

Urokinase-Type Plasminogen Activator Receptor Regulates Prosurvival and Angiogenic Properties of Cardiac Mesenchymal Stromal Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
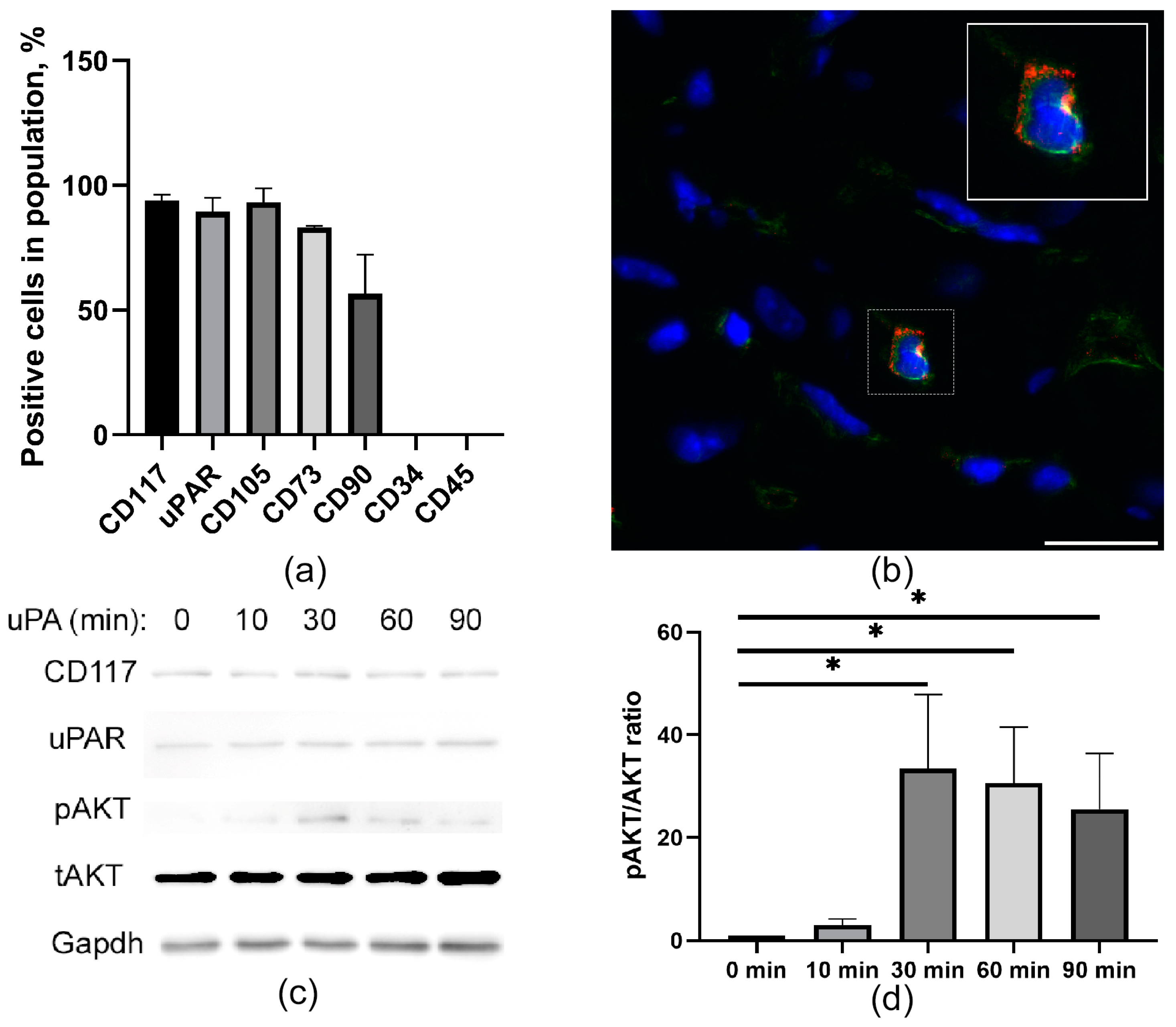
2.1. CD117+ MPCs Express an uPAR Receptor
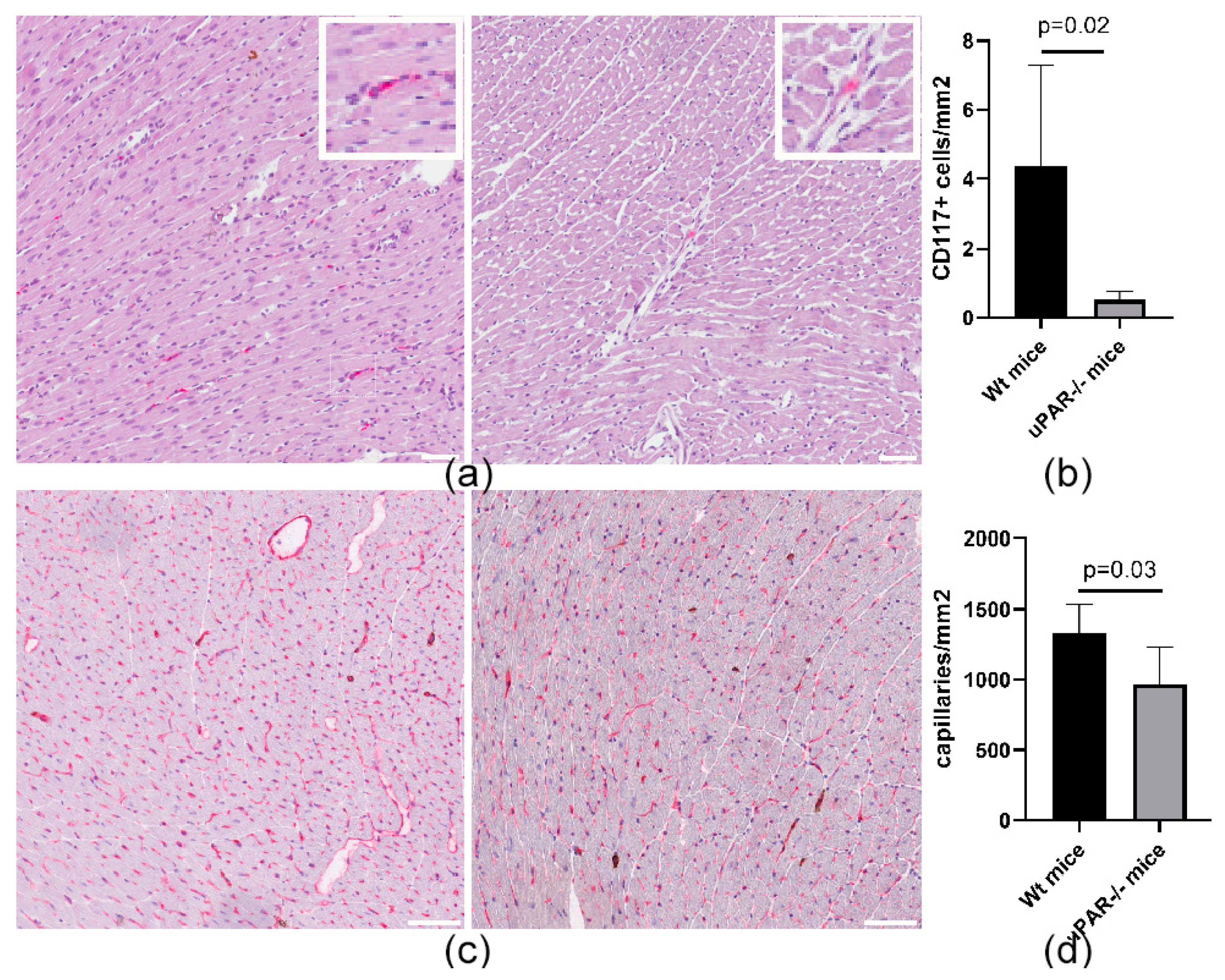
2.2. uPAR−/− Mice Are Characterized by a Reduced Number of CD117+ MPCs in the Heart, Compared to Wild-Type (uPAR+/+) Animals
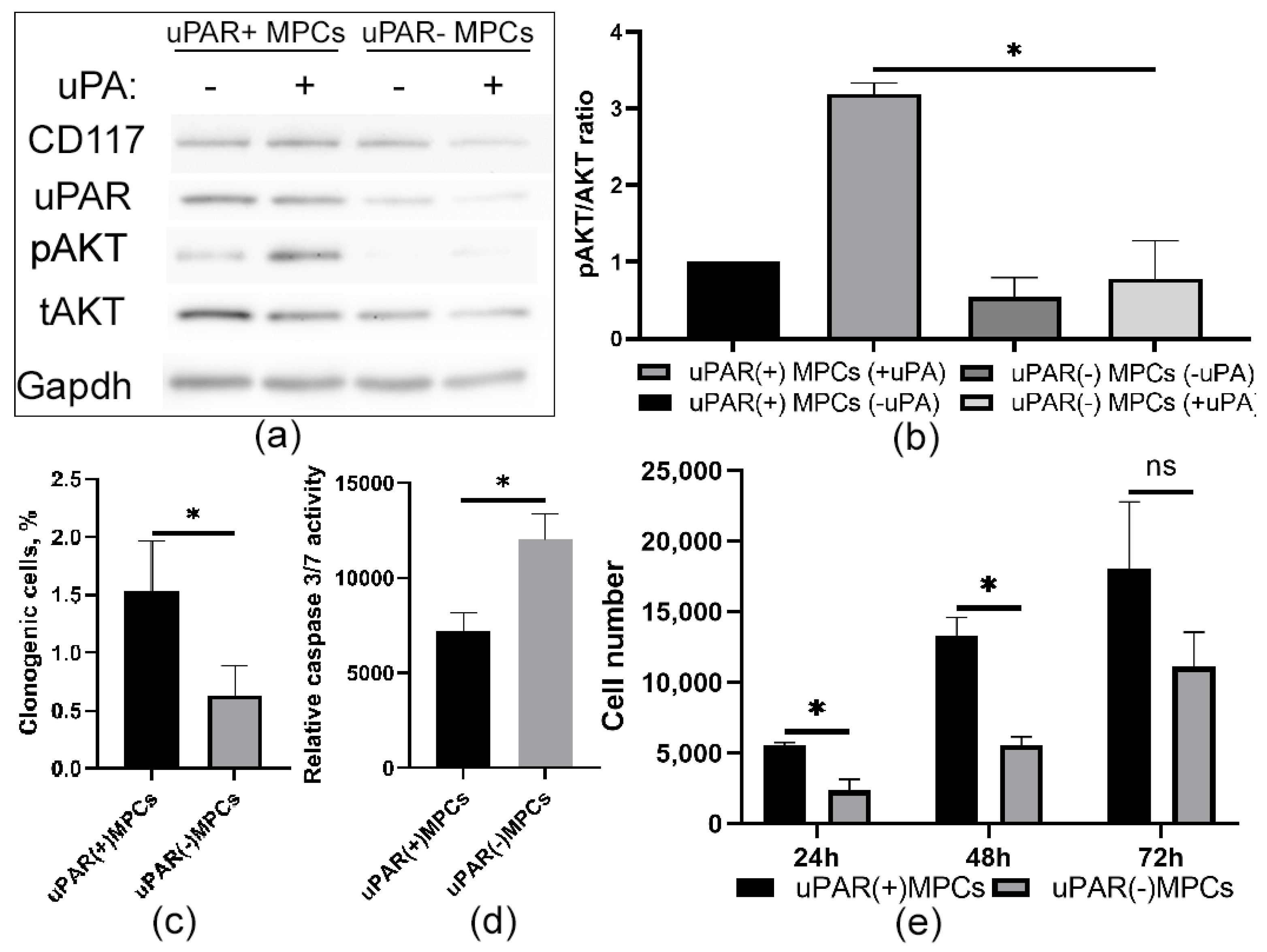
2.3. Knockdown of uPAR Is Accompanied by a Decrease in Prosurvival Properties in MPCs
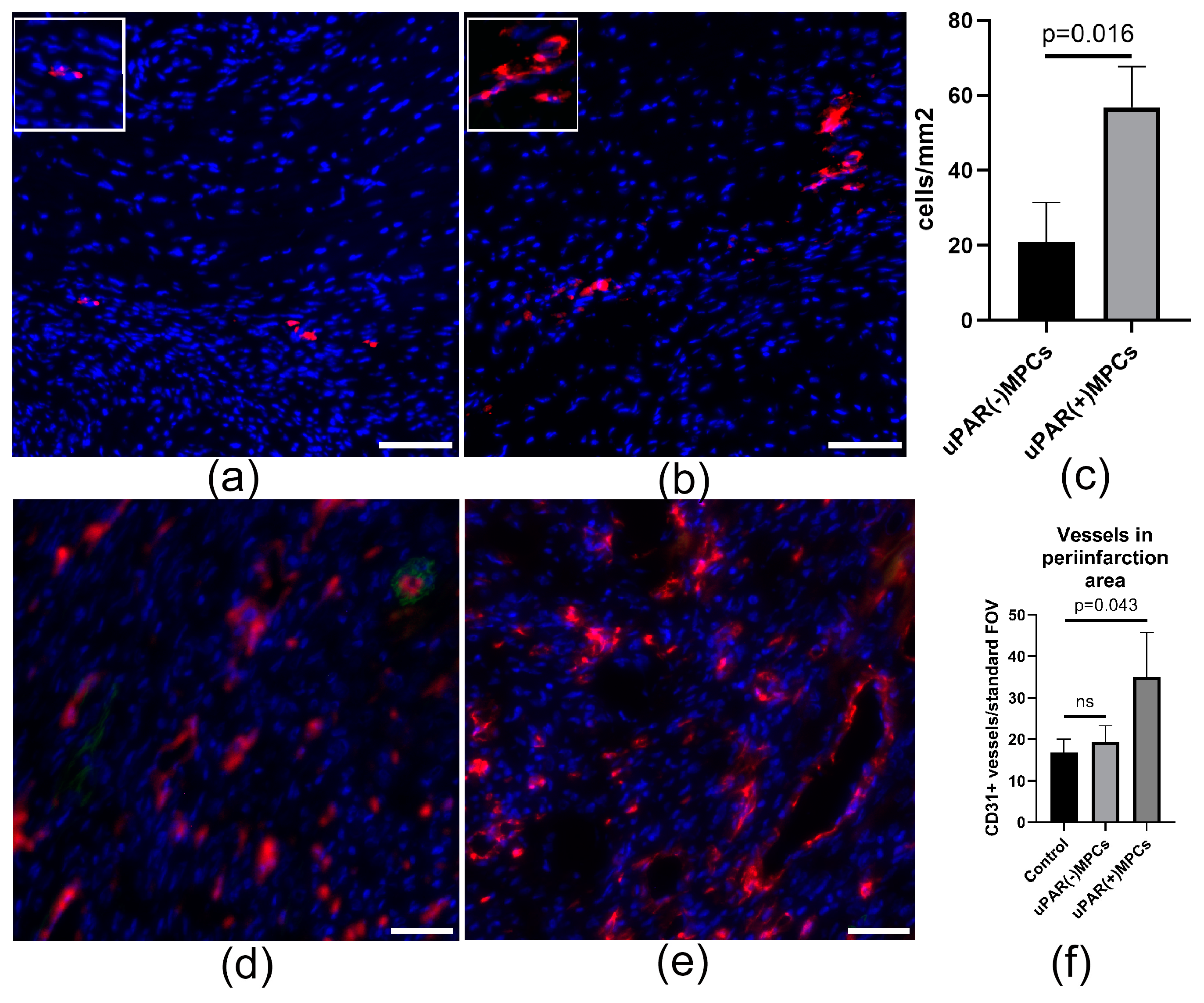
2.4. uPAR Knockdown Reduces MPCs Posttransplantation Retention and Decreases Reparative Angiogenesis Activity after Myocardial Infarction
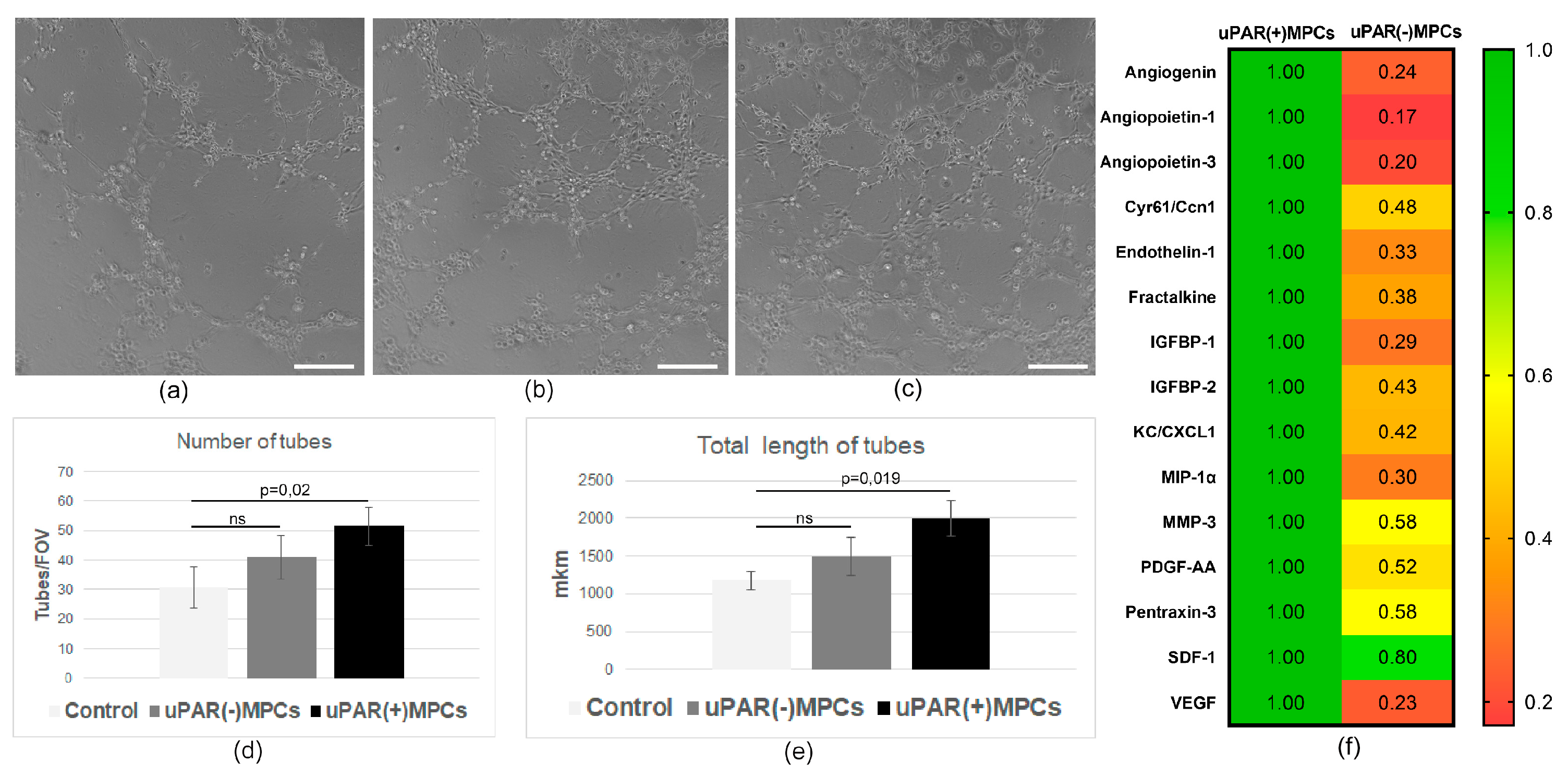
2.5. uPAR−/− MPCs Showed Reduced Proangiogenic Secretory Activity and Limited Ability to Stimulate Angiogenesis In Vivo
3. Discussion
4. Materials and Methods
4.1. Animals and Ethics Statement
4.2. CD117+ MPCs Isolation
4.3. uPAR Downregulation in MPCs
4.4. Clonogenic Assay
4.5. MTT—Assay
4.6. Caspase Assay
4.7. Western Blot Protocol
4.8. Flow Cytometry
4.9. Myocardial Infarction Model and Cell Transplantation
4.10. Posttransplantstion MPCs Retention and Angiogenesis Analysis
4.11. Immunohistochemical Analysis
4.12. MPCs Secretion Analysis
4.13. Mouse Neonatal Cardiac Endothelial Cell Isolation and Tube Assay
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Urbanek, K.; Cesselli, D.; Rota, M.; Nascimbene, A.; De Angelis, A.; Hosoda, T.; Bearzi, C.; Boni, A.; Bolli, R.; Kajstura, J.; et al. Stem cell niches in the adult mouse heart. Proc. Natl. Acad. Sci. USA 2006, 103, 9226–9231. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, N.; Smith, A.J.; Waring, C.D.; Hasan, M.K.; Miyamoto, S.; Matsuoka, R.; Ellison, G.M. c-kitpos GATA-4 High Rat Cardiac Stem Cells Foster Adult Cardiomyocyte Survival through IGF-1 Paracrine Signalling. PLoS ONE 2010, 5, e14297. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef] [PubMed]
- Vicinanza, C.; Aquila, I.; Scalise, M.; Cristiano, F.; Marino, F.; Cianflone, E.; Mancuso, T.; Marotta, P.; Sacco, W.; Lewis, F.C.; et al. Adult cardiac stem cells are multipotent and robustly myogenic: C-kit expression is necessary but not sufficient for their identification. Cell Death Differ. 2017, 24, 2101–2116. [Google Scholar] [CrossRef] [PubMed]
- Ellison, G.M.; Torella, D.; Dellegrottaglie, S.; Perez-Martinez, C.; Perez de Prado, A.; Vicinanza, C.; Purushothaman, S.; Galuppo, V.; Iaconetti, C.; Waring, C.D.; et al. Endogenous cardiac stem cell activation by insulin-like growth factor-1/hepatocyte growth factor intracoronary injection fosters survival and regeneration of the infarcted pig heart. J. Am. Coll. Cardiol. 2011, 58, 977–986. [Google Scholar] [CrossRef]
