
Characterizing Early Cardiac Metabolic Programming via 30% Maternal Nutrient Reduction during Fetal Development in a Non-Human Primate Model

 ,
,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
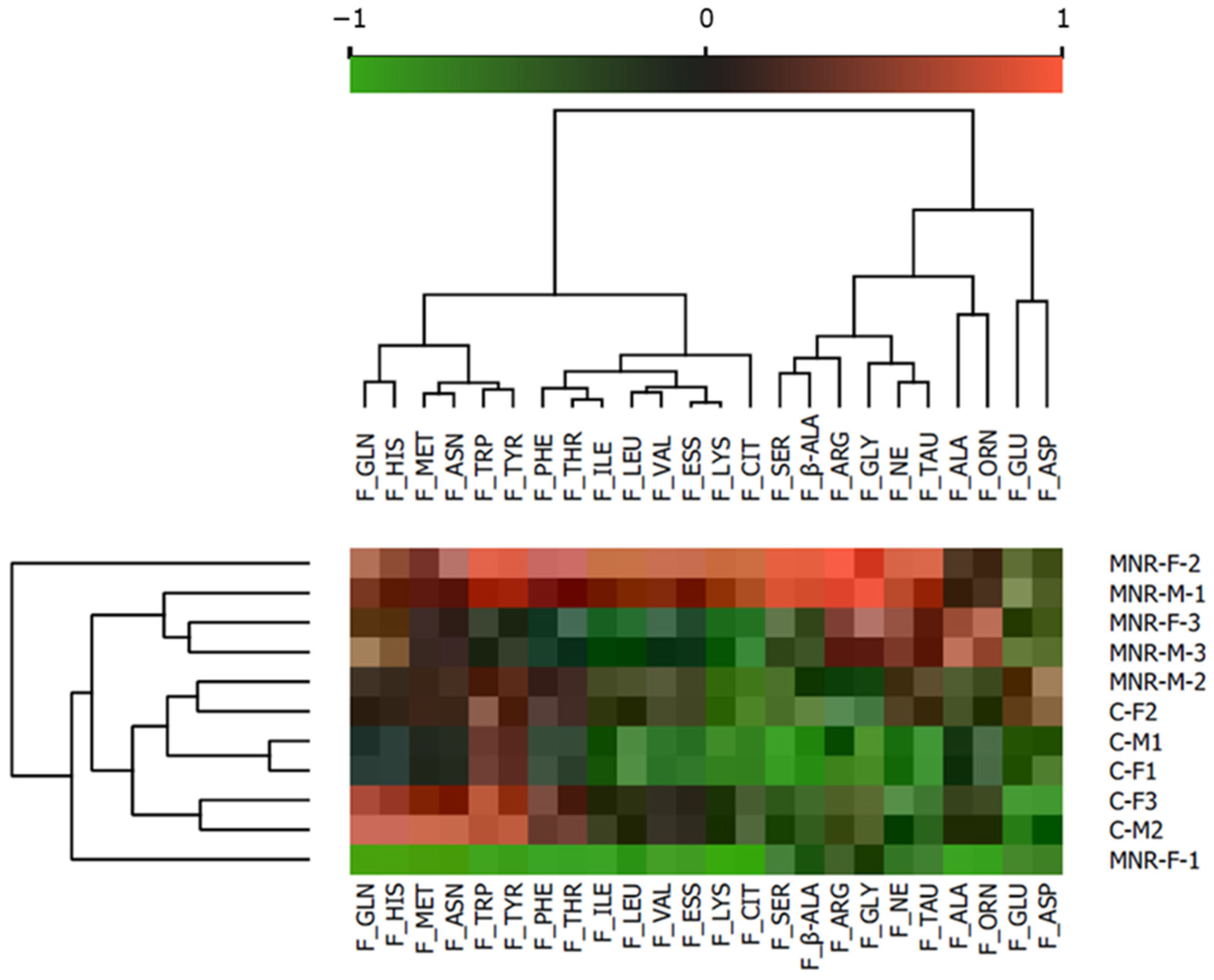
2.1. MNR-Induced Maternal and Fetal Morphometrics Alterations Accompained by Fetal Blood Biochemistry Changes
2.2. No Alterations Were Detected in Cardiac Mitochondrial DNA Copy Number
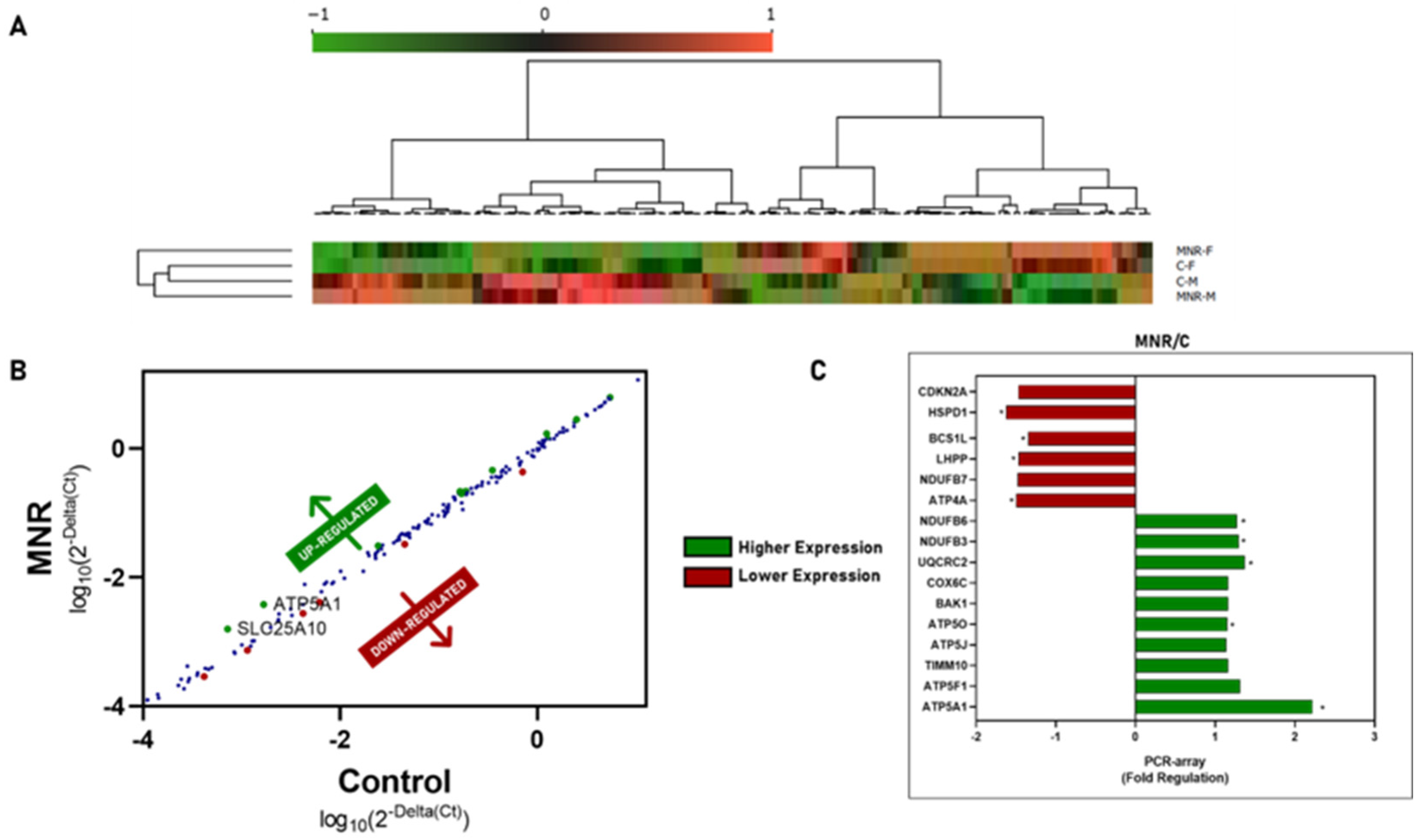
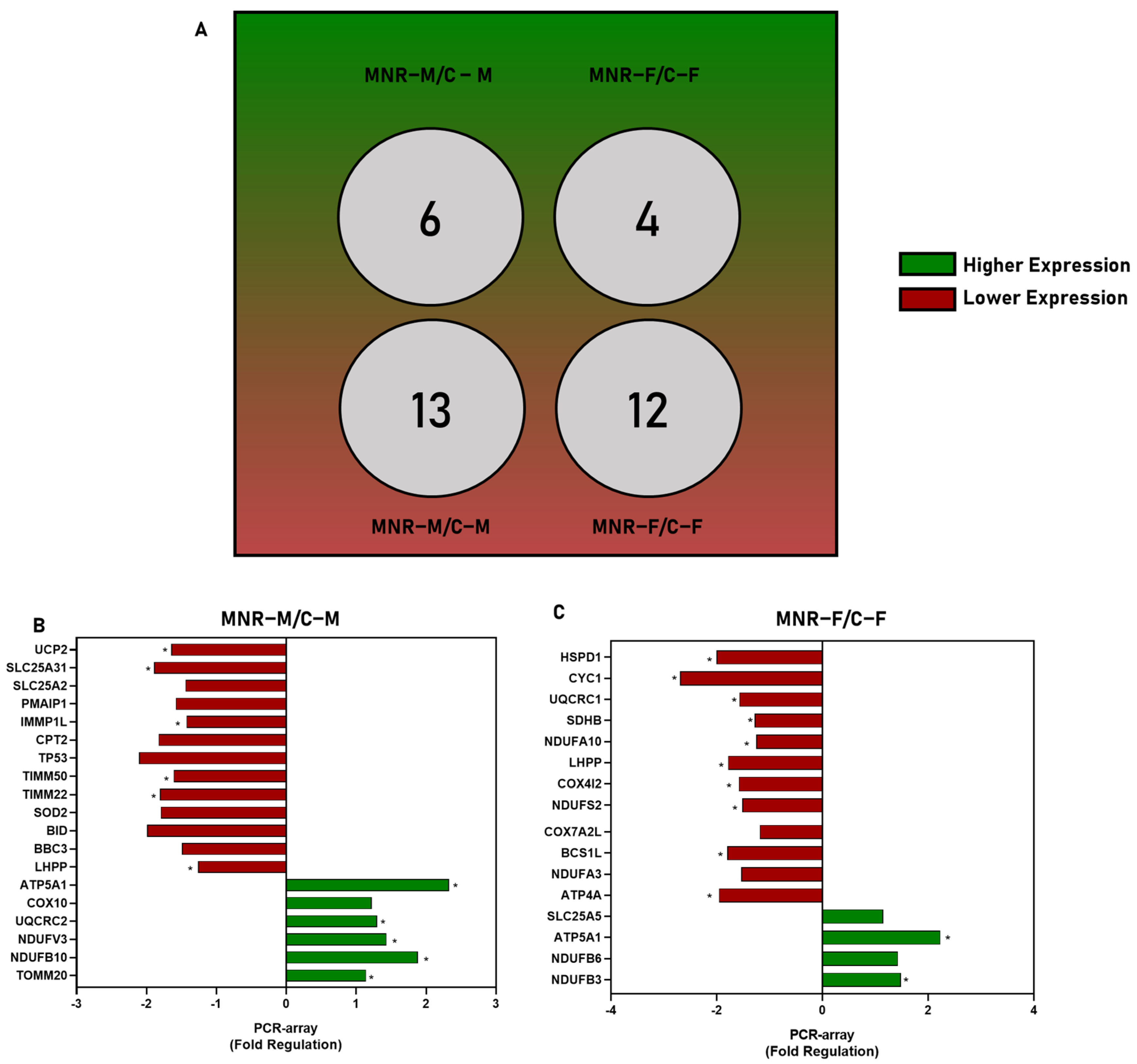
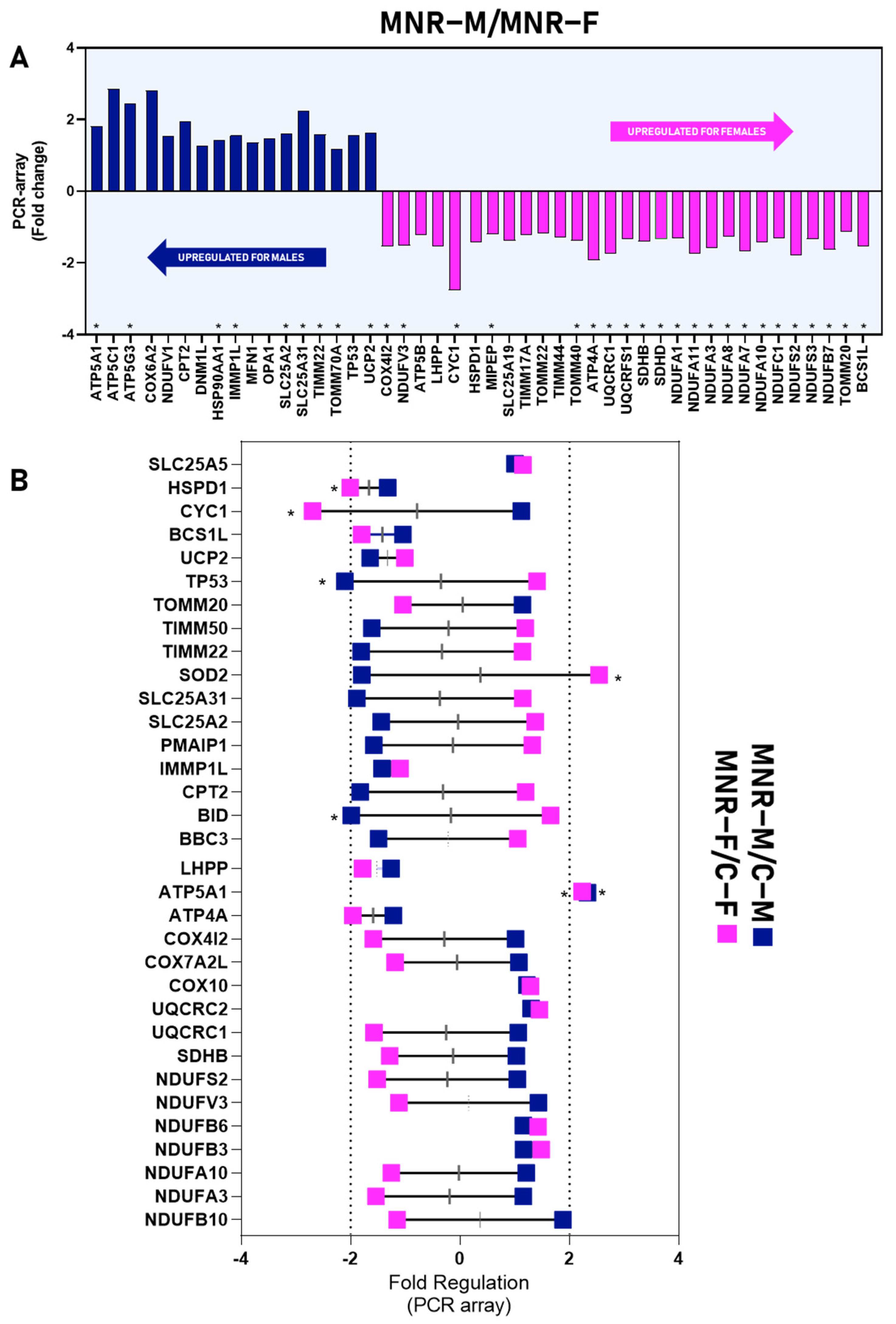
2.3. MNR-Induced Mitochondrial Transcriptional Alteration in the Fetal Cardiac Left Ventricle at 0.5 Gestation
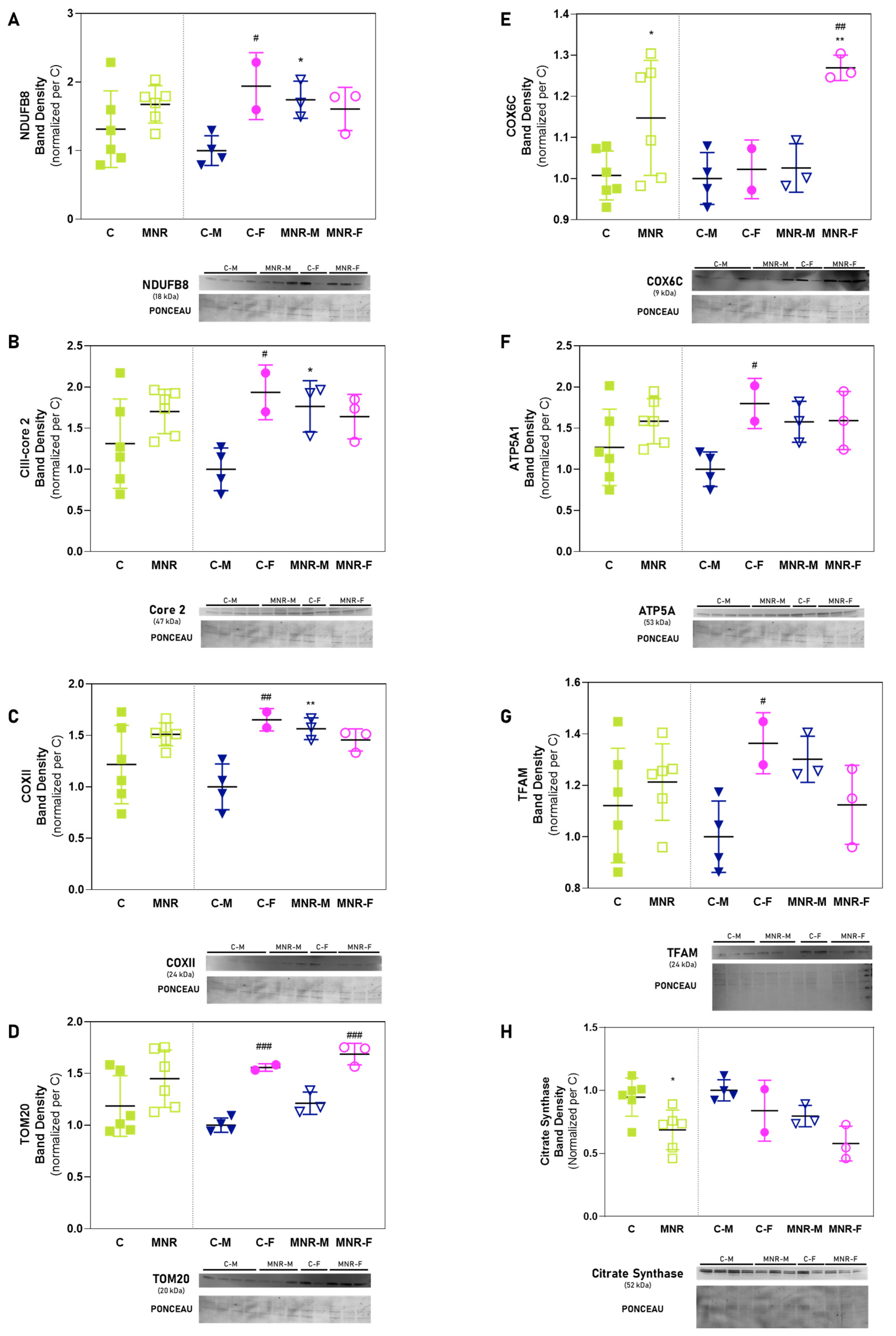
2.4. MNR-Induced Mitochondrial Protein Modulation in the Fetal Cardiac Left Ventricle at 0.5 Gestation
3. Discussion
4. Materials and Methods
4.1. Animal Care and In Vivo Procedures
4.1.1. Animal Care and Maintenance
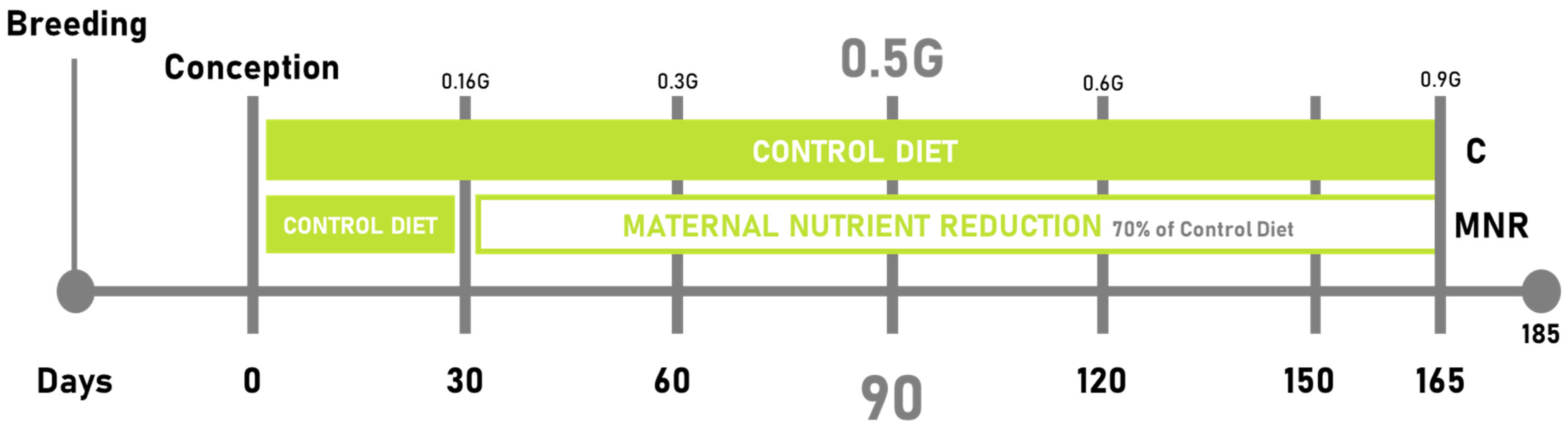
4.1.2. Experimental Design
4.1.3. Cesarean Section, Fetal and Maternal Morphometry, and Blood Sampling
4.2. Analysis of mtDNA Copy Number via Quantitative Real-Time PCR
4.3. Gene Expression Analysis by PCR Array
4.4. Protein Analyses via Western Blotting
4.5. Data Analysis and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersson, C.; Vasan, R.S. Epidemiology of Cardiovascular Disease in Young Individuals. Nat. Rev. Cardiol. 2018, 15, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Flora, G.D.; Nayak, M.K. A Brief Review of Cardiovascular Diseases, Associated Risk Factors and Current Treatment Regimes. Curr. Pharm. Des. 2019, 25, 4063–4084. [Google Scholar] [CrossRef] [PubMed]
- Gerdts, E.; Regitz-Zagrosek, V. Sex Differences in Cardiometabolic Disorders. Nat. Med. 2019, 25, 1657–1666. [Google Scholar] [CrossRef]
- Grilo, L.F.; Tocantins, C.; Diniz, M.S.; Gomes, R.M.; Oliveira, P.J.; Matafome, P.; Pereira, S.P. Metabolic Disease Programming: From Mitochondria to Epigenetics, Glucocorticoid Signalling and Beyond. Eur. J. Clin. Invest. 2021, 51. [Google Scholar] [CrossRef]
- Diniz, M.S.; Grilo, L.F.; Tocantins, C.; Falcão-Pires, I.; Pereira, S.P. Made in the Womb: Maternal Programming of Offspring Cardiovascular Function by an Obesogenic Womb. Metabolites 2023, 13, 845. [Google Scholar] [CrossRef]
- McDonald, T.J.; Wu, G.; Nijland, M.J.; Jenkins, S.L.; Nathanielsz, P.W.; Jansson, T. Effect of 30% Nutrient Restriction in the First Half of Gestation on Maternal and Fetal Baboon Serum Amino Acid Concentrations. Br. J. Nutr. 2013, 109, 1382–1388. [Google Scholar] [CrossRef]
- Kesavan, K.; Devaskar, S.U. Intrauterine Growth Restriction: Postnatal Monitoring and Outcomes. Pediatr. Clin. N. Am. 2019, 66, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Deter, R.L.; Lee, W.; Dicker, P.; Tully, E.C.; Cody, F.; Malone, F.D.; Flood, K.M. Growth patterns and cardiovascular abnormalities in small for gestational age fetuses: 1. Pattern characteristics. J. Matern. Neonatal Med. 2019, 34, 3029–3038. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Wong, F.Y.; Horne, R.S.C.; Yiallourou, S.R. Intrauterine Growth Restriction: Impact on Cardiovascular Development and Function throughout Infancy. Pediatr. Res. 2016, 79, 821–830. [Google Scholar] [CrossRef]
- Jonker, S.S.; Kamna, D.; LoTurco, D.; Kailey, J.; Brown, L.D. IUGR Impairs Cardiomyocyte Growth and Maturation in Fetal Sheep. J. Endocrinol. 2018, 239, 253–265. [Google Scholar] [CrossRef]
- Zogopoulos, G.; Nathanielsz, P.; Hendy, G.; Goodyer, C. The Baboon: A Model for the Study of Primate Growth Hormone Receptor Gene Expression during Development. J. Mol. Endocrinol. 1999, 23, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.P.; Tavares, L.C.; Duarte, A.I.; Baldeiras, I.; Cunha-Oliveira, T.; Martins, J.D.; Santos, M.S.; Maloyan, A.; Moreno, A.J.; Cox, L.A.; et al. Sex-Dependent Vulnerability of Fetal Nonhuman Primate Cardiac Mitochondria to Moderate Maternal Nutrient Reduction. Clin Sci (Lond) 2021, 135, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; McDonald, T.J.; Wu, G.; Nijland, M.J.; Nathanielsz, P.W. Intrauterine Growth Restriction Alters Term Fetal Baboon Hypothalamic Appetitive Peptide Balance. J. Endocrinol. 2013, 217, 275–282. [Google Scholar] [CrossRef]
- Kuo, A.H.; Li, C.; Li, J.; Huber, H.F.; Nathanielsz, P.W.; Clarke, G.D. Cardiac Remodelling in a Baboon Model of Intrauterine Growth Restriction Mimics Accelerated Ageing. J. Physiol. 2017, 595, 1093–1110. [Google Scholar] [CrossRef] [PubMed]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Vringer, E.; Tait, S.W.G. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2022, 30, 304–312. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, X.; Hu, Q.; Wu, J.; Wang, G.; Hong, Z.; Ren, J. Mitochondrial DNA in liver inflammation and oxidative stress. Life Sci. 2019, 236, 116464. [Google Scholar] [CrossRef]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mt DNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef] [PubMed]
- Tocantins, C.; Diniz, M.S.; Grilo, L.F.; Pereira, S.P. The Birth of Cardiac Disease: Mechanisms Linking Gestational Diabetes Mellitus and Early Onset of Cardiovascular Disease in Offspring. WIREs Mech. Dis. 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- A Mohamed, T.M.; Abouleisa, R.; Hill, B.G. Metabolic Determinants of Cardiomyocyte Proliferation. Stem Cells 2022, 40, 458–467. [Google Scholar] [CrossRef]
