

Rational Design, Synthesis and Binding Affinity Studies of Anthraquinone Derivatives Conjugated to Gonadotropin-Releasing Hormone (GnRH) Analogues towards Selective Immunosuppression of Hormone-Dependent Cancer

 , , ,
, , ,  , ,
, ,  , , ,
, , , 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Rational Design of Mitoxantrone–GnRH Analogues
2.2. Synthesis of Anthraquinone and Mitoxantrone-GnRH Analogues
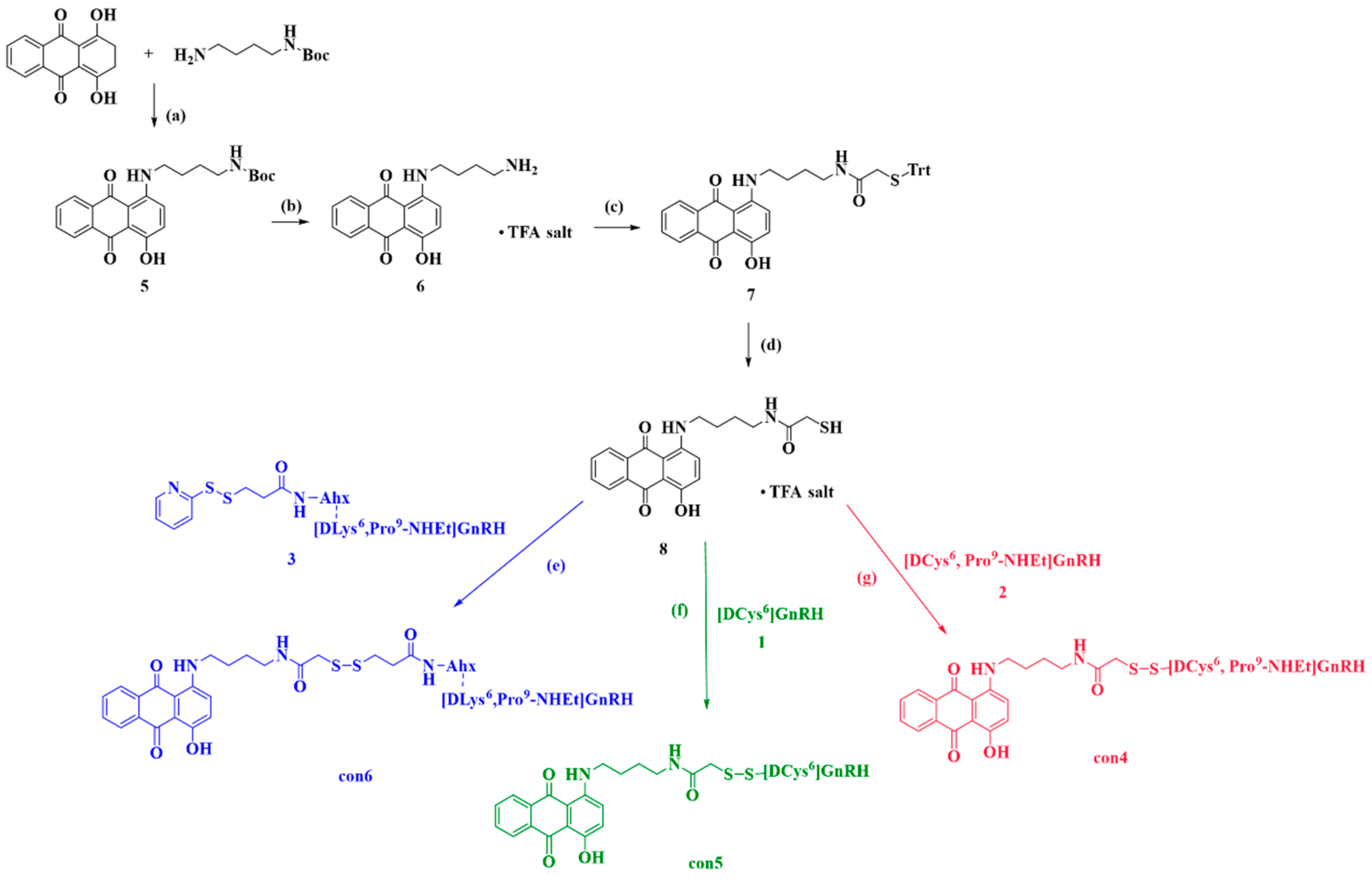
2.2.1. Synthesis of Anthraquinone—GnRH Conjugates con4; con5; con6
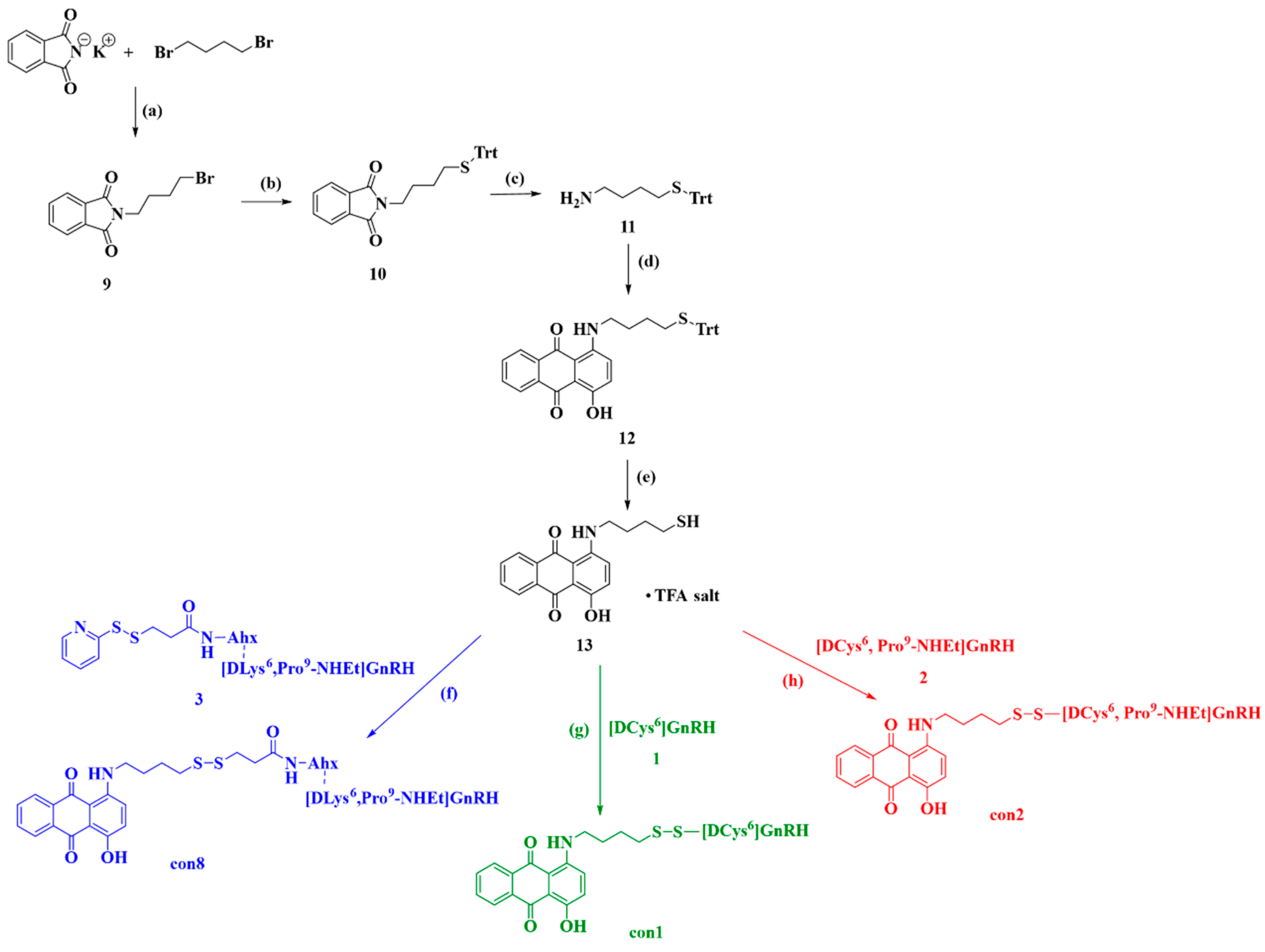
2.2.2. Synthesis of Anthraquinone—GnRH Conjugates con1; con2; con8
2.2.3. Synthesis of Anthraquinone—GnRH Conjugates con7; con3
2.3. Conformational Studies of con7 and con3
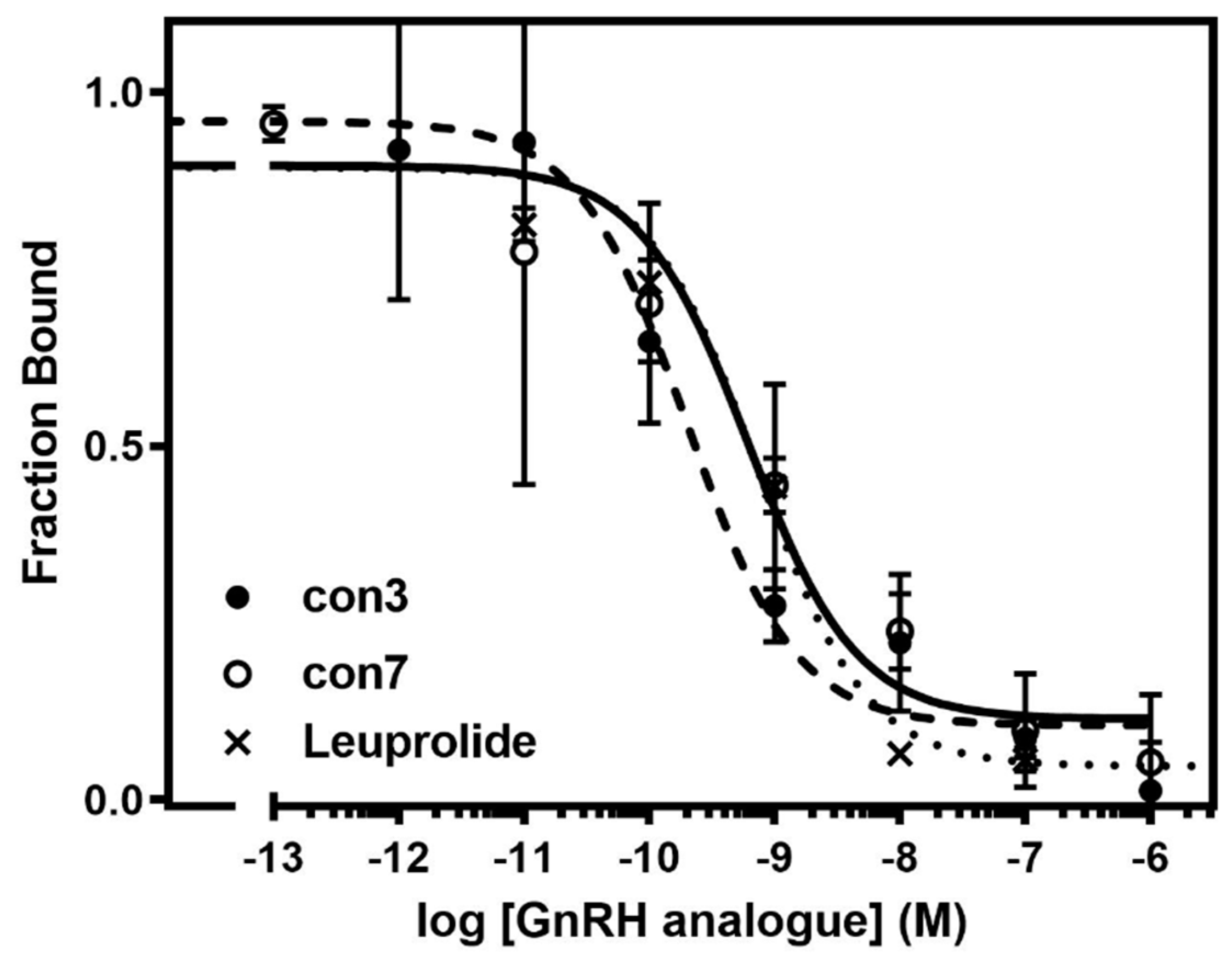
2.4. Binding Affinity Studies of Synthesized Conjugates
2.5. Disulfides Reduction of con7 and Insulin by EcoTrx1
3. Materials and Methods
3.1. Synthesis of Anthraquinone and Mitoxantrone-GnRH Analogues
3.1.1. Synthesis of Anthraquinone—GnRH Conjugate con4
3.1.2. Synthesis of Anthraquinone—GnRH Conjugate con5
3.1.3. Synthesis of Anthraquinone—GnRH Conjugate con6
3.1.4. Synthesis of Anthraquinone—GnRH Conjugate con1
3.1.5. Synthesis of Anthraquinone—GnRH Conjugate con2
3.1.6. Synthesis of Anthraquinone—GnRH Conjugate con8
3.1.7. Synthesis of Mitoxantrone—GnRH Conjugate con7
3.1.8. Synthesis of Mitoxantrone—GnRH Conjugate con3
3.2. Conformational Studies of con7 and con3
3.3. GnRH Receptor Binding Assay of Synthesized Analogues
3.4. Disulfide Bond Reduction Assays by EcoTrx1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schally, A.V.; Arimura, A.; Kastin, A.J.; Matsuo, H.; Baba, Y.; Redding, T.W.; Nair, R.M.; Debeljuk, L.; White, W.F. Gonadotropin-Releasing Hormone: One Polypeptide Regulates of Luteinizing and Follicle-Stimulating Hormones. Science 1979 1971, 173, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- McCann, S.M.; Dhariwal, A.P.S. Hypothalamic Factors Which Influence Gonadotrophin Secretion. Trans. N. Y. Acad. Sci. 1964, 27, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Clarke, I.J.; Pompolo, S. Synthesis and Secretion of GnRH. Anim. Reprod. Sci. 2005, 88, 29–55. [Google Scholar] [CrossRef]
- Schally, A.V.; Arimura, A.; Baba, Y.; Nair, R.M.; Matsuo, H.; Redding, T.W.; Debeljuk, L. Isolation and Properties of the FSH and LH-Releasing Hormone. Biochem. Biophys. Res. Commun. 1971, 43, 393–399. [Google Scholar] [CrossRef]
- Simoni, M.; Weinbauer, G.F.; Gromoll, J.; Nieschlag, E. Role of FSH in Male Gonadal Function. Ann. Endocrinol. 1999, 60, 102–106. [Google Scholar]
- Simoni, M.; Gromoll, J.; Nieschlag, E. The Follicle-Stimulating Hormone Receptor: Biochemistry, Molecular Biology, Physiology, and Pathophysiology. Endocr. Rev. 1997, 18, 739–773. [Google Scholar]
- Richards, J.S. Maturation of Ovarian Follicles: Actions and Interactions of pituitary and Ovarian Hormones on Follicular Cell Differentiation. Physiol. Rev. 1980, 60, 51–89. [Google Scholar] [CrossRef]
- Howles, C.M. Role of LH and FSH in Ovarian Function. Mol. Cell. Endocrinol. 2000, 161, 25–30. [Google Scholar] [CrossRef]