- Smith, A.J.; Lewis, F.C.; Aquila, I.; Waring, C.D.; Nocera, A.; Agosti, V.; Nadal-Ginard, B.; Torella, D.; Ellison, G.M. Isolation and characterization of resident endogenous c-Kit+ cardiac stem cells from the adult mouse and rat heart. Nat. Protoc. 2014, 9, 1662–1681. [Google Scholar] [CrossRef]
- Bearzi, C.; Rota, M.; Hosoda, T.; Tillmanns, J.; Nascimbene, A.; De Angelis, A.; Yasuzawa-Amano, S.; Trofimova, I.; Siggins, R.W.; Lecapitaine, N.; et al. Human cardiac stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 14068–14073. [Google Scholar] [CrossRef]
- Carr, C.A.; Stuckey, D.J.; Tan, J.J.; Tan, S.C.; Gomes, R.S.; Camelliti, P.; Messina, E.; Giacomello, A.; Ellison, G.M.; Clarke, K. Cardiosphere-derived cells improve function in the infarcted rat heart for at least 16 weeks-an MRI study. PLoS ONE 2011, 6, e25669. [Google Scholar] [CrossRef]
- Ellison, G.M.; Vicinanza, C.; Smith, A.J.; Aquila, I.; Leone, A.; Waring, C.D.; Henning, B.J.; Stirparo, G.G.; Papait, R.; Scarfò, M.; et al. Adult c-kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 2013, 154, 827–842. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Tseliou, E.; Sun, B.; Cheng, K.; Malliaras, K.; Makkar, R.R.; Marbán, E. Therapeutic efficacy of cardiosphere-derived cells in a transgenic mouse model of non-ischaemic dilated cardiomyopathy. Eur. Heart J. 2015, 36, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Chugh, A.R.; Beache, G.M.; Loughran, J.H.; Mewton, N.; Elmore, J.B.; Kajstura, J.; Pappas, P.; Tatooles, A.; Stoddard, M.F.; Lima, J.A.; et al. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: The SCIPIO trial: Surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation 2012, 126, S54–S64. [Google Scholar] [CrossRef] [PubMed]
- Kulandavelu, S.; Karantalis, V.; Fritsch, J.; Hatzistergos, K.E.; Loescher, V.Y.; McCall, F.; Wang, B.; Bagno, L.; Golpanian, S.; Wolf, A.; et al. Pim1 Kinase Overexpression Enhances ckit(+) Cardiac Stem Cell Cardiac Repair Following Myocardial Infarction in Swine. J. Am. Coll. Cardiol. 2016, 68, 2454–2464. [Google Scholar] [CrossRef]
- Barrère-Lemaire, S.; Vincent, A.; Jorgensen, C.; Piot, C.; Nargeot, J.; Djouad, F. Mesenchymal Stromal Cells for Improvement of Cardiac Function Following Acute Myocardial Infarction: A Matter of Timing. Physiol. Rev. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Phinney, D.G. Functional Heterogeneity of Mesenchymal Stem Cells: Implications for Cell Therapy. J. Cell. Biochem. 2012, 113, 2806–2812. [Google Scholar] [CrossRef] [PubMed]
