function of mammalian MSRs: Enzymes for repair, protection and regulation. J. Nutr. Biochem. 2005, 1703, 239–247. [Google Scholar]
- Bin, P.; Huang, R.; Zhou, X. Oxidation Resistance of the Sulfur Amino Acids: Methionine and Cysteine. Biomed. Res. Int. 2017, 2017, 9584932. [Google Scholar] [CrossRef]
- Jiang, B.; Moskovitz, J. The Functions of the Mammalian Methionine Sulfoxide Reductase System and Related Diseases. Antioxidants 2018, 7, 122. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Stadtman, T.C.; Hatfield, D.L.; Jeang, K.T. Levels of major selenoproteins in T cells decrease during HIV infection and low molecular mass selenium compounds increase. Proc. Natl. Acad. Sci. USA 1999, 96, 835–839. [Google Scholar] [CrossRef]
- Lourenço dos Santos, S.; Petropoulos, I.; Friguet, B. The Oxidized Protein Repair Enzymes Methionine Sulfoxide Reductases and Their Roles in Protecting against Oxidative Stress, in Ageing and in Regulating Protein Function. Antioxidants 2018, 7, 191. [Google Scholar] [CrossRef]
- Hung, R.J.; Spaeth, C.S.; Yesilyurt, H.G.; Terman, J.R. SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 2013, 15, 1445–1454. [Google Scholar] [CrossRef]
- Zhang, Y.; Roh, Y.J.; Han, S.J.; Park, I.; Lee, H.M.; Ok, Y.S.; Lee, B.C.; Lee, S.R. Role of Selenoproteins in Redox Regulation of Signaling and the Antioxidant System: A Review. Antioxidants 2020, 9, 383. [Google Scholar] [CrossRef]
- Jia, Y.; Zhou, J.; Liu, H.; Huang, K. Effect of methionine sulfoxide reductase B1 (SelR) gene silencing on peroxynitrite-induced F-actin disruption in human lens epithelial cells. Biochem. Biophys. Res. Commun. 2014, 443, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Liu, H.; Zhou, J.; Huang, K. Selenoprotein R Protects Human Lens Epithelial Cells against D-Galactose-Induced Apoptosis by Regulating Oxidative Stress and Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2016, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Lee, S.G.; Choo, M.K.; Kim, J.H.; Lee, H.M.; Kim, S.; Fomenko, D.E.; Kim, H.Y.; Park, J.M.; Gladyshev, V.N. Selenoprotein MsrB1 promotes anti-inflammatory cytokine gene expression in macrophages and controls immune response in vivo. Sci. Rep. 2017, 7, 5119. [Google Scholar] [CrossRef] [PubMed]
- Achilli, C.; Ciana, A.; Minetti, G. Brain, immune system and selenium: A starting point for a new diagnostic marker for Alzheimer’s disease? Oxid. Med. Cell. Longev. 2018, 138, 223–226. [Google Scholar] [CrossRef]
- He, Q.; Li, H.; Meng, F.; Sun, X.; Feng, X.; Chen, J.; Li, L.; Liu, J. Methionine Sulfoxide Reductase B1 Regulates Hepatocellular Carcinoma Cell Proliferation and Invasion via the Mitogen-Activated Protein Kinase Pathway and Epithelial-Mesenchymal Transition. Oxid. Med. Cell. Longev. 2018, 2018, 5287971. [Google Scholar] [CrossRef]
- Li, H.; He, Q.; Meng, F.; Feng, X.; Chen, J.; Li, L.; Liu, J. Methionine sulfoxide reductase B1 regulates proliferation and invasion by affecting mitogen-activated protein kinase pathway and epithelial-mesenchymal transition in u2os cells. Biochem. Biophys. Res. Commun. 2018, 496, 806–813. [Google Scholar] [CrossRef]
- Fomenko, D.E.; Novoselov, S.V.; Natarajan, S.K.; Lee, B.C.; Koc, A.; Carlson, B.A.; Lee, T.H.; Kim, H.Y.; Hatfield, D.L.; Gladyshev, V.N. MsrB1 (methionine-R-sulfoxide reductase 1) knock-out mice: Roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J. Biol. Chem. 2009, 284, 5986–5993. [Google Scholar] [CrossRef]