- Richards, J.S. Hormonal Control of Gene Expression in the Ovary. Endocr. Rev. 1994, 15, 725–751. [Google Scholar] [CrossRef]
- Wildt, L.; Häusler, A.; Marshall, G.; Hutchison, J.S.; Plant, T.M.; Belchetz, P.E.; Knobil, E. Frequency and Amplitude of Gonadotropin-Releasing Hormone and Gonadotropin Secretion in the Rhesus Monkey. Endocrinology 1981, 109, 376–385. [Google Scholar] [CrossRef]
- Nett, T.M.; Turzillo, A.M.; Baratta, M.; Rispoli, L.A. Pituitary Effects of Steroid Hormones on Secretion Of-Stimulating Hormone and Luteinizing Hormone. Domest. Anim. Endocrinol. 2002, 23, 33–42. [Google Scholar] [CrossRef]
- Ferris, H.A.; Shupnik, M.A. Mechanisms for Pulsatile Regulation of the Gonadotropin Subunit by GNRH1. Biol. Reprod. 2006, 74, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Savoy-Moore, R.T.; Swartz, K.H. Several GnRH Stimulation Frequencies Differentially Release FSH and LH from Isolated, Perfused Rat Anterior Pituitary Cells. In Regulation of Ovarian and Testicular Function; Mahesh Virendra, B., Dhindsa, D.S., Anderson, E., Kalra, S.P., Eds.; Springer: Boston, MA, USA, 1987; pp. 641–645. ISBN 978-1-4684-5395-9. [Google Scholar]
- Schwanzel-Fukuda, M.; Bick, D.; Pfaff, D.W. Luteinizing Hormone-Releasing Hormone (LHRH)-Expressing Cells Do Not Migrate Normally in an Inherited Hypogonadal (Kallmann) Syndrome. Mol. Brain Res. 1989, 6, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.M.; Mitnick, M.; Chieffo, V. In Vitro Biosynthesis of TSH- and LH-Releasing Factors by the Human Placenta. Am. J. Obstet. Gynecol. 1975, 121, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Nagy, A. Chemotherapy Targeted to Cancers through Tumoral Hormone. Trends Endocrinol. Metab. 2004, 15, 300–310. [Google Scholar] [CrossRef]
- Brantley, K.D.; Hankinson, S.E.; Heather Eliassen, A. Hormones and Cancer. In Encyclopedia of Cancer, 3rd ed.; Boffetta, P., Hainaut, P., Eds.; Academic Press: Oxford, UK, 2019; pp. 237–253. ISBN 978-0-12-812485-7. [Google Scholar]
- Cheung, L.W.T.; Wong, A.S.T. Gonadotropin-Releasing Hormone: GnRH Receptor Signaling in extrapituitary Tissues. FEBS J. 2008, 275, 5479–5495. [Google Scholar] [CrossRef]
- Naor, Z. Signaling by G-Protein-Coupled Receptor (GPCR): Studies on the GnRH Receptor. Front. Neuroendocrinol. 2009, 30, 10–29. [Google Scholar] [CrossRef]
- Liu, S.V.; Schally, A.V.; Hawes, D.; Xiong, S.; Fazli, L.; Gleave, M.; Cai, J.; Groshen, S.; Brands, F.; Engel, J.; et al. Expression of Receptors for Luteinizing Hormone-Releasing (LH-RH) in Prostate Cancers Following Therapy with LH-RH Agonists. Clin. Cancer Res. 2010, 16, 4675–4680. [Google Scholar] [CrossRef]
- Yano, T.; Pinski, J.; Radulovic, S.; Schally, A. V Inhibition of Human Epithelial Ovarian Cancer Cell Growth in vitro by Agonistic and Antagonistic Analogues of Luteinizing-Releasing Hormone. Proc. Natl. Acad. Sci. USA 1994, 91, 1701–1705. [Google Scholar] [CrossRef]
- Yan, W.; Cheng, L.; Wang, W.; Wu, C.; Yang, X.; Du, X.; Ma, L.; Qi, S.; Wei, Y.; Lu, Z.; et al. Structure of the Human Gonadotropin-Releasing Hormone Receptor GnRH1R Reveals an Unusual Ligand Binding Mode. Nat. Commun. 2020, 11, 5287. [Google Scholar] [CrossRef]