- Tormin, A.; Brune, J.C.; Olsson, E.; Valcich, J.; Neuman, U.; Olofsson, T.; Jacobsen, S.E.; Scheding, S. Characterization of Bone Marrow-Derived Mesenchymal Stromal Cells (MSC) Based on Gene Expression Profiling of Functionally Defined MSC Subsets. Cytotherapy 2009, 11, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.; Wang, S.; Zhou, Y.; Li, H.; Wu, Y. Mesenchymal Stem Cell Subpopulations: Phenotype, Property and Therapeutic Potential. Cell. Mol. Life Sci. 2016, 73, 3311–3321. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y. The uPA/uPAR System Orchestrates the Inflammatory Response, Vascular Homeostasis, and Immune System in Fibrosis Progression. Int. J. Mol. Sci. 2023, 24, 1796. [Google Scholar] [CrossRef]
- Dergilev, K.V.; Stepanova, V.V.; Beloglazova, I.B.; Tsokolayev, Z.I.; Parfenova, E.V. Multifaced Roles of the Urokinase System in the Regulation of Stem Cell Niches. Acta Nat. 2018, 10, 19–32. [Google Scholar] [CrossRef]
- Blasi, F.; Sidenius, N. The urokinase receptor: Focused cell surface proteolysis, cell adhesion and signaling. FEBS Lett. 2010, 584, 1923–1930. [Google Scholar] [CrossRef]
- Kwaan, H.C. The Role of Fibrinolytic System in Health and Disease. Int. J. Mol. Sci. 2022, 23, 5262. [Google Scholar] [CrossRef]
- Carriero, M.V.; Del Vecchio, S.; Capozzoli, M.; Franco, P.; Fontana, L.; Zannetti, A.; Botti, G.; D’Aiuto, G.; Salvatore, M.; Stoppelli, M.P. Urokinase receptor interacts with alpha(v)beta5 vitronectin receptor, promoting urokinase-dependent cell migration in breast cancer. Cancer Res. 1999, 59, 5307–5314. [Google Scholar] [PubMed]
- Saldanha, R.G.; Molloy, M.P.; Bdeir, K.; Cines, D.B.; Song, X.; Uitto, P.M.; Weinreb, P.H.; Violette, S.M.; Baker, M.S. Proteomic identification of lynchpin urokinase plasminogen activator receptor protein interactions associated with epithelial cancer malignancy. J. Proteome Res. 2007, 6, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.I.; Rao, N.K.; Xu, H.; Wei, Y.; Majdic, O.; Ronne, E.; Kobzik, L.; Chapman, H.A. Mac-1 (CD11b/CD18) and the urokinase receptor (CD87) form a functional unit on monocytic cells. Blood 1996, 88, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Mizukami, I.; Todd, R.F., 3rd; Petty, H.R. Urokinase-type plasminogen activator receptors associate with beta1 and beta3 integrins of fibrosarcoma cells: Dependence on extracellular matrix components. Cancer Res. 1997, 57, 1682–1689. [Google Scholar]
- Gude, N.A.; Sussman, M.A. Chasing c-Kit through the heart: Taking a broader view. Pharmacol. Res. 2018, 127, 110–115. [Google Scholar] [CrossRef]