- Kim, K.Y.; Kwak, G.H.; Singh, M.P.; Gladyshev, V.N.; Kim, H.Y. Selenoprotein MsrB1 deficiency exacerbates acetaminophen-induced hepatotoxicity via increased oxidative damage. Arch. Biochem. Biophys. 2017, 634, 69–75. [Google Scholar] [CrossRef]
- Olson, G.E.; Winfrey, V.P.; Hill, K.E.; Burk, R.F. Megalin Mediates Selenoprotein P Uptake by Kidney Proximal Tubule Epithelial Cells. J. Biol. Chem. 2008, 283, 6854–6860. [Google Scholar] [CrossRef]
- Olson, G.E.; Winfrey, V.P.; NagDas, S.K.; Hill, K.E.; Burk, R.F. Apolipoprotein E Receptor-2 (ApoER2) Mediates Selenium Uptake from Selenoprotein P by the Mouse Testis. J. Biol. Chem. 2007, 282, 12290–12297. [Google Scholar] [CrossRef]
- Behne, D.; Hilmert, H.; Scheid, S.; Gessner, H.; Elger, W. Evidence for Specific Selenium Target Tissues and New Biologically Important Selenoproteins. Biochim. Biophys. Acta. 1988, 966, 12–21. [Google Scholar] [CrossRef]
- Nakayama, A.; Hill, K.E.; Austin, L.M.; Motley, A.K.; Burk, R.F. All Regions of Mouse Brain Are Dependent on Selenoprotein P for Maintenance of Selenium. J. Nutr. 2007, 137, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E.; Olson, G.E.; Weeber, E.J.; Motley, A.K.; Winfrey, V.P.; Austin, L.M. Deletion of Apolipoprotein E Receptor-2 in Mice Lowers Brain Selenium and Causes Severe Neurological Dysfunction and Death When a Low-Selenium Diet Is Fed. J. Neurosci. 2007, 27, 6207–6211. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. Deletion of Selenoprotein P Alters Distribution of Selenium in the Mouse. J. Biol. Chem. 2003, 278, 13640–13646. [Google Scholar] [CrossRef]
- Schomburg, L.; Schweizer, U.; Holtmann, B.; Flohe, R.L.; Sendtner, M.; Köhrle, J. Gene Disruption Discloses Role of Selenoprotein P in Selenium Delivery to Target Tissues. Biochem. J. 2003, 370 Pt 2, 397–402. [Google Scholar] [CrossRef]
- Pitts, M.W.; Raman, A.V.; Hashimoto, A.C.; Todorovic, C.; Nichols, R.A.; Berry, M.J. Deletion of Selenoprotein P Results in Impaired Function of Parvalbumin Interneurons and Alterations in Fear Learning and Sensorimotor Gating. Neuroscience 2012, 208, 58–68. [Google Scholar] [CrossRef]
- Gong, T.; Torres, D.J.; Berry, M.J.; Pitts, M.W. Hypothalamic Redox Balance and Leptin Signaling-Emerging Role of Selenoproteins. Free Radic. Biol. Med. 2018, 127, 172–181. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dogaru, C.B.; Muscurel, C.; Duță, C.; Stoian, I. “Alphabet” Selenoproteins: Their Characteristics and Physiological Roles. Int. J. Mol. Sci. 2023, 24, 15992. https://doi.org/10.3390/ijms242115992
Dogaru CB, Muscurel C, Duță C, Stoian I. “Alphabet” Selenoproteins: Their Characteristics and Physiological Roles. International Journal of Molecular Sciences. 2023; 24(21):15992. https://doi.org/10.3390/ijms242115992
Chicago/Turabian StyleDogaru, Carmen Beatrice, Corina Muscurel, Carmen Duță, and Irina Stoian. 2023. "“Alphabet” Selenoproteins: Their Characteristics and Physiological Roles" International Journal of Molecular Sciences 24, no. 21: 15992. https://doi.org/10.3390/ijms242115992
APA StyleDogaru, C. B., Muscurel, C., Duță, C., & Stoian, I. (2023). “Alphabet” Selenoproteins: Their Characteristics and Physiological Roles. International Journal of Molecular Sciences, 24(21), 15992. https://doi.org/10.3390/ijms242115992