
Combination of On-Line and Off-Line Two-Dimensional Liquid Chromatography-Mass Spectrometry for Comprehensive Characterization of mAb Charge Variants and Precise Instructions for Rapid Process Development

Abstract
:1. Introduction
2. Results
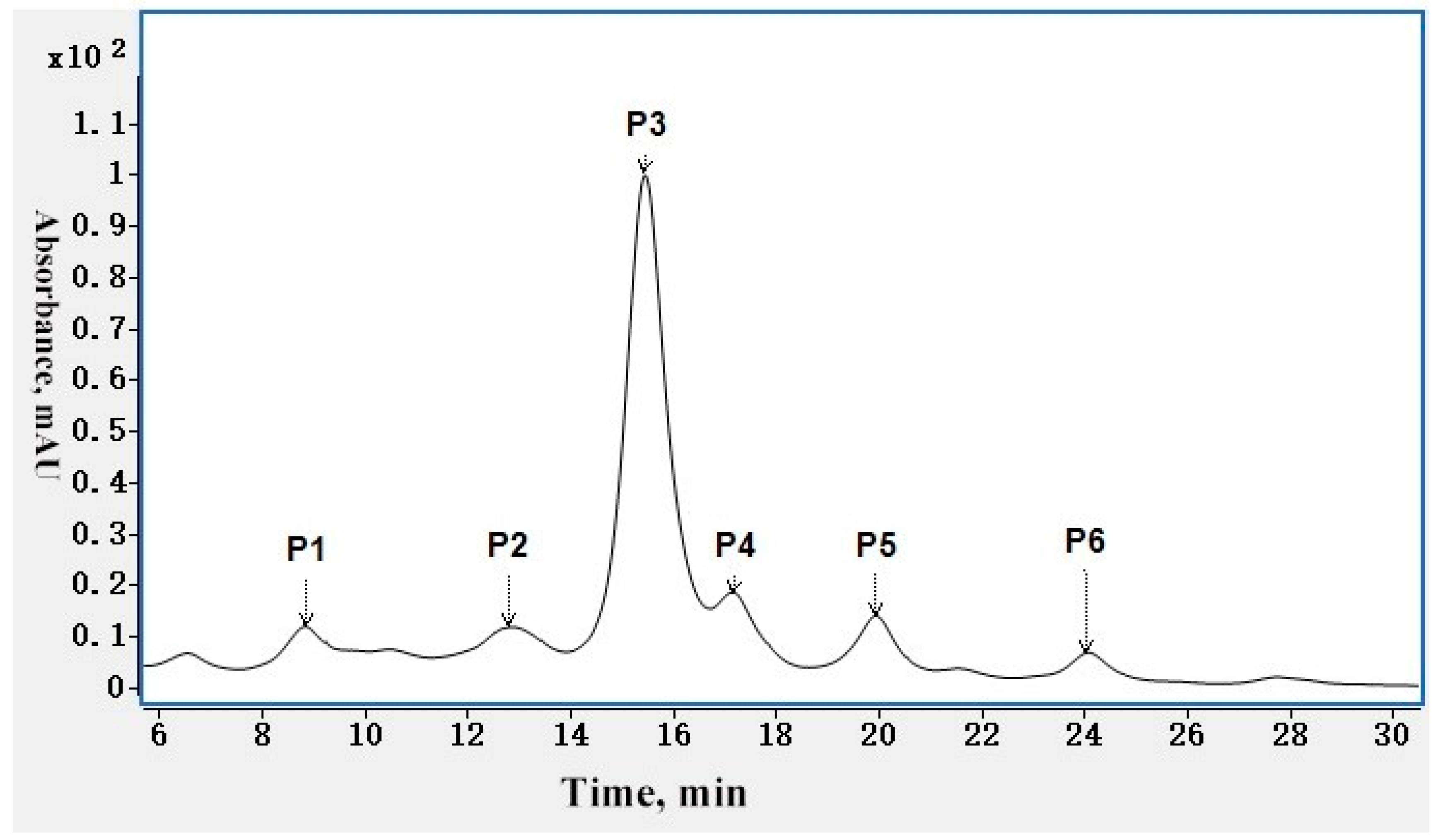
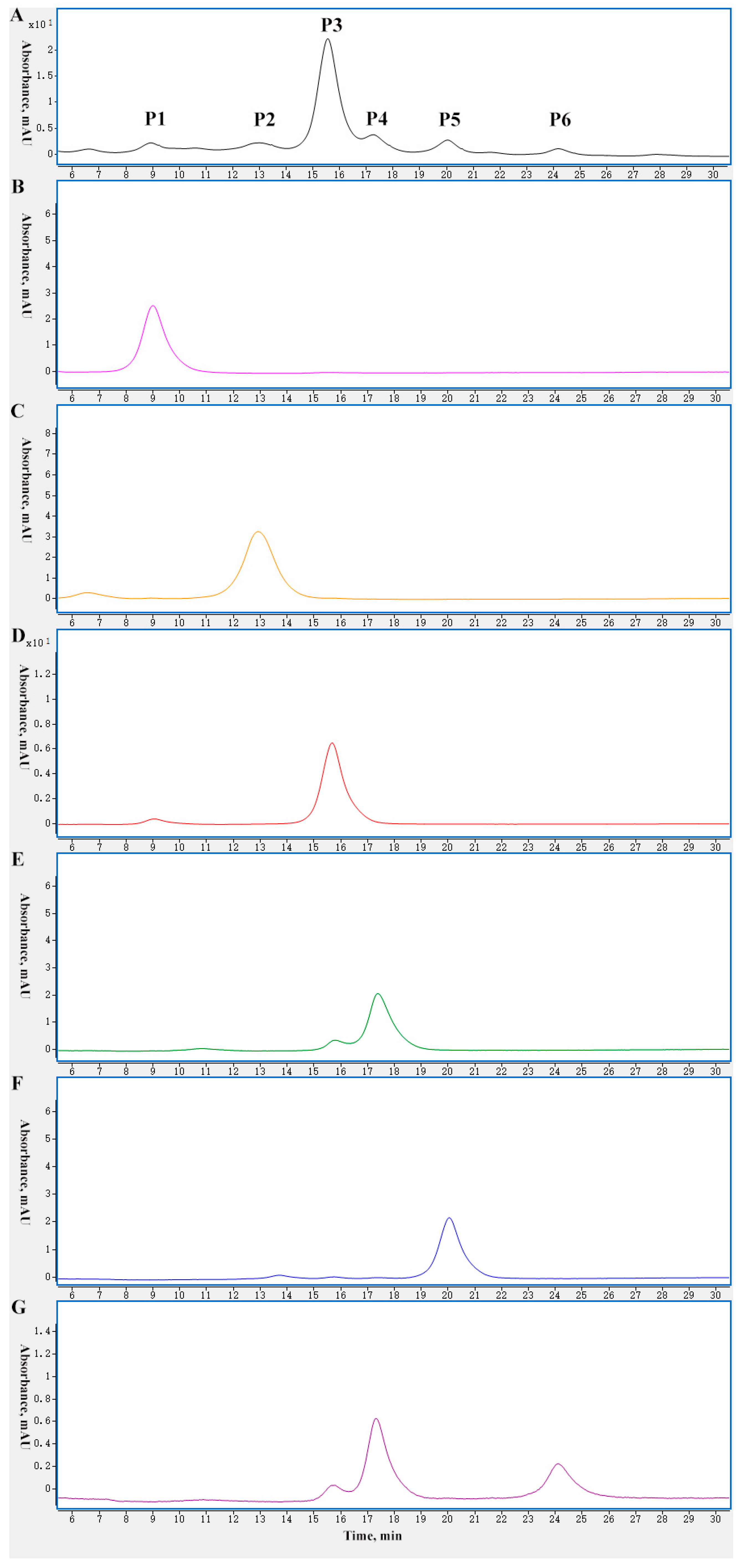
2.1. Charge Variant Profile of mAb 1 by CEX Liquid Chromatography