- Bolli, R.; Mitrani, R.D.; Hare, J.M.; Pepine, C.J.; Perin, E.C.; Willerson, J.T.; Traverse, J.H.; Henry, T.D.; Yang, P.C.; Murphy, M.P.; et al. A Phase II study of autologous mesenchymal stromal cells and c-kit positive cardiac cells, alone or in combination, in patients with ischaemic heart failure: The CCTRN CONCERT-HF trial. Eur. J. Heart Fail. 2021, 23, 661–674. [Google Scholar] [CrossRef]
- Sanada, F.; Kim, J.; Czarna, A.; Chan, N.Y.-K.; Signore, S.; Ogórek, B.; Isobe, K.; Wybieralska, E.; Borghetti, G.; Pesapane, A.; et al. c-Kit–positive cardiac stem cells nested in hypoxic niches are activated by stem cell factor reversing the aging myopathy. Circ. Res. 2014, 114, 41–55. [Google Scholar] [CrossRef]
- Toru, H. C-kit-positive cardiac stem cells and myocardial regeneration. Am. J. Cardiovasc. Dis. 2012, 21, 58. [Google Scholar]
- Urbanek, K.; Rota, M.; Cascapera, S.; Bearzi, C.; Nascimbene, A.; De Angelis, A.; Hosoda, T.; Chimenti, S.; Baker, M.; Limana, F.; et al. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ. Res. 2005, 97, 663–673. [Google Scholar] [CrossRef]
- Renko, O.; Tolonen, A.M.; Rysä, J.; Magga, J.; Mustonen, E.; Ruskoaho, H.; Serpi, R. SDF1 gradient associates with the distribution of c-Kit+ cardiac cells in the heart. Sci. Rep. 2018, 8, 1160. [Google Scholar] [CrossRef]
- Klimovich, P.S.; Semina, E.V.; Karagyaur, M.N.; Rysenkova, K.D.; Sysoeva, V.Y.; Mironov, N.A.; Sagaradze, G.D.; Az’muko, A.A.; Popov, V.S.; Rubina, K.A.; et al. Urokinase receptor regulates nerve regeneration through its interaction with α5β1-integrin. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 125, 110008. [Google Scholar] [CrossRef] [PubMed]
- Dergilev, K.V.; Tsokolaeva, Z.I.; Beloglazova, I.; Ratner, E.; Molokotina, Y.; Parfenova, E.V. Angiogenic properties of myocardial c-kit+ cells. Genes Cells 2018, 13, 120751. [Google Scholar] [CrossRef]
- Zubkova, E.S.; Beloglazova, I.B.; Makarevich, P.I.; Boldyreva, M.A.; Sukhareva, O.Y.; Shestakova, M.V.; Dergilev, K.V.; Parfyonova, Y.V.; Menshikov, M.Y. Regulation of Adipose Tissue Stem Cells Angiogenic Potential by Tumor Necrosis Factor-Alpha. J. Cell. Biochem. 2016, 117, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Arnaoutova, I.; George, J.; Kleinman, H.K.; Benton, G. The endothelial cell tube formation assay on basement membrane turns 20: State of the science and the art. Angiogenesis 2009, 12, 267–274. [Google Scholar] [CrossRef]
- Liu, J.; Wu, P.; Wang, H.; Wang, Y.; Du, Y.; Cheng, W.; Xu, Z.; Zhou, N.; Wang, L.; Yang, Z. Necroptosis Induced by Ad-HGF Activates Endogenous C-Kit+ Cardiac Stem Cells and Promotes Cardiomyocyte Proliferation and Angiogenesis in the Infarcted Aged Heart. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 40, 847–860. [Google Scholar] [CrossRef]