- Torres-Cuevas, I.; Parra-Llorca, A.; Sánchez-Illana, Á.; Nuñez-Ramiro, A.; Kuligowski, J.; Cháfer-Pericás, C.; Cernada, M.; Escobar, J.; Vento, M. Oxygen and oxidative stress in the perinatal period. Redox Biol. 2017, 12, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Díaz, M.; Nicolás-Ávila, J.Á.; Cordero, M.D.; Hidalgo, A. Mitochondrial Adaptations in the Growing Heart. Trends in Endocrinology & Metabolism 2020, 31, 308–319. [Google Scholar] [CrossRef]
- May-Panloup, P.; Boguenet, M.; Hachem, H.; Bouet, P.-E.; Reynier, P. Embryo and Its Mitochondria. Antioxidants 2021, 10, 139. [Google Scholar] [CrossRef]
- Pohjoismäki, J.L.; Goffart, S. The role of mitochondria in cardiac development and protection. Free. Radic. Biol. Med. 2017, 106, 345–354. [Google Scholar] [CrossRef]
- Piquereau, J.; Ventura-Clapier, R. Maturation of Cardiac Energy Metabolism During Perinatal Development. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perez-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial Oxidative Metabolism Is Required for the Cardiac Differentiation of Stem Cells. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, S60–S67. [Google Scholar] [CrossRef]
- Sahara, M.; Santoro, F.; Sohlmér, J.; Zhou, C.; Witman, N.; Leung, C.Y.; Mononen, M.; Bylund, K.; Gruber, P.; Chien, K.R. Population and Single-Cell Analysis of Human Cardiogenesis Reveals Unique LGR5 Ventricular Progenitors in Embryonic Outflow Tract. Dev. Cell 2019, 48, 475–490.e7. [Google Scholar] [CrossRef]
- Männer, J. When Does the Human Embryonic Heart Start Beating? A Review of Contemporary and Historical Sources of Knowledge about the Onset of Blood Circulation in Man. J. Cardiovasc. Dev. Dis. 2022, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Bazer, F.W.; Cudd, T.A.; Meininger, C.J.; Spencer, T.E. Maternal Nutrition and Fetal Development. J. Nutr. 2004, 134, 2169–2172. [Google Scholar] [CrossRef]
- Jain, S.; Maheshwari, A.; Jain, S.K. Maternal Nutrition and Fetal/Infant Development. Clin. Perinatol. 2022, 49, 313–330. [Google Scholar] [CrossRef]
- Lindsay, K.L.; Buss, C.; Wadhwa, P.D.; Entringer, S. The Interplay Between Nutrition and Stress in Pregnancy: Implications for Fetal Programming of Brain Development. Biol. Psychiatry 2018, 85, 135–149. [Google Scholar] [CrossRef]
- Ghaly, A.; Maki, Y.; Nygard, K.; Hammond, R.; Hardy, D.B.; Richardson, B.S. Maternal nutrient restriction in guinea pigs leads to fetal growth restriction with increased brain apoptosis. Pediatr. Res. 2018, 85, 105–112. [Google Scholar] [CrossRef]
- Heasman, L.; Clarke, L.; Stephenson, T.J.; Symonds, M.E. The influence of maternal nutrient restriction in early to mid-pregnancy on placental and fetal development in sheep. Proc. Nutr. Soc. 1999, 58, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Kovalskys, I.; Mazza, C.S. 3.17 Infants Born Small for Gestational Age at Term. In Pediatric Nutrition in Practice; Karger: Basel, Switzerland, 2022; pp. 338–344. [Google Scholar] [CrossRef]
- Wilcox, A.J.; Cortese, M.; McConnaughey, D.R.; Moster, D.; Basso, O. The limits of small-for-gestational-age as a high-risk category. Eur. J. Epidemiol. 2021, 36, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Christian, P. Fetal Growth Restriction and Preterm as Determinants of Child Growth in the First Two Years and Potential Interventions. In International Nutrition: Achieving Millennium Goals and Beyond; Karger: Basel, Switzerland, 2014; pp. 81–91. [Google Scholar] [CrossRef]
- Sandoval, C.; Wu, G.; Smith, S.B.; Dunlap, K.A.; Satterfield, M.C. Maternal Nutrient Restriction and Skeletal Muscle Development: Consequences for Postnatal Health. In Amino Acids in Nutrition and Health; Springer: Cham, Switzerland, 2020; pp. 153–165. [Google Scholar] [CrossRef]
- Albu, A.; Anca, A.; Horhoianu, V.; Horhoianu, I. Predictive factors for intrauterine growth restriction. J. Med. Life 2014, 7, 165–171. [Google Scholar]
- Dapkekar, P.; Bhalerao, A.; Kawathalkar, A.; Vijay, N. Risk Factors Associated With Intrauterine Growth Restriction: A Case-Control Study. Cureus 2023, 15, e40178. [Google Scholar] [CrossRef]
- Menendez-Castro, C.; Rascher, W.; Hartner, A. Intrauterine Growth Restriction—Impact on Cardiovascular Diseases Later in Life. Mol. Cell Pediatr. 2018, 5. [Google Scholar] [CrossRef]
- Dorn, G.W.; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Minerva Anestesiol. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Economides, D.L.; Nicolaides, K.H. Blood Glucose and Oxygen Tension Levels in Small-for-Gestational-Age Fetuses. Am. J. Obstet. Gynecol. 1989, 160, 385–389. [Google Scholar] [CrossRef]
- Smoak, I.W. Hypoglycemia and embryonic heart development. Front. Biosci. 2002, 7, d307–d318. [Google Scholar] [CrossRef] [PubMed]
- Smoak, I.W.; Branch, S. Glut-1 expression and its response to hypoglycemia in the embryonic mouse heart. Anat. Embryol. 2000, 201, 327–333. [Google Scholar] [CrossRef]
- Tan, C.M.J.; Lewandowski, A.J. The Transitional Heart: From Early Embryonic and Fetal Development to Neonatal Life. Fetal Diagn. Ther. 2020, 47, 373–386. [Google Scholar] [CrossRef]