- Flanagan, C.A.; Zhou, W.; Chi, L.; Yuen, T.; Rodic V and Robertson, D.; Johnson, M.; Holland, P.; Millar R P and Weinstein, H.; Mitchell, R.; Sealfon, S.C. The Functional Microdomain in Transmembrane Helices 2 and 7 Expression, Activation, and Coupling Pathways of the gonadotropin-Releasing Hormone Receptor. J. Biol. Chem. 1999, 274, 28880–28886. [Google Scholar] [CrossRef] [PubMed]
- Tzoupis, H.; Nteli, A.; Platts, J.; Mantzourani, E.; Tselios, T. Refinement of the Gonadotropin Releasing Hormone Receptor I Homology Model by Applying Molecular Dynamics. J. Mol. Graph. Model. 2019, 89, 147–155. [Google Scholar] [CrossRef]
- Limonta, P.; Manea, M. Gonadotropin-Releasing Hormone Receptors as Molecular Targets in Prostate Cancer: Current Options and emerging Strategies. Cancer Treat. Rev. 2013, 39, 647–663. [Google Scholar] [CrossRef]
- Emons, G.; Schally, A.V. The Use of Luteinizing Hormone Releasing Hormone Agonists and antagonists in Gynaecological Cancers. Hum. Reprod. 1994, 9, 1364–1379. [Google Scholar] [CrossRef]
- Redding, T.W.; Kastin, A.J.; Gonzales-Barcena, D.; Coy, D.H.; Coy, E.J.; Schalch, D.S.; Schally, A.V. The Half-Life, Metabolism and Excretion of Tritiated Luteinizing-Releasing Hormone (LH-RH) in Man. J. Clin. Endocrinol. Metab. 1973, 37, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Karten, M.J.; Rivier, J.E. Gonadotropin-Releasing Hormone Analog Design. Structure-Function Studies Toward the Development of Agonists and Antagonists: Rationale and Perspective. Endocr. Rev. 1986, 7, 44–66. [Google Scholar] [CrossRef] [PubMed]
- Fujino, M.; Kobayashi, S.; Obayashi, M.; Fukuda, T.; Shinagawa, S.; Yamazaki, I.; Nakayama, R.; White, W.F.; Rippel, R.H. Syntheses and Biological Activities of Analogs of Luteinizing Hormone Releasing Hormone (LH-RH). Biochem. Biophys. Res. Commun. 1972, 49, 698–705. [Google Scholar] [CrossRef]
- Monahan, M.W.; Amoss, M.S.; Anderson, H.A.; Vale, W. Synthetic Analogs of the Hypothalamic Luteinizing Hormone Releasing Factor with Increased Agonist or Antatonist Properties. Biochemistry 1973, 12, 4616–4620. [Google Scholar] [CrossRef]
- Meyer, J.D.; Manning, M.C.; Vander Velde, D.G. Characterization of the Solution Conformations of Leuprolide. J. Pept. Res. 2002, 60, 159–168. [Google Scholar] [CrossRef]
- Wilson, A.C.; Meethal, S.V.; Bowen, R.L.; Atwood, C.S. Leuprolide Acetate: A Drug of Diverse Clinical Applications. Expert Opin. Investig. Drugs 2007, 16, 1851–1863. [Google Scholar] [CrossRef]
- Fujino, M.; Fukuda, T.; Shinagawa, S.; Kobayashi, S.; Yamazaki, I.; Nakayama, R.; Seely, J.H.; White, W.F.; Rippel, R.H. Synthetic Analogs of Luteinizing Hormone Releasing Hormone (LH-RH) Substituted in Position 6 and 10. Biochem. Biophys. Res. Commun. 1974, 60, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Su, F.; Liu, Y.; Yang, Y.; Cao, Y.; Qiu, J.; Wang, Y.; Zhang, L.; Wang, J.; Cao, X. The Pharmacokinetics of Buserelin after Intramuscular Administration in Pigs and Cows. BMC Vet. Res. 2022, 18, 136. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, A.; Labrie, F.; Lemay, A.; Caron, S.; Raynaud, J.P. Inhibitory Effects of a Single Intranasal Administration of [d-Ser(TBU)6, Des-Gly-NH210]LHRK Ethylamide, a Potent LHRK Agonist, on Serum Steroid Levels in Normal Adult Men. J. Steroid Biochem. 