- Bolli, R.; Tang, X.L.; Guo, Y.; Li, Q. After the storm: An objective appraisal of the efficacy of c-kit+ cardiac progenitor cells in preclinical models of heart disease. Can. J. Physiol. Pharmacol. 2021, 99, 129–139. [Google Scholar] [CrossRef]
- Marino, F.; Scalise, M.; Cianflone, E.; Mancuso, T.; Aquila, I.; Agosti, V.; Torella, M.; Paolino, D.; Mollace, V.; Nadal-Ginard, B.; et al. Role of c-Kit in Myocardial Regeneration and Aging. Front. Endocrinol. 2019, 10, 371. [Google Scholar] [CrossRef]
- Dergilev, K.; Tsokolaeva, Z.; Makarevich, P.; Beloglazova, I.; Zubkova, E.; Boldyreva, M.; Ratner, E.; Dyikanov, D.; Menshikov, M.; Ovchinnikov, A.; et al. C-Kit Cardiac Progenitor Cell Based Cell Sheet Improves Vascularization and Attenuates Cardiac Remodeling following Myocardial Infarction in Rats. BioMed Res. Int. 2018, 2018, 3536854. [Google Scholar] [CrossRef]
- Stavropoulou, A.; Philippou, A.; Halapas, A.; Sourla, A.; Pissimissis, N.; Koutsilieris, M. uPA, uPAR and TGFβ1 expression during early and late post myocardial infarction period in rat myocardium. In Vivo 2010, 24, 647–652. [Google Scholar]
- Creemers, E.; Cleutjens, J.; Smits, J.; Heymans, S.; Moons, L.; Collen, D.; Daemen, M.; Carmeliet, P. Disruption of the plasminogen gene in mice abolishes wound healing after myocardial infarction. Am. J. Pathol. 2000, 156, 1865–1873. [Google Scholar] [CrossRef]
- Herold, J.; Heidrich, F.M.; Quick, S.; Loehn, T.; Ibrahim, K.; Mahlmann, A.; Youssef, A. Influence of the plasminogen activator system on necrosis in acute myocardial infarction: Analysis of urokinase- and urokinase receptor-knockout mouse models. Am. J. Transl. Res. 2019, 11, 3629–3636. [Google Scholar]
- Heymans, S.; Luttun, A.; Nuyens, D.; Theilmeier, G.; Creemers, E.; Moons, L.; Dyspersin, G.D.; Cleutjens, J.P.; Shipley, M.; Angellilo, A.; et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 1999, 5, 1135–1142. [Google Scholar] [CrossRef]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef]
- Chaurasia, P.; Aguirre-Ghiso, J.A.; Liang, O.D.; Gardsvoll, H.; Ploug, M.; Ossowski, L. A region in urokinase plasminogen receptor domain III controlling a functional association with alpha5beta1 integrin and tumor growth. J. Biol. Chem. 2006, 281, 14852–14863. [Google Scholar] [CrossRef]
- Degryse, B.; Resnati, M.; Czekay, R.P.; Loskutoff, D.J.; Blasi, F. Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: Generation of a new integrin inhibitor. J. Biol. Chem. 2005, 280, 24792–24803. [Google Scholar] [CrossRef] [PubMed]
- Eden, G.; Archinti, M.; Furlan, F.; Murphy, R.; Degryse, B. The urokinase receptor interactome. Curr. Pharm. Des. 2011, 17, 1874–1889. [Google Scholar] [CrossRef] [PubMed]