- Garbern, J.C.; Lee, R.T. Mitochondria and Metabolic Transitions in Cardiomyocytes: Lessons from Development for Stem Cell-Derived Cardiomyocytes. Stem Cell Res. Ther. 2021, 12, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.M.; Larsen, N.K.; Thankam, F.G.; Agrawal, D.K. Fetal Cardiomyocyte Phenotype, Ketone Body Metabolism, and Mitochondrial Dysfunction in the Pathology of Atrial Fibrillation. Mol. Cell Biochem. 2021, 476, 1165. [Google Scholar] [CrossRef]
- Zhao, Q.; Sun, Q.; Zhou, L.; Liu, K.; Jiao, K. Complex Regulation of Mitochondrial Function during Cardiac Development. J. Am. Heart Assoc. 2019, 8. [Google Scholar] [CrossRef]
- Watanabe, M.; Jafri, A.; Fisher, S.A. Apoptosis Is Required for the Proper Formation of the Ventriculo-Arterial Connections. Dev. Biol. 2001, 240, 274–288. [Google Scholar] [CrossRef]
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W.; Scorrano, L. Mitochondrial Fusion Directs Cardiomyocyte Differentiation via Calcineurin and Notch Signaling. Science 2013, 342, 734–737. [Google Scholar] [CrossRef]
- Hsiao, C.P.; Chen, M.K.; Veigl, M.L.; Ellis, R.; Cooney, M.; Daly, B.; Hoppel, C. Relationships between Expression of BCS1L, Mitochondrial Bioenergetics, and Fatigue among Patients with Prostate Cancer. Cancer Manag. Res. 2019, 11, 6703–6717. [Google Scholar] [CrossRef]
- Beeckmans, S. Some structural and regulatory aspects of citrate synthase. Int. J. Biochem. 1984, 16, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, B.; Thrush, A.B.; Quizi, J.; Antoun, G.; McIntosh, N.; Al-Dirbashi, O.Y.; Patti, M.E.; Harper, M.E. Undernutrition during Pregnancy in Mice Leads to Dysfunctional Cardiac Muscle Respiration in Adult Offspring. Biosci. Rep. 2015, 35, 1–10. [Google Scholar] [CrossRef]
- TL, L.; CR, T.; CL, Y.; YP, W.; SH, K. Estrogen Receptor-β in Mitochondria: Implications for Mitochondrial Bioenergetics and Tumorigenesis. Ann. N. Y Acad. Sci. 2015, 1350, 52–60. [Google Scholar] [CrossRef]
- Gonzalez-Tendero, A.; Torre, I.; Garcia-Canadilla, P.; Crispi, F.; García-García, F.; Dopazo, J.; Bijnens, B.; Gratacós, E. Intrauterine Growth Restriction Is Associated with Cardiac Ultrastructural and Gene Expression Changes Related to the Energetic Metabolism in a Rabbit Model. Am. J. Physiol. Heart Circ. Physiol. 2013, 305. [Google Scholar] [CrossRef][Green Version]
- Guitart-Mampel, M.; Gonzalez-Tendero, A.; Niñerola, S.; Morén, C.; Catalán-Garcia, M.; González-Casacuberta, I.; Juárez-Flores, D.L.; Ugarteburu, O.; Matalonga, L.; Cascajo, M.V.; et al. Cardiac and Placental Mitochondrial Characterization in a Rabbit Model of Intrauterine Growth Restriction. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1157–1167. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New Emerging Biological Function and Therapeutic Target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, A.E.T.S.; Bassaneze, V.; Forni, M.F.; Keusseyan, A.A.; Kowaltowski, A.J.; Krieger, J.E. Early Postnatal Cardiomyocyte Proliferation Requires High Oxidative Energy Metabolism. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Bisaccia, G.; Ricci, F.; Gallina, S.; Di Baldassarre, A.; Ghinassi, B. Mitochondrial Dysfunction and Heart Disease: Critical Appraisal of an Overlooked Association. Int. J. Mol. Sci. 2021, 22, 614. [Google Scholar] [CrossRef] [PubMed]
- Stamerra, C.A.; Di Giosia, P.; Giorgini, P.; Ferri, C.; Sukhorukov, V.N.; Sahebkar, A. Mitochondrial Dysfunction and Cardiovascular Disease: Pathophysiology and Emerging Therapies. Oxid. Med. Cell. Longev. 2022, 2022, 9530007. [Google Scholar] [CrossRef]
- Elias, A.A.; Maki, Y.; Matushewski, B.; Nygard, K.; Regnault, T.R.H.; Richardson, B.S. Maternal Nutrient Restriction in Guinea Pigs Leads to Fetal Growth Restriction with Evidence for Chronic Hypoxia. Pediatr. Res. 2017, 82, 141–147. [Google Scholar] [CrossRef]
- Radford, B.N.; Han, V.K.M. Evidence of Increased Hypoxia Signaling in Fetal Liver from Maternal Nutrient Restriction in Mice. Pediatr. Res. 2020, 87, 450–455. [Google Scholar] [CrossRef]
- Natarajan, B.; Arige, V.; Khan, A.A.; Reddy, S.S.; Barthwal, M.K.; Mahapatra, N.R. Hypoxia-Mediated Regulation of Mitochondrial Transcription Factors in Renal Epithelial Cells: Implications for Hypertensive Renal Physiology. Hypertens. Res. 2021, 44, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-S.; Zhou, Y.-N.; Li, L.; Li, S.-F.; Long, D.; Chen, X.-L.; Zhang, J.-B.; Feng, L.; Li, Y.-P. HIF-1α Protects against Oxidative Stress by Directly Targeting Mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA Damage and Reactive Oxygen Species in Neurodegenerative Disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Schlabritz-Loutsevitch, N.E.; Howell, K.; Rice, K.; Glover, E.J.; Nevill, C.H.; Jenkins, S.L.; Cummins, L.B.; Frost, P.A.; McDonald, T.J.; Nathanielsz, P.W. Development of a System for Individual Feeding of Baboons Maintained in an Outdoor Group Social Environment. J. Med. Primatol. 2004, 33, 117–126. [Google Scholar] [CrossRef]
- Cox, L.A.; Nijland, M.J.; Gilbert, J.S.; Schlabritz-Loutsevitch, N.E.; Hubbard, G.B.; McDonald, T.J.; Shade, R.E.; Nathanielsz, P.W. Effect of 30 per Cent Maternal Nutrient Restriction from 0.16 to 0.5 Gestation on Fetal Baboon Kidney Gene Expression. J. Physiol. 2006, 572, 67–85. [Google Scholar] [CrossRef]