1980, 13, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Muller, V.; Ortmann, O.; Grossmann, G.; Trautner, U.; Stuckrad, B.; Schulz, K.; Schally, A. Luteinizing Hormone-Releasing Hormone Agonist Triptorelin Signal Transduction and Mitogenic Activity of epidermal Growth Factor in Human Ovarian and Endometrial Cancer Lines. Int. J. Oncol. 1996, 9, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Letassy, N.A.; Thompson, D.F.; Britton, M.L.; Suda Sr, R.R. Nafarelin Acetate: A Gonadotropin-Releasing Hormone Agonist for the Treatment of Endometriosis. DICP 1990, 24, 1204–1209. [Google Scholar] [CrossRef]
- Chrisp, P.; Goa, K.L. Nafarelin. Drugs 1990, 39, 523–551. [Google Scholar] [CrossRef]
- Mizutani, T.; Sakata, M.; Terakawa, N. Effect of Gonadotropin-Releasing Hormone Agonists, Nafarelin, Buserelin, and Leuprolide, on Experimentally Induced in the Rat. Int. J. Fertil. Menopausal Stud. 1995, 40, 106–111. [Google Scholar]
- Jonat, W.; Kaufmann, M.; Sauerbrei, W.; Blamey, R.; Cuzick, J.; Namer, M.; Fogelman, I.; de Haes, J.C.; de Matteis, A.; Stewart, A.; et al. Goserelin versus Cyclophosphamide, Methotrexate, and fluorouracil as Adjuvant Therapy in Premenopausal Patients with node-Positive Breast Cancer: The Zoladex Early Breast Cancer Association Study. J. Clin. Oncol. 2002, 20, 4628–4635. [Google Scholar] [CrossRef]
- Ferguson, S.S. Evolving Concepts in G Protein-Coupled Receptor Endocytosis: The Role in Receptor Desensitization and Signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar]
- Söderhäll, J.A.; Polymeropoulos, E.E.; Paulini, K.; Günther, E.; Kühne, R. Antagonist and Agonist Binding Models of the Human-Releasing Hormone Receptor. Biochem. Biophys. Res. Commun. 2005, 333, 568–582. [Google Scholar] [CrossRef]
- Conn, P.M.; Crowley, W.F., Jr. Gonadotropin-Releasing Hormone and Its Analogs. Annu. Rev. Med. 1994, 45, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.R.; Caunt, C.J.; Armstrong Stephen P and McArdle, C.A. Agonist-Induced Internalization and Downregulation of gonadotropin-Releasing Hormone Receptors. Am. J. Physiol. Cell Physiol. 2009, 297, C591–C600. [Google Scholar] [CrossRef] [PubMed]
- Knobil, E. The Neuroendocrine Control of the Menstrual Cycle. Recent Prog. Horm. Res. 1980, 36, 53–88. [Google Scholar]
- Janáky, T.; Juhász, A.; Bajusz, S.; Csernus, V.; Srkalovic, G.; Bokser, L.; Milovanovic, S.; Redding T W and Rékási, Z.; Nagy, A. Analogues of Luteinizing Hormone-Releasing Hormone Containing Groups. Proc. Natl. Acad. Sci. USA 1992, 89, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.; Gunnarsson, K.; Iatropoulos, M.; Brughera, M. Toxicological Testing of Cytotoxic Drugs (Review). Int. J. Oncol. 2001, 19, 1021–1028. [Google Scholar] [CrossRef]
- Karampelas, T.; Argyros, O.; Sayyad, N.; Spyridaki, K.; Pappas, C.; Morgan, K.; Kolios, G.; Millar, R.P.; Liapakis, G.; Tzakos, A.G.; et al. GnRH-Gemcitabine Conjugates for the Treatment of Androgen-Independent Prostate Cancer: Pharmacokinetic Enhancements Combined with Targeted Drug Delivery. Bioconjug. Chem. 2014, 25, 813–823. [Google Scholar] [CrossRef][Green Version]
- Schally, A.V.; Nagy, A. Cancer Chemotherapy Based on Targeting of Cytotoxic Peptide Conjugates to Their Receptors on Tumors. Eur. J. Endocrinol. 1999, 141, 1–14. [Google Scholar] [CrossRef]
- Li, S.; Zhao, H.; Chang, X.; Wang, J.; Zhao, E.; Yin, Z.; Mao, X.; Deng, S.; Hao, T.; Wang, H.; et al. Synthesis, in Vitro Stability, and Antiproliferative Effect of d-Cysteine Modified GnRH-Doxorubicin Conjugates. J. Pept. Sci. 2019, 25, e3135. [Google Scholar] [CrossRef]
- Vrettos, E.I.; Mező, G.; Tzakos, A.G. On the Design Principles of Peptide–Drug Conjugates for Targeted Drug Delivery to the Malignant Tumor Site. Beilstein J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef]
- Szepeshazi, K.; Schally, A.V.; Keller, G.; Block, N.L.; Benten, D.; Halmos, G.; Szalontay, L.; Vidaurre, I.; Jaszberenyi, M.; Rick, F.G. Receptor-Targeted Therapy of Human Experimental Urinary Bladder with Cytotoxic LH-RH Analog AN-152 [AEZS- 108]. Oncotarget 2012, 3, 686–699. [Google Scholar] [CrossRef][Green Version]
- Bajo, A.M.; Schally, A.V.; Halmos, G.; Nagy, A. Targeted Doxorubicin-Containing Luteinizing Hormone-Releasing Analogue AN-152 Inhibits the Growth of doxorubicin-Resistant MX-1 Human Breast Cancers. Clin. Cancer Res. 2003, 9, 3742–3748. [Google Scholar] [PubMed]
- Engel, J.B.; Keller, G.; Schally, A.V.; Nagy, A.; Chism, D.D.; Halmos, G. Effective Treatment of Experimental Human Endometrial cancers with Targeted Cytotoxic Luteinizing Hormone-Releasing Hormone AN-152 and AN-207. Fertil. Steril. 2005, 83 (Suppl. S1), 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Kaufmann, M.; Gorchev, G.; Tsekova, V.; Gründker, C.; Günthert, A.R.; Hanker, L.C.; Velikova, M.; Sindermann, H.; Engel, J.; et al. Dose Escalation and Pharmacokinetic Study of AEZS-108(AN-152), an LHRH Agonist Linked to Doxorubicin, in women with LHRH Receptor-Positive Tumors. Gynecol. Oncol. 2010, 119, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Gründker, C.; Völker, P.; Griesinger, F.; Ramaswamy, A.; Nagy, A.; Schally, A.V.; Emons, G. Antitumor Effects of the Cytotoxic Luteinizing Hormone-Releasing Analog AN-152 on Human Endometrial and Ovarian Cancers into Nude Mice. Am. J. Obstet. Gynecol. 2002, 187, 528–537. [Google Scholar] [CrossRef]
- Günthert, A.R.; Gründker, C.; Bongertz, T.; Nagy, A.; Schally, A.V.; Emons, G. Induction of Apoptosis by AN-152, a Cytotoxic Analog of luteinizing Hormone-Releasing Hormone (LHRH), in LHRH-R Human Breast Cancer Cells Is Independent of Multidrug-1 (MDR-1) System. Breast Cancer Res. Treat. 2004, 87, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Gorchev, G.; Sehouli, J.; Wimberger, P.; Stähle, A.; Hanker, L.; Hilpert, F.; Sindermann, H.; Gründker Carsten and Harter, P. Efficacy and Safety of AEZS-108 (INN: Zoptarelin Doxorubicin) an LHRH Agonist Linked to Doxorubicin in Women with platinum Refractory or Resistant Ovarian Cancer Expressing Receptors: A Multicenter Phase II Trial of the ago-Study Group (AGO GYN 5). Gynecol. Oncol. 2014, 133, 427–432. [Google Scholar] [CrossRef]