- Montuori, N.; Cosimato, V.; Rinaldi, L.; Rea, V.E.; Alfano, D.; Ragno, P. uPAR regulates pericellular proteolysis through a mechanism involving integrins and fMLF-receptors. Thromb. Haemost. 2013, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, R.; Zhao, L.; Zhang, X.; Xu, T.; Cui, M. Basing on uPAR-binding fragment to design chimeric antigen receptors triggers antitumor efficacy against uPAR expressing ovarian cancer cells. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 117, 109173. [Google Scholar] [CrossRef] [PubMed]
- Semina, E.V.; Rubina, K.A.; Shmakova, A.A.; Rysenkova, K.D.; Klimovich, P.S.; Aleksanrushkina, N.A.; Sysoeva, V.Y.; Karagyaur, M.N.; Tkachuk, V.A. Downregulation of uPAR promotes urokinase translocation into the nucleus and epithelial to mesenchymal transition in neuroblastoma. J. Cell. Physiol. 2020, 235, 6268–6286. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, P.B.; Hodjat, M.; Haller, H.; Dumler, I.; Kiyan, Y. Loss of urokinase receptor sensitizes cells to DNA damage and delays DNA repair. PLoS ONE 2014, 9, e101529. [Google Scholar] [CrossRef] [PubMed]
- Vajravelu, B.N.; Hong, K.U.; Al-Maqtari, T.; Cao, P.; Keith, M.C.; Wysoczynski, M.; Zhao, J.; Moore, J.B.t.; Bolli, R. C-Kit Promotes Growth and Migration of Human Cardiac Progenitor Cells via the PI3K-AKT and MEK-ERK Pathways. PLoS ONE 2015, 10, e0140798. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Fibbi, G.; Cinelli, M.; Guiducci, S.; Del Rosso, A.; Margheri, F.; Serratì, S.; Pucci, M.; Kahaleh, B.; Fan, P.; et al. Matrix metalloproteinase 12–dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2004, 10, 3275–3285. [Google Scholar] [CrossRef]
- Giusti, B.; Fibbi, G.; Margheri, F.; Serratì, S.; Rossi, L.; Poggi, F.; Abbate, R. A model of anti-angiogenesis: Differential transcriptosome profiling of microvascular endothelial cells from diffuse systemic sclerosis patients. Arthritis Res. Ther. 2006, 8, R115. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Allanore, Y.; Revillod, L.; Fatini, C.; Guiducci, S.; Cuomo, G.; Bonino, C.; Riccieri, V.; Bazzichi, L.; Liakouli, V.; et al. A genetic variation located in the promoter region of the UPAR (CD87) gene is associated with the vascular complications of systemic sclerosis. Arthritis Rheum. 2011, 63, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Dergilev, K.V.; Beloglazova, I.B.; Tsokolaeva, Z.I.; Vasilets, Y.D.; Parfenova, E.V. Deficiency of Urokinase-Type Plasminogen Activator Receptor Is Associated with the Development of Perivascular Fibrosis in Mouse Heart. Bull. Exp. Biol. Med. 2022, 173, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Beloglazova, I.; Stepanova, V.; Zubkova, E.; Dergilev, K.; Koptelova, N.; Tyurin-Kuzmin, P.A.; Dyikanov, D.; Plekhanova, O.; Cines, D.B.; Mazar, A.P.; et al. Mesenchymal stromal cells enhance self-assembly of a HUVEC tubular network through uPA-uPAR/VEGFR2/integrin/NOTCH crosstalk. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2022, 1869, 119157. [Google Scholar] [CrossRef]