- Machado, N.G.; Baldeiras, I.; Pereira, G.C.; Pereira, S.P.; Oliveira, P.J. Sub-Chronic Administration of Doxorubicin to Wistar Rats Results in Oxidative Stress and Unaltered Apoptotic Signaling in the Lung. Chem. Biol. Interact. 2010, 188, 478–486. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Maternal and Fetal Morphological Parameters at 0.5 Gestation | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Maternal | |||||||||||||||||
| Combination | Mothers of Male Fetuses | Mothers of Female Fetuses | Mann–Whitney U Test | ||||||||||||||
| C | MNR | C | MNR | C | MNR | Diet | Male C vs. MNR | Female C vs. MNR | C M vs. F | MNR M vs. F | |||||||
| (n = 11) | (n = 11) | (n = 6) | (n = 6) | (n = 5) | (n = 5) | ||||||||||||
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | p−Value | p-Value | p-Value | p-Value | p-Value | |
| Body mass at pre-conception (kg) | 13.90 | 0.46 | 14.26 | 0.61 | 13.45 | 0.71 | 14.79 | 1.00 | 14.34 | 0.59 | 13.61 | 0.60 | – | – | – | – | – |
| Body mass at Cs (kg) | 13.79 | 0.46 | 13.54 | 0.65 | 13.41 | 0.75 | 14.07 | 1.07 | 14.18 | 0.55 | 12.90 | 0.65 | – | – | – | – | – |
| Body mass variation (kg) | −0.11 | 0.12 | −0.72 | 0.15 | −0.05 | 0.20 | −0.72 | 0.20 | −0.16 | 0.15 | −0.71 | 0.24 | 0.008 | – | 0.047 | – | – |
| Age (year) | 10.79 | 0.94 | 9.86 | 0.82 | 11.27 | 1.83 | 9.84 | 1.45 | 10.30 | 0.72 | 9.88 | 0.74 | – | – | – | – | – |
| Fetal | |||||||||||||||||
| Combined Sex | Male | Female | Mann–Whitney U Test | ||||||||||||||
| C | MNR | C | MNR | C | MNR | Diet | Male C vs. MNR | Female C vs. MNR | C M vs. F | MNR M vs. F | |||||||
| (n = 11) | (n = 11) | (n = 6) | (n = 6) | (n = 5) | (n = 5) | ||||||||||||
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | p-Value | p-Value | p-Value | p-Value | p-Value | |
| Body mass (g) | 101.14 | 3.25 | 101.19 | 2.96 | 103.32 | 5.55 | 103.67 | 4.69 | 98.52 | 2.98 | 98.22 | 3.35 | – | – | – | – | – |
| Heart mass (g) | 0.85 | 0.21 | 0.81 | 0.19 | 0.59 | 0.07 | 0.93 | 0.34 | 1.12 | 0.41 | 0.66 | 0.05 | – | – | – | – | – |
| HW/BW (×1000) | 8.31 | 2.01 | 7.84 | 1.59 | 5.49 | 0.61 | 8.82 | 2.96 | 11.14 | 3.72 | 6.66 | 0.33 | – | – | – | 0.047 | – |
| Body length (cm) | 17.41 | 0.31 | 17.52 | 0.27 | 17.00 | 0.41 | 17.50 | 0.29 | 17.90 | 0.43 | 17.54 | 0.52 | – | – | – | – | – |
| BMI (kg/m2) | 3.35 | 0.12 | 3.31 | 0.13 | 3.58 | 0.15 | 3.40 | 0.22 | 3.09 | 0.13 | 3.20 | 0.12 | – | – | – | 0.045 | – |
| Chest circ (cm) | 58.77 | 11.92 | 52.32 | 12.39 | 35.25 | 16.64 | 48.75 | 17.77 | 87.00 | 1.22 | 56.60 | 19.04 | – | – | – | – | – |
| Waist circ (cm) | 49.77 | 10.02 | 44.95 | 10.79 | 30.42 | 14.10 | 42.50 | 15.35 | 73.00 | 2.55 | 47.90 | 16.82 | – | – | – | – | – |
| Hip circ (cm) | 64.31 | 8.31 | 68.33 | 1.67 | 50.67 | 21.88 | 70.00 | 2.89 | 72.50 | 2.24 | 66.67 | 1.67 | – | – | – | – | – |
| Femur (cm) | 3.47 | 0.09 | 3.20 | 0.11 | 3.28 | 0.10 | 3.33 | 0.17 | 3.70 | 0.09 | 3.04 | 0.13 | – | – | 0.014 | 0.021 | – |
| Comparison of Maternal and Fetal Blood Biochemical Parameters at 0.5 G | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Maternal | |||||||||||||||||
| Combination | Mothers of Male Fetuses | Mothers of Female Fetuses | Mann–Whitney U Test | ||||||||||||||
| C | MNR | C | MNR | C | MNR | Diet | Male C vs. MNR | Female C vs. MNR | C M vs. F | MNR M vs. F | |||||||
| (n = 8) | (n = 6) | (n = 3) | (n = 3) | (n = 5) | (n = 3) | ||||||||||||
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | p-Value | p-Value | p-Value | p-Value | p-Value | |
| Hemoglobin (g/dL) | 12.61 | 0.34 | 12.90 | 0.44 | 12.73 | 0.46 | 13.27 | 0.15 | 12.54 | 0.51 | 12.53 | 0.90 | – | – | – | – | – |
| Glucose | 74.38 | 9.63 | 85.83 | 10.63 | 55.67 | 2.91 | 97.67 | 13.22 | 85.60 | 13.15 | 74.00 | 15.82 | – | 0.05 | 0.04 | ||
| BUN | 10.25 | 0.67 | 7.50 | 0.43 | 8.67 | 0.67 | 7.33 | 0.67 | 11.20 | 0.73 | 7.67 | 0.67 | 0.01 | – | 0.02 | 0.04 | |
| BUN/CREAT Ratio | 11.59 | 1.40 | 8.37 | 0.61 | 12.30 | 3.86 | 7.13 | 0.43 | 11.16 | 0.95 | 9.60 | 0.40 | – | – | – | – | 0.04 |
| Cholesterol | 51.63 | 5.64 | 42.17 | 1.70 | 44.33 | 9.26 | 40.00 | 2.00 | 56.00 | 7.11 | 44.33 | 2.40 | – | – | – | – | – |
| Total Protein | 6.70 | 0.19 | 6.68 | 0.16 | 6.73 | 0.30 | 6.73 | 0.33 | 6.68 | 0.27 | 6.63 | 0.15 | – | – | – | – | – |
| Sodium | 140.00 | 0.98 | 142.83 | 1.01 | 138.67 | 1.86 | 143.67 | 1.76 | 140.80 | 1.11 | 142.00 | 1.15 | 0.05 | – | – | – | – |
| Potassium | 3.59 | 0.10 | 3.92 | 0.21 | 3.37 | 0.19 | 4.03 | 0.18 | 3.72 | 0.09 | 3.80 | 0.42 | – | 0.05 | – | – | – |
| Triglycerides | 28.88 | 3.36 | 30.00 | 6.12 | 26.67 | 4.63 | 33.67 | 11.67 | 30.20 | 4.90 | 26.33 | 6.12 | – | – | – | – | – |
| Fetal | |||||||||||||||||
| Combined Sex | Male | Female | Mann–Whitney U Test | ||||||||||||||
| C | MNR | C | MNR | C | MNR | Diet | Male C vs. MNR | Female C vs. MNR | C M vs. F | MNR M vs. F | |||||||
| (n = 8) | (n = 6) | (n = 3) | (n = 3) | (n = 5) | (n = 3) | ||||||||||||
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | p-Value | P-Value | p-Value | p-Value | p-Value | |
| Hemoglobin (g/dL) | 11.08 | 0.21 | 11.77 | 0.15 | 11.37 | 0.09 | . | . | 10.65 | 0.35 | 11.77 | 0.15 | 0.04 | – | – | – | – |
| Glucose | 47.14 | 4.35 | 39.00 | 7.44 | 34.50 | 7.50 | 43.33 | 15.84 | 52.20 | 3.43 | 34.67 | 2.73 | – | – | 0.04 | – | – |
| BUN | 11.00 | 0.58 | 8.00 | 0.52 | 9.50 | 0.50 | 7.33 | 0.67 | 11.60 | 0.60 | 8.67 | 0.67 | 0.00 | – | 0.02 | 0.03 | – |
| BUN/CREAT Ratio | 13.97 | 0.96 | 9.97 | 0.98 | 13.35 | 3.35 | 8.50 | 0.76 | 14.22 | 0.88 | 11.43 | 1.43 | 0.04 | – | – | – | – |
| Cholesterol | 65.57 | 4.06 | 58.50 | 3.43 | 65.00 | 3.00 | 58.00 | 4.93 | 65.80 | 5.80 | 59.00 | 5.86 | – | – | – | – | – |
| Total Protein | 2.60 | 0.06 | 2.65 | 0.04 | 2.60 | 0.00 | 2.67 | 0.03 | 2.60 | 0.09 | 2.63 | 0.09 | – | – | – | – | – |
| Sodium | 137.71 | 1.66 | 141.40 | 0.40 | 134.50 | 6.50 | 141.00 | 0.00 | 139.00 | 0.32 | 141.67 | 0.67 | 0.01 | – | 0.02 | – | – |
| Potassium | 4.03 | 0.28 | 4.36 | 0.40 | 3.50 | 0.20 | 4.20 | 0.90 | 4.24 | 0.35 | 4.47 | 0.50 | – | – | – | – | – |
| Triglycerides | 36.43 | 4.94 | 28.83 | 5.53 | 37.00 | 8.00 | 25.00 | 4.04 | 36.20 | 6.69 | 32.67 | 11.05 | – | – | – | – | – |
| Gene Expression Fold Regulation and p-Value | |||
|---|---|---|---|
| Group | Gene | Fold Regulation | p-Value |
| MNR vs. C | CDKN2A | – | 0.0558 |
| HSPD1 | −1.6229 | 0.0097 | |
| BCS1L | −1.3446 | 0.0089 | |
| LHPP | −1.4705 | 0.0026 | |
| NDUFB7 | – | 0.0587 | |
| ATP4A | −1.5006 | 0.0199 | |
| NDUFB6 | 1.2714 | 0.0474 | |
| NDUFB3 | 1.2952 | 0.0125 | |
| UQCRC2 | 1.3692 | 0.0274 | |
| COX6C | – | 0.0714 | |
| BAK1 | – | 0.0856 | |
| ATP5O | 1.151 | 0.0373 | |
| ATP5J | – | 0.0588 | |
| TIMM10 | – | 0.0762 | |
| ATP5F1 | – | 0.0595 | |
| ATP5A1 | 2.2 | 0.0089 | |
| MNR-M vs. C-M | UCP2 | −1.6445 | 0.0155 |
| SLC25A31 | −1.8892 | 0.0416 | |
| SLC25A2 | – | 0.0677 | |
| PMAIP1 | – | 0.0770 | |
| IMMP1L | −1.4269 | 0.0070 | |
| CPT2 | – | 0.0958 | |
| TP53 | – | 0.0577 | |
| TIMM50 | −1.6130 | 0.0319 | |
| TIMM22 | −1.8063 | 0.0223 | |
| SOD2 | – | 0.0620 | |
| BID | – | 0.0759 | |
| BBC3 | – | 0.0848 | |
| LHPP | −1.2608 | 0.0369 | |
| ATP5A1 | 2.3 | 0.0004 | |
| COX10 | – | 0.0827 | |
| UQCRC2 | 1.2991 | 0.0298 | |
| NDUFV3 | 1.4313 | 0.0382 | |
| NDUFB10 | 1.8811 | 0.0035 | |
| TOMM20 | 1.1394 | 0.0481 | |
| MNR-F vs. C-F | HSPD1 | −2.0 | 0.0108 |
| CYC1 | −2.7 | 0.0039 | |
| UQCRC1 | 1.5758 | 0.0089 | |
| SDHB | −1.2856 | 0.0066 | |
| NDUFA10 | −1.2561 | 0.0236 | |
| LHPP | −1.7836 | 0.0142 | |
| COX4I2 | −1.5859 | 0.0211 | |
| NDUFS2 | −1.5158 | 0.0022 | |
| COX7A2L | – | 0.0671 | |
| BCS1L | −1.7991 | 0.0015 | |
| NDUFA3 | – | 0.0942 | |
| ATP4A | −1.9599 | 0.0182 | |
| SLC25A5 | – | 0.0868 | |
| ATP5A1 | 2.2 | 0.0419 | |
| NDUFB6 | – | 0.0974 | |
| NDUFB3 | 1.4818 | 0.0207 | |
| MNR-M vs. MNR-F | ATP5A1 | 1.8097 | 0.0446 |
| ATP5C1 | – | 0.0996 | |
| ATP5G3 | 2.4323 | 0.0173 | |
| COX6A2 | – | 0.0668 | |
| NDUFV1 | – | 0.0624 | |
| CPT2 | – | 0.0539 | |
| DNM1L | – | 0.0999 | |
| HSPD90AA1 | 1.4362 | 0.0029 | |
| IMMP1L | 1.5493 | 0.0042 | |
| MFN1 | – | 0.0924 | |
| OPA1 | – | 0.0655 | |
| SLC25A2 | 1.5927 | 0.0175 | |
| SLC25A31 | 2.2323 | 0.0154 | |
| TIMM22 | 1.5782 | 0.0229 | |
| TOMM70A | 1.1683 | 0.0102 | |
| TP53 | – | 0.0850 | |
| UCP2 | 1.6187 | 0.0465 | |
| COX4I2 | −1.5388 | 0.0368 | |
| NDUFV3 | −1.5110 | 0.0201 | |
| ATP5B | – | 0.0588 | |
| LHPP | −1.5396 | 0.0255 | |
| CYC1 | −2.7650 | 0.0036 | |
| HSPD1 | – | 0.0591 | |
| MIPEP | −1.1997 | 0.0007 | |
| SLC25A19 | – | 0.0997 | |
| TIMM17A | – | 0.0961 | |
| TOMM22 | – | 0.0823 | |
| TIMM44 | – | 0.0885 | |
| TOMM40 | −1.3842 | 0.0427 | |
| ATP4A | −1.9241 | 0.0034 | |
| UQCRC1 | −1.7358 | 0.0011 | |
| UQCRFS1 | −1.3300 | 0.0213 | |
| SDHB | −1.4021 | 0.0053 | |
| SDHD | −1.3393 | 0.0046 | |
| NDUFA1 | −1.3118 | 0.0246 | |
| NDUFA11 | −1.7337 | 0.0439 | |
| NDUFA3 | −1.5965 | 0.0239 | |
| NDUFA8 | −1.2592 | 0.0271 | |
| NDUFA7 | −1.6862 | 0.004 | |
| NDUFA10 | −1.4186 | 0.0113 | |
| NDUFC1 | −1.3032 | 0.0121 | |
| NDUFS2 | −1.7828 | 0.0039 | |
| NDUFS3 | −1.3326 | 0.0118 | |
| NDUFB7 | −1.6299 | 0.0388 | |
| TOMM20 | −1.1220 | 0.0320 | |
| BCS1L | −1.5516 | 0.0052 | |
| Position | Symbol | Refseq | Description |
|---|---|---|---|
| A01 | ATP12A | NM_001676 | ATPase, H+/K+ transporting, nongastric, alpha polypeptide |
| A02 | ATP4A | NM_000704 | ATPase, H+/K+ exchanging, alpha polypeptide |
| A03 | ATP4B | NM_000705 | ATPase, H+/K+ exchanging, beta polypeptide |
| A04 | ATP5A1 | NM_004046 | ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1 |
| A05 | ATP5B | NM_001686 | ATP synthase, H+ transporting, mitochondrial F1 complex, beta polypeptide |
| A06 | ATP5C1 | NM_005174 | ATP synthase, H+ transporting, mitochondrial F1 complex, gamma polypeptide 1 |
| A07 | ATP5F1 | NM_001688 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit B1 |
| A08 | ATP5G1 | NM_005175 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit C1 |
| A09 | ATP5G2 | NM_001002031 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit C2 |
| A10 | ATP5G3 | NM_001689 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit C3 |
| A11 | ATP5H | NM_006356 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit d |
| A12 | ATP5I | NM_007100 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit E |
| B01 | ATP5J | NM_001685 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit F6 |
| B02 | ATP5J2 | NM_004889 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit F2 |
| B03 | ATP5L | NM_006476 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit G |
| B04 | ATP5O | NM_001697 | ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit |
| B05 | ATP6V0A2 | NM_012463 | ATPase, H+ transporting, lysosomal V0 subunit a2 |
| B06 | ATP6V0D2 | NM_152565 | ATPase, H+ transporting, lysosomal 38 kDa, V0 subunit d2 |
| B07 | ATP6V1C2 | NM_144583 | ATPase, H+ transporting, lysosomal 42 kDa, V1 subunit C2 |
| B08 | ATP6V1E2 | NM_080653 | ATPase, H+ transporting, lysosomal 31 kDa, V1 subunit E2 |
| B09 | ATP6V1G3 | NM_133262 | ATPase, H+ transporting, lysosomal 13 kDa, V1 subunit G3 |
| B10 | BCS1L | NM_004328 | BCS1-like (S. cerevisiae) |
| B11 | COX4I1 | NM_001861 | Cytochrome c oxidase subunit IV isoform 1 |
| B12 | COX4I2 | NM_032609 | Cytochrome c oxidase subunit IV isoform 2 |
| C01 | COX5A | NM_004255 | Cytochrome c oxidase subunit Va |
| C02 | COX5B | NM_001862 | Cytochrome c oxidase subunit Vb |
| C03 | COX6A1 | NM_004373 | Cytochrome c oxidase subunit VIa polypeptide 1 |
| C04 | COX6A2 | NM_005205 | Cytochrome c oxidase subunit VIa polypeptide 2 |
| C05 | COX6B1 | NM_001863 | Cytochrome c oxidase subunit Vib polypeptide 1 |
| C06 | COX6B2 | NM_144613 | Cytochrome c oxidase subunit VIb polypeptide 2 |
| C07 | COX6C | NM_004374 | Cytochrome c oxidase subunit Vic |
| C08 | COX7A2 | NM_001865 | Cytochrome c oxidase subunit VIIa polypeptide 2 |
| C09 | COX7A2L | NM_004718 | Cytochrome c oxidase subunit VIIa polypeptide 2 like |
| C10 | COX7B | NM_001866 | Cytochrome c oxidase subunit VIIb |
| C11 | COX8A | NM_004074 | Cytochrome c oxidase subunit VIIIA |
| C12 | COX8C | NM_182971 | Cytochrome c oxidase subunit VIIIC |
| D01 | CYC1 | NM_001916 | Cytochrome c-1 |
| D02 | LHPP | NM_022126 | Phospholysine phosphohistidine inorganic pyrophosphate phosphatase |
| D03 | NDUFA1 | NM_004541 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 1 |
| D04 | NDUFA10 | NM_004544 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 10 |
| D05 | NDUFA11 | NM_175614 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 11 |
| D06 | NDUFA2 | NM_002488 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 2 |
| D07 | NDUFA3 | NM_004542 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 3 |
| D08 | NDUFA4 | NM_002489 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4 |
| D09 | NDUFA5 | NM_005000 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 5 |
| D10 | NDUFA6 | NM_002490 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 6 |
| D11 | NDUFA7 | NM_005001 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 7 |
| D12 | NDUFA8 | NM_014222 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 8 |
| E01 | NDUFAB1 | NM_005003 | NADH dehydrogenase (ubiquinone) 1, alpha/beta subcomplex, 1 |
| E02 | NDUFB10 | NM_004548 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 10 |
| E03 | NDUFB2 | NM_004546 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 2 |
| E04 | NDUFB3 | NM_002491 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 3 |
| E05 | NDUFB4 | NM_004547 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 4 |
| E06 | NDUFB5 | NM_002492 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 5 |
| E07 | NDUFB6 | NM_182739 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 6 |
| E08 | NDUFB7 | NM_004146 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 7 |
| E09 | NDUFB8 | NM_005004 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 8 |
| E10 | NDUFB9 | NM_005005 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 9 |
| E11 | NDUFC1 | NM_002494 | NADH dehydrogenase (ubiquinone) 1, subcomplex unknown, 1 |
| E12 | NDUFC2 | NM_004549 | NADH dehydrogenase (ubiquinone) 1, subcomplex unknown, 2 |
| F01 | NDUFS1 | NM_005006 | NADH dehydrogenase (ubiquinone) Fe-S protein 1 |
| F02 | NDUFS2 | NM_004550 | NADH dehydrogenase (ubiquinone) Fe-S protein 2 |
| F03 | NDUFS3 | NM_004551 | NADH dehydrogenase (ubiquinone) Fe-S protein 3 |
| F04 | NDUFS4 | NM_002495 | NADH dehydrogenase (ubiquinone) Fe-S protein 4 |
| F05 | NDUFS5 | NM_004552 | NADH dehydrogenase (ubiquinone) Fe-S protein 5 |
| F06 | NDUFS6 | NM_004553 | NADH dehydrogenase (ubiquinone) Fe-S protein 6 |
| F07 | NDUFS7 | NM_024407 | NADH dehydrogenase (ubiquinone) Fe-S protein 7 |
| F08 | NDUFS8 | NM_002496 | NADH dehydrogenase (ubiquinone) Fe-S protein 8 |
| F09 | NDUFV1 | NM_007103 | NADH dehydrogenase (ubiquinone) flavoprotein 1 |
| F10 | NDUFV2 | NM_021074 | NADH dehydrogenase (ubiquinone) flavoprotein 2 |
| F11 | NDUFV3 | NM_021075 | NADH dehydrogenase (ubiquinone) flavoprotein 3 |
| F12 | OXA1L | NM_005015 | Oxidase (cytochrome c) assembly 1-like |
| G01 | PPA1 | NM_021129 | Pyrophosphatase (inorganic) 1 |
| G02 | PPA2 | NM_176869 | Pyrophosphatase (inorganic) 2 |
| G03 | SDHA | NM_004168 | Succinate dehydrogenase complex, subunit A, flavoprotein (Fp) |
| G04 | SDHB | NM_003000 | Succinate dehydrogenase complex, subunit B, iron sulfur (Ip) |
| G05 | SDHC | NM_003001 | Succinate dehydrogenase complex, subunit C, integral membrane protein |
| G06 | SDHD | NM_003002 | Succinate dehydrogenase complex, subunit D, integral membrane protein |
| G07 | UQCR11 | NM_006830 | Ubiquinol-cytochrome c reductase, complex III subunit XI |
| G08 | UQCRC1 | NM_003365 | Ubiquinol-cytochrome c reductase core protein I |
| G09 | UQCRC2 | NM_003366 | Ubiquinol-cytochrome c reductase core protein II |
| G10 | UQCRFS1 | NM_006003 | Ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1 |
| G11 | UQCRH | NM_006004 | Ubiquinol-cytochrome c reductase hinge protein |
| G12 | UQCRQ | NM_014402 | Ubiquinol-cytochrome c reductase, complex III subunit VII, 9.5 kDa |
| H01 | B2M | NM_004048 | Beta-2-microglobulin |
| H02 | HPRT1 | NM_000194 | Hypoxanthine phosphoribosyltransferase 1 |
| H03 | RPL13A | NM_012423 | Ribosomal protein L13a |
| H04 | GAPDH | NM_002046 | Glyceraldehyde-3-phosphate dehydrogenase |
| H05 | ACTB | NM_001101 | Actin, beta |
| H06 | HGDC | SA_00105 | Human Genomic DNA Contamination |
| H07 | RTC | SA_00104 | Reverse Transcription Control |
| H08 | RTC | SA_00104 | Reverse Transcription Control |
| H09 | RTC | SA_00104 | Reverse Transcription Control |
| H10 | PPC | SA_00103 | Positive PCR Control |
| H11 | PPC | SA_00103 | Positive PCR Control |
| H12 | PPC | SA_00103 | Positive PCR Control |
| Position | Symbol | Refseq | Description |
|---|---|---|---|
| A01 | AIFM2 | NM_032797 | Apoptosis-inducing factor, mitochondrion-associated, 2 |
| A02 | AIP | NM_003977 | Aryl hydrocarbon receptor-interacting protein |
| A03 | BAK1 | NM_001188 | BCL2-antagonist/killer 1 |
| A04 | BBC3 | NM_014417 | BCL2 binding component 3 |
| A05 | BCL2 | NM_000633 | B-cell CLL/lymphoma 2, apoptosis regulator |
| A06 | BCL2L1 | NM_138578 | BCL2-like 1, apoptosis regulator BCLX |
| A07 | BID | NM_001196 | BH3-interacting domain death agonist |
| A08 | BNIP3 | NM_004052 | BCL2/adenovirus E1B 19kDa-interacting protein 3, pro-apoptotic factor |
| A09 | CDKN2A | NM_000077 | Cyclin-dependent kinase inhibitor 2A, inhibits CDK4 |
| A10 | COX10 | NM_001303 | COX10 cytochrome c oxidase assembly protein homolog |
| A11 | COX18 | NM_173827 | COX18 cytochrome c oxidase assembly homolog |
| A12 | CPT1B | NM_004377 | Carnitine palmitoyltransferase 1B |
| B01 | CPT2 | NM_000098 | Carnitine palmitoyltransferase 2 |
| B02 | DNAJC19 | NM_145261 | DnaJ (Hsp40) homolog, subfamily C, member 19, TIMM14 |
| B03 | DNM1L | NM_005690 | Dynamin 1-like, mitochondrial and peroxisomal division |
| B04 | FIS1 | NM_016068 | Mitochondrial fission 1 protein homolog |
| B05 | TIMM10B | NM_012192 | Translocase of inner mitochondrial membrane 10 homolog B |
| B06 | GRPEL1 | NM_025196 | GrpE-like 1, mitochondrial protein import |
| B07 | HSP90AA1 | NM_001017963 | Heat shock protein 90kDa alpha, class A member 1, folding of target proteins |
| B08 | HSPD1 | NM_002156 | Heat shock 60kDa protein 1, chaperonin family, folding and assembly of proteins |
| B09 | IMMP1L | NM_144981 | Mitochondrial inner membrane protease subunit 1-like |
| B10 | IMMP2L | NM_032549 | Mitochondrial inner membrane protease Subunit 2-like |
| B11 | LRPPRC | NM_133259 | Leucine-rich PPR-motif containing, cytoskeletal organization and vesicular transport |
| B12 | MFN1 | NM_033540 | Mitofusin 1, mediator of mitochondrial fusion |
| C01 | MFN2 | NM_014874 | Mitofusin 2, mediator of mitochondrial fusion |
| C02 | MIPEP | NM_005932 | Mitochondrial intermediate peptidase, maturation of OXPHOS-related proteins |
| C03 | MSTO1 | NM_018116 | Misato homolog 1, mitochondrial distribution and morphology regulator |
| C04 | MTX2 | NM_006554 | Metaxin 2, mitochondrial outer membrane import complex protein 2 |
| C05 | NEFL | NM_006158 | Neurofilament, light polypeptide, protein phosphatase 1 |
| C06 | OPA1 | NM_130837 | Optic atrophy 1, mitochondrial dynamin-like GTPase, related to mitochondrial network |
| C07 | PMAIP1 | NM_021127 | Phorbol-12-myristate-13-acetate-induced protein 1, related to activation of caspases and apoptosis |
| C08 | RHOT1 | NM_018307 | Ras homolog gene family, member T1, mitochondrial GTPase involved in mitochondrial trafficking |
| C09 | RHOT2 | NM_138769 | Ras homolog gene family, member T2, mitochondrial GTPase involved in mitochondrial trafficking |
| C10 | SFN | NM_006142 | Stratifin |
| C11 | SH3GLB1 | NM_016009 | SH3-domain GRB2-like endophilin B1, Bax-interacting Factor 1, apoptotic signaling pathway |
| C12 | SLC25A1 | NM_005984 | Solute carrier family 25 (mitochondrial carrier; citrate transporter), member 1 |
| D01 | SLC25A10 | NM_012140 | Solute carrier family 25 (mitochondrial carrier; dicarboxylate transporter), member 10 |
| D02 | SLC25A12 | NM_003705 | Solute carrier Family 25 (aspartate/glutamate carrier), member 12, calcium carrier |
| D03 | SLC25A13 | NM_014251 | Solute carrier Family 25 (aspartate/glutamate carrier), member 13 |
| D04 | SLC25A14 | NM_003951 | Solute carrier family 25 (mitochondrial carrier), member 14, UCP5 |
| D05 | SLC25A15 | NM_014252 | Solute carrier family 25 (mitochondrial carrier; ornithine transporter) member 15 |
| D06 | SLC25A16 | NM_152707 | Solute carrier family 25 (mitochondrial carrier), member 16 |
| D07 | SLC25A17 | NM_006358 | Solute carrier family 25 (mitochondrial carrier; peroxisomal membrane protein), member 17 |
| D08 | SLC25A19 | NM_021734 | Solute carrier family 25 (mitochondrial thiamine pyrophosphate carrier), member 19 |
| D09 | SLC25A2 | NM_031947 | Solute carrier family 25 (mitochondrial carrier; ornithine transporter) member 2, ORNT2 |
| D10 | SLC25A20 | NM_000387 | Solute carrier family 25 (carnitine/acylcarnitine translocase), member 20 |
| D11 | SLC25A21 | NM_030631 | Solute carrier family 25 (mitochondrial oxodicarboxylate carrier), member 21 |
| D12 | SLC25A22 | NM_024698 | Solute carrier family 25 (mitochondrial carrier: glutamate), member 22 |
| E01 | SLC25A23 | NM_024103 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 23 |
| E02 | SLC25A24 | NM_013386 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 24 |
| E03 | SLC25A25 | NM_052901 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 25 |
| E04 | SLC25A27 | NM_004277 | Solute carrier family 25, member 27, UCP4 |
| E05 | SLC25A3 | NM_002635 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 3 |
| E06 | SLC25A30 | NM_001010875 | Solute carrier family 25, member 30 |
| E07 | SLC25A31 | NM_031291 | Solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 31, ANT4 |
| E08 | SLC25A37 | NM_016612 | Solute carrier family 25, (mitochondrial iron transporter), member 37 |
| E09 | SLC25A4 | NM_001151 | Solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 4, ANT1 |
| E10 | SLC25A5 | NM_001152 | Solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 5, ANT2 |
| E11 | SOD1 | NM_000454 | Superoxide dismutase 1, soluble, Cu/Zn superoxide dismutase |
| E12 | SOD2 | NM_000636 | Superoxide dismutase 2, mitochondrial, Fe/Mn superoxide dismutase |
| F01 | STARD3 | NM_006804 | StAR-related lipid transfer (START) domain containing 3, lipid trafficking protein |
| F02 | TAZ | NM_000116 | Tafazzin |
| F03 | TIMM10 | NM_012456 | Translocase of inner mitochondrial membrane 10 homolog (yeast) |
| F04 | TIMM17A | NM_006335 | Translocase of inner mitochondrial membrane 17 homolog A (yeast) |
| F05 | TIMM17B | NM_005834 | Translocase of inner mitochondrial membrane 17 homolog B (yeast) |
| F06 | TIMM22 | NM_013337 | Translocase of inner mitochondrial membrane 22 homolog (yeast) |
| F07 | TIMM23 | NM_006327 | Translocase of inner mitochondrial membrane 23 homolog (yeast) |
| F08 | TIMM44 | NM_006351 | Translocase of inner mitochondrial membrane 44 homolog (yeast) |
| F09 | TIMM50 | NM_001001563 | Translocase of inner mitochondrial membrane 50 homolog (S. cerevisiae) |
| F10 | TIMM8A | NM_004085 | Translocase of inner mitochondrial membrane 8 homolog A (yeast) |
| F11 | TIMM8B | NM_012459 | Translocase of inner mitochondrial membrane 8 homolog B (yeast) |
| F12 | TIMM9 | NM_012460 | Translocase of inner mitochondrial membrane 9 homolog (yeast) |
| G01 | TOMM20 | NM_014765 | Translocase of outer mitochondrial membrane 20 homolog (yeast) |
| G02 | TOMM22 | NM_020243 | Translocase of outer mitochondrial membrane 22 homolog (yeast) |
| G03 | TOMM34 | NM_006809 | Translocase of outer mitochondrial membrane 34 |
| G04 | TOMM40 | NM_006114 | Translocase of outer mitochondrial membrane 40 homolog (yeast) |
| G05 | TOMM40L | NM_032174 | Translocase of outer mitochondrial membrane 40 homolog (yeast)-like |
| G06 | TOMM70A | NM_014820 | Translocase of outer mitochondrial membrane 70 homolog A (S. cerevisiae) |
| G07 | TP53 | NM_000546 | Tumor protein p53, P53 tumor suppressor |
| G08 | TSPO | NM_000714 | Translocator protein (18kDa), transport of cholesterol |
| G09 | UCP1 | NM_021833 | Uncoupling protein 1 (mitochondrial, proton carrier), SLC25A7, proton leak |
| G10 | UCP2 | NM_003355 | Uncoupling protein 2 (mitochondrial, proton carrier), SLC25A8, proton leak |
| G11 | UCP3 | NM_003356 | Uncoupling protein 3 (mitochondrial, proton carrier), SLC25A9, proton leak |
| G12 | UXT | NM_004182 | Ubiquitously expressed transcript |
| H01 | B2M | NM_004048 | Beta-2-microglobulin |
| H02 | HPRT1 | NM_000194 | Hypoxanthine phosphoribosyltransferase 1 |
| H03 | RPL13A | NM_012423 | Ribosomal protein L13a |
| H04 | GAPDH | NM_002046 | Glyceraldehyde-3-phosphate dehydrogenase |
| H05 | ACTB | NM_001101 | Actin, beta |
| H06 | HGDC | SA_00105 | Human Genomic DNA Contamination |
| H07 | RTC | SA_00104 | Reverse Transcription Control |
| H08 | RTC | SA_00104 | Reverse Transcription Control |
| H09 | RTC | SA_00104 | Reverse Transcription Control |
| H10 | PPC | SA_00103 | Positive PCR Control |
| H11 | PPC | SA_00103 | Positive PCR Control |
| H12 | PPC | SA_00103 | Positive PCR Control |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, S.P.; Diniz, M.S.; Tavares, L.C.; Cunha-Oliveira, T.; Li, C.; Cox, L.A.; Nijland, M.J.; Nathanielsz, P.W.; Oliveira, P.J. Characterizing Early Cardiac Metabolic Programming via 30% Maternal Nutrient Reduction during Fetal Development in a Non-Human Primate Model. Int. J. Mol. Sci. 2023, 24, 15192. https://doi.org/10.3390/ijms242015192
Pereira SP, Diniz MS, Tavares LC, Cunha-Oliveira T, Li C, Cox LA, Nijland MJ, Nathanielsz PW, Oliveira PJ. Characterizing Early Cardiac Metabolic Programming via 30% Maternal Nutrient Reduction during Fetal Development in a Non-Human Primate Model. International Journal of Molecular Sciences. 2023; 24(20):15192. https://doi.org/10.3390/ijms242015192
Chicago/Turabian StylePereira, Susana P., Mariana S. Diniz, Ludgero C. Tavares, Teresa Cunha-Oliveira, Cun Li, Laura A. Cox, Mark J. Nijland, Peter W. Nathanielsz, and Paulo J. Oliveira. 2023. "Characterizing Early Cardiac Metabolic Programming via 30% Maternal Nutrient Reduction during Fetal Development in a Non-Human Primate Model" International Journal of Molecular Sciences 24, no. 20: 15192. https://doi.org/10.3390/ijms242015192
APA StylePereira, S. P., Diniz, M. S., Tavares, L. C., Cunha-Oliveira, T., Li, C., Cox, L. A., Nijland, M. J., Nathanielsz, P. W., & Oliveira, P. J. (2023). Characterizing Early Cardiac Metabolic Programming via 30% Maternal Nutrient Reduction during Fetal Development in a Non-Human Primate Model. International Journal of Molecular Sciences, 24(20), 15192. https://doi.org/10.3390/ijms242015192