- Miller, D.S.; Scambia, G.; Bondarenko, I.; Westermann, A.M.; Oaknin, A.; Oza, A.M.; Lisyanskaya, A.S.; Vergote, I.; Wenham, R.M.; Temkin, S.M.; et al. ZoptEC: Phase III Randomized Controlled Study Comparing Zoptarelin with Doxorubicin as Second Line Therapy for Locally Advanced, Recurrent, or Metastatic Endometrial Cancer (NCT01767155). J. Clin. Oncol. 2018, 36, 5503. [Google Scholar] [CrossRef]
- Nagy, A.; Plonowski, A.; Schally, A.V. Stability of Cytotoxic Luteinizing Hormone-Releasing Hormone Conjugate (AN-152) Containing Doxorubicin 14-O-Hemiglutarate in Mouse and Human Serum in Vitro: Implications for the Design of Preclinical Studies. Proc. Natl. Acad. Sci. USA 2000, 97, 829–834. [Google Scholar] [CrossRef]
- Limonta, P.; Montagnani Marelli, M.; Mai, S.; Motta, M.; Martini, L.; Moretti, R.M. GnRH Receptors in Cancer: From Cell Biology to Novel Targeted Strategies. Endocr. Rev. 2012, 33, 784–811. [Google Scholar] [CrossRef]
- Murdock, K.C.; Child, R.G.; Fabio, P.F.; Angier, R.B.; Wallace, R.E.; Durr, F.E.; Citarella, R. V Antitumor Agents. 1. 1,4-Bis[(Aminoalkyl)Amino]-9,10-Anthracenediones. J. Med. Chem. 1979, 22, 1024–1030. [Google Scholar] [CrossRef]
- Kapuscinski, J.; Darzynkiewicz, Z. Relationship between the Pharmacological Activity of Antitumor Drugs Ametantrone and Mitoxantrone (Novatrone) and Their Ability to Condense Nucleic Acids. Proc. Natl. Acad. Sci. USA 1986, 83, 6302–6306. [Google Scholar] [CrossRef]
- Fox, E.J. Mechanism of Action of Mitoxantrone. Neurology 2004, 63, S15–S18. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Durr, F.E. Development of Mitoxantrone. Investig. New Drugs 1985, 3, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.O. The Role of Mitoxantrone in Non-Hodgkin’s Lymphoma. Oncology 2002, 16, 490–502, 507–508, 511–512, 517. [Google Scholar]
- Cheng, C.C.; Zee-Cheng, R.K. The Design, Synthesis and Development of a New Class of Potent Anthraquinones. Prog. Med. Chem. 1983, 20, 83–118. [Google Scholar]
- Landys, K.; Borgstrom, S.; Andersson, T.; Noppa, H. Mitoxantrone as a First-Line Treatment of Advanced Breast Cancer. Investig. New Drugs 1985, 3, 133–137. [Google Scholar] [CrossRef]
- van Dodewaard-de Jong, J.M.; Santegoets, S.J.; de Ven, P.M.; Versluis, J.; Verheul Henk M W and de Gruijl, T.D.; Gerritsen, W.R.; van den Eertwegh, A.J.M. Improved Efficacy of Mitoxantrone in Patients With-Resistant Prostate Cancer after Vaccination with GM-CSF-Transduced Allogeneic Prostate Cancer Cells. Oncoimmunology 2016, 5, e1105431. [Google Scholar] [CrossRef] [PubMed]
- Conner, C.S. Mitoxantrone: A Replacement for Doxorubicin? Drug Intell. Clin. Pharm. 1984, 18, 479–480. [Google Scholar] [CrossRef]
- Neidhart, J.A.; Gochnour, D.; Roach, R.; Hoth, D.; Young, D. A Comparison of Mitoxantrone and Doxorubicin in Breast Cancer. J. Clin. Oncol. 1986, 4, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.C.; Allegra, J.C.; Woodcock, T.; Wolff, S.; Bryan, S.; Cartwright, K.; Dukart, G.; Henry, D. Randomized Clinical Trial Comparing Mitoxantrone with Doxorubicin in Previously Treated Patients with Metastatic Breast Cancer. J. Clin. Oncol. 1989, 7, 560–571. [Google Scholar] [CrossRef]
- Damiani, R.M.; Moura, D.J.; Viau, C.M.; Caceres, R.A.; Henriques, J.A.P.; Saffi, J. Pathways of Cardiac Toxicity: Comparison between Chemotherapeutic Doxorubicin and Mitoxantrone. Arch. Toxicol. 2016, 90, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Cebula, M.; Moolla, N.; Capovilla, A.; Arnér, E.S.J. The Rare TXNRD1_v3 (“v3”) Splice Variant of Human Reductase 1 Protein Is Targeted to Membrane Rafts by N-Acylation and Induces Filopodia Independently of Its Redox Site Integrity. J. Biol. Chem. 2013, 288, 10002–10011. [Google Scholar] [CrossRef] [PubMed]
- Powis, G.; Mustacich, D.; Coon, A. The Role of the Redox Protein Thioredoxin in Cell Growth and Cancer. Free Radic. Biol. Med. 2000, 29, 312–322. [Google Scholar] [CrossRef]
- Tapeinou, A.; Giannopoulou, E.; Simal, C.; Hansen, B.E.; Kalofonos, H.; Apostolopoulos, V.; Vlamis-Gardikas, A.; Tselios, T. Design, Synthesis and Evaluation of an Anthraquinone Derivative to Myelin Basic Protein Immunodominant (MBP85-99) Epitope: Towards Selective Immunosuppression. Eur. J. Med. Chem. 2018, 143, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Lown, J.W. Anthracycline and Anthraquinone Anticancer Agents: Current and Recent Developments. Pharmacol. Ther. 1993, 60, 185–214. [Google Scholar] [CrossRef]
- Krishnamoorthy, C.R.; Yen, S.F.; Smith, J.C.; Lown, J.W.; Wilson, W.D. Stopped-Flow Kinetic Analysis of the Interaction of Anthraquinone Drugs with Calf Thymus DNA, Poly[d(G-C)].Poly[d(G-C)], and Poly[d(A-T)].Poly[d(A-T)]. Biochemistry 1986, 25, 5933–5940. [Google Scholar] [CrossRef]
- Nishio, A.; Uyeki, E.M. Cellular Uptake and Inhibition of DNA Synthesis by dihydroxyanthraquinone and Two Analogues. Cancer Res. 1983, 43, 1951–1956. [Google Scholar]
- Laimou, D.K.; Katsara, M.; Matsoukas, M.T.I.; Apostolopoulos, V.; Troganis, A.N.; Tselios, T.V. Structural Elucidation of Leuprolide and Its Analogues in Solution: Insight into Their Bioactive Conformation. Amino Acids 2010, 39, 1147–1160. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a Visualization System for Exploratory research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Tzoupis, H.; Nteli, A.; Androutsou, M.-E.; Tselios, T. Molecular Dynamics Simulations Studies of Gonadotropin Releasing Hormone (GnRH) and GnRH Receptor. J. Peptide Sci. 2018, 24, S129–S130. [Google Scholar]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T.; Darden, T.; Duke, R.; Gohlke, H.; et al. Amber 14; University of California: San Fransisco, CA, USA, 2014. [Google Scholar]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Chapter 41—Advances in Electronic Structure Theory: GAMESS a Decade Later. In Theory and Applications of Computational Chemistry; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. ISBN 978-0-444-51719-7. [Google Scholar]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Pople, J.A.; Gill, P.M.W.; Johnson, B.G. Kohn—Sham Density-Functional Theory within a Finite Basis Set. Chem. Phys. Lett. 1992, 199, 557–560. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the Parameterization of the Local Correlation Functional. What Is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Laimou, D.; Katsila, T.; Matsoukas, J.; Schally, A.; Gkountelias, K.; Liapakis, G.; Tamvakopoulos, C.; Tselios, T. Rationally Designed Cyclic Analogues of Luteinizing Hormone-Releasing Hormone: Enhanced Enzymatic Stability and Biological Properties. Eur. J. Med. Chem. 2012, 58, 237–247. [Google Scholar] [CrossRef]
- Luthman, M.; Holmgren, A. Rat Liver Thioredoxin and Thioredoxin Reductase: Purification Characterization. Biochemistry 1982, 21, 6628–6633. [Google Scholar] [CrossRef]
- Sflakidou, E.; Leonidis, G.; Foroglou, E.; Siokatas, C.; Sarli, V. Recent Advances in Natural Product-Based Hybrids as Anti-Cancer Agents. Molecules 2022, 27, 6632. [Google Scholar] [CrossRef]
- Engel, J.; Emons, G.; Pinski, J.; Schally, A.V. AEZS-108: A Targeted Cytotoxic Analog of LHRH for the Treatment of Cancers Positive for LHRH Receptors. Expert Opin. Investig. Drugs 2012, 21, 891–899. [Google Scholar] [CrossRef]







 ) or presence of TrxR (
) or presence of TrxR ( ) and (B) insulin in the absence (
) and (B) insulin in the absence ( ) or presence of TrxR (
) or presence of TrxR ( ).
) or presence of TrxR () and (B) insulin in the absence () or presence of TrxR ().
).
) or presence of TrxR () and (B) insulin in the absence () or presence of TrxR ().

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biniari, G.; Markatos, C.; Nteli, A.; Tzoupis, H.; Simal, C.; Vlamis-Gardikas, A.; Karageorgos, V.; Pirmettis, I.; Petrou, P.; Venihaki, M.; et al. Rational Design, Synthesis and Binding Affinity Studies of Anthraquinone Derivatives Conjugated to Gonadotropin-Releasing Hormone (GnRH) Analogues towards Selective Immunosuppression of Hormone-Dependent Cancer. Int. J. Mol. Sci. 2023, 24, 15232. https://doi.org/10.3390/ijms242015232
Biniari G, Markatos C, Nteli A, Tzoupis H, Simal C, Vlamis-Gardikas A, Karageorgos V, Pirmettis I, Petrou P, Venihaki M, et al. Rational Design, Synthesis and Binding Affinity Studies of Anthraquinone Derivatives Conjugated to Gonadotropin-Releasing Hormone (GnRH) Analogues towards Selective Immunosuppression of Hormone-Dependent Cancer. International Journal of Molecular Sciences. 2023; 24(20):15232. https://doi.org/10.3390/ijms242015232
Chicago/Turabian StyleBiniari, Georgia, Christos Markatos, Agathi Nteli, Haralambos Tzoupis, Carmen Simal, Alexios Vlamis-Gardikas, Vlasios Karageorgos, Ioannis Pirmettis, Panagiota Petrou, Maria Venihaki, and et al. 2023. "Rational Design, Synthesis and Binding Affinity Studies of Anthraquinone Derivatives Conjugated to Gonadotropin-Releasing Hormone (GnRH) Analogues towards Selective Immunosuppression of Hormone-Dependent Cancer" International Journal of Molecular Sciences 24, no. 20: 15232. https://doi.org/10.3390/ijms242015232
APA StyleBiniari, G., Markatos, C., Nteli, A., Tzoupis, H., Simal, C., Vlamis-Gardikas, A., Karageorgos, V., Pirmettis, I., Petrou, P., Venihaki, M., Liapakis, G., & Tselios, T. (2023). Rational Design, Synthesis and Binding Affinity Studies of Anthraquinone Derivatives Conjugated to Gonadotropin-Releasing Hormone (GnRH) Analogues towards Selective Immunosuppression of Hormone-Dependent Cancer. International Journal of Molecular Sciences, 24(20), 15232. https://doi.org/10.3390/ijms242015232