- Long, D.; Yang, J.; Wu, X.; Gui, Y.; Yu, L. Urokinase-type plasminogen activator protects human umbilical vein endothelial cells from apoptosis in sepsis. Int. J. Clin. Exp. Pathol. 2019, 12, 77. [Google Scholar]
- Prager, G.W.; Mihaly, J.; Brunner, P.M.; Koshelnick, Y.; Hoyer-Hansen, G.; Binder, B.R. Urokinase mediates endothelial cell survival via induction of the X-linked inhibitor of apoptosis protein. Blood J. Am. Soc. Hematol. 2009, 113, 1383–1390. [Google Scholar] [CrossRef]
- Song, T.; Meng, S.; Xu, S.T.; Jin, S.J.; Zeng, Q.Z.; Gu, G.J. The overexpression of uPA promotes the proliferation and fibrinolytic activity of human umbilical vein endothelial cells. Int. J. Clin. Exp. Pathol. 2019, 12, 2959. [Google Scholar]
- Balsara, R.D.; Merryman, R.; Virjee, F.; Northway, C.; Castellino, F.J.; Ploplis, V.A. A deficiency of uPAR alters endothelial angiogenic function and cell morphology. Vasc. Cell 2011, 3, 10. [Google Scholar] [CrossRef]
- Reuning, U.; Sperl, S.; Kopitz, C.; Kessler, H.; Kruger, A.; Schmitt, M.; Magdolen, V. Urokinase-type plasminogen activator (uPA) and its receptor (uPAR): Development of antagonists of uPA/uPAR interaction and their effects in vitro and in vivo. Curr. Pharm. Des. 2003, 9, 1529–1543. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.A.; Prager, G.W.; Mihaly-Bison, J.; Uhrin, P.; Sunzenauer, S.; Binder, B.R.; Schütz, G.J.; Freissmuth, M.; Breuss, J.M. VEGF-induced endothelial cell migration requires urokinase receptor (uPAR)-dependent integrin redistribution. Cardiovasc. Res. 2012, 94, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Poettler, M.; Unseld, M.; Mihaly-Bison, J.; Uhrin, P.; Koban, F.; Binder, B.R.; Prager, G.W. The urokinase receptor (CD87) represents a central mediator of growth factor-induced endothelial cell migration. Thromb. Haemost. 2012, 108, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Bertolino, G.M.; Maumus, M.; Jorgensen, C.; Noël, D. Therapeutic Potential in Rheumatic Diseases of Extracellular Vesicles Derived from Mesenchymal Stromal Cells. Nat. Rev. Rheumatol. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Biagioni, A.; Laurenzana, A.; Menicacci, B.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Guasti, D.; Paoli, P.; Serratì, S.; Mocali, A.; et al. UPAR-Expressing Melanoma Exosomes Promote Angiogenesis by VE-Cadherin, EGFR and UPAR Overexpression and Rise of ERK1,2 Signaling in Endothelial Cells. Cell. Mol. Life Sci. 2021, 78, 3057–3072. [Google Scholar] [CrossRef] [PubMed]
- Montuori, N.; Carriero, M.V.; Salzano, S.; Rossi, G.; Ragno, P. The Cleavage of the Urokinase Receptor Regulates Its Multiple Functions. J. Biol. Chem. 2002, 277, 46932–46939. [Google Scholar] [CrossRef]
- Blasi, F.; Carmeliet, P. UPAR: A Versatile Signalling Orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef]
- Nusrat, A.R.; Chapman, H.A. An Autocrine Role for Urokinase in Phorbol Ester-Mediated Differentiation of Myeloid Cell Lines. J. Clin. Investig. 1991, 87, 1091–1097. [Google Scholar] [CrossRef]
- Resnati, M.; Pallavicini, I.; Wang, J.M.; Oppenheim, J.; Serhan, C.N.; Romano, M.; Blasi, F. The Fibrinolytic Receptor for Urokinase Activates the G Protein-Coupled Chemotactic Receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. USA 2002, 99, 1359–1364. [Google Scholar] [CrossRef]
- Genua, M.; D’Alessio, S.; Cibella, J.; Gandelli, A.; Sala, E.; Correale, C.; Spinelli, A.; Arena, V.; Malesci, A.; Rutella, S.; et al. The Urokinase Plasminogen Activator Receptor (UPAR) Controls Macrophage Phagocytosis in Intestinal Inflammation. Gut 2015, 64, 589–600. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dergilev, K.; Tsokolaeva, Z.; Goltseva, Y.; Beloglazova, I.; Ratner, E.; Parfyonova, Y. Urokinase-Type Plasminogen Activator Receptor Regulates Prosurvival and Angiogenic Properties of Cardiac Mesenchymal Stromal Cells. Int. J. Mol. Sci. 2023, 24, 15554. https://doi.org/10.3390/ijms242115554
Dergilev K, Tsokolaeva Z, Goltseva Y, Beloglazova I, Ratner E, Parfyonova Y. Urokinase-Type Plasminogen Activator Receptor Regulates Prosurvival and Angiogenic Properties of Cardiac Mesenchymal Stromal Cells. International Journal of Molecular Sciences. 2023; 24(21):15554. https://doi.org/10.3390/ijms242115554
Chicago/Turabian StyleDergilev, Konstantin, Zoya Tsokolaeva, Yulia Goltseva, Irina Beloglazova, Elizaveta Ratner, and Yelena Parfyonova. 2023. "Urokinase-Type Plasminogen Activator Receptor Regulates Prosurvival and Angiogenic Properties of Cardiac Mesenchymal Stromal Cells" International Journal of Molecular Sciences 24, no. 21: 15554. https://doi.org/10.3390/ijms242115554
APA StyleDergilev, K., Tsokolaeva, Z., Goltseva, Y., Beloglazova, I., Ratner, E., & Parfyonova, Y. (2023). Urokinase-Type Plasminogen Activator Receptor Regulates Prosurvival and Angiogenic Properties of Cardiac Mesenchymal Stromal Cells. International Journal of Molecular Sciences, 24(21), 15554. https://doi.org/10.3390/ijms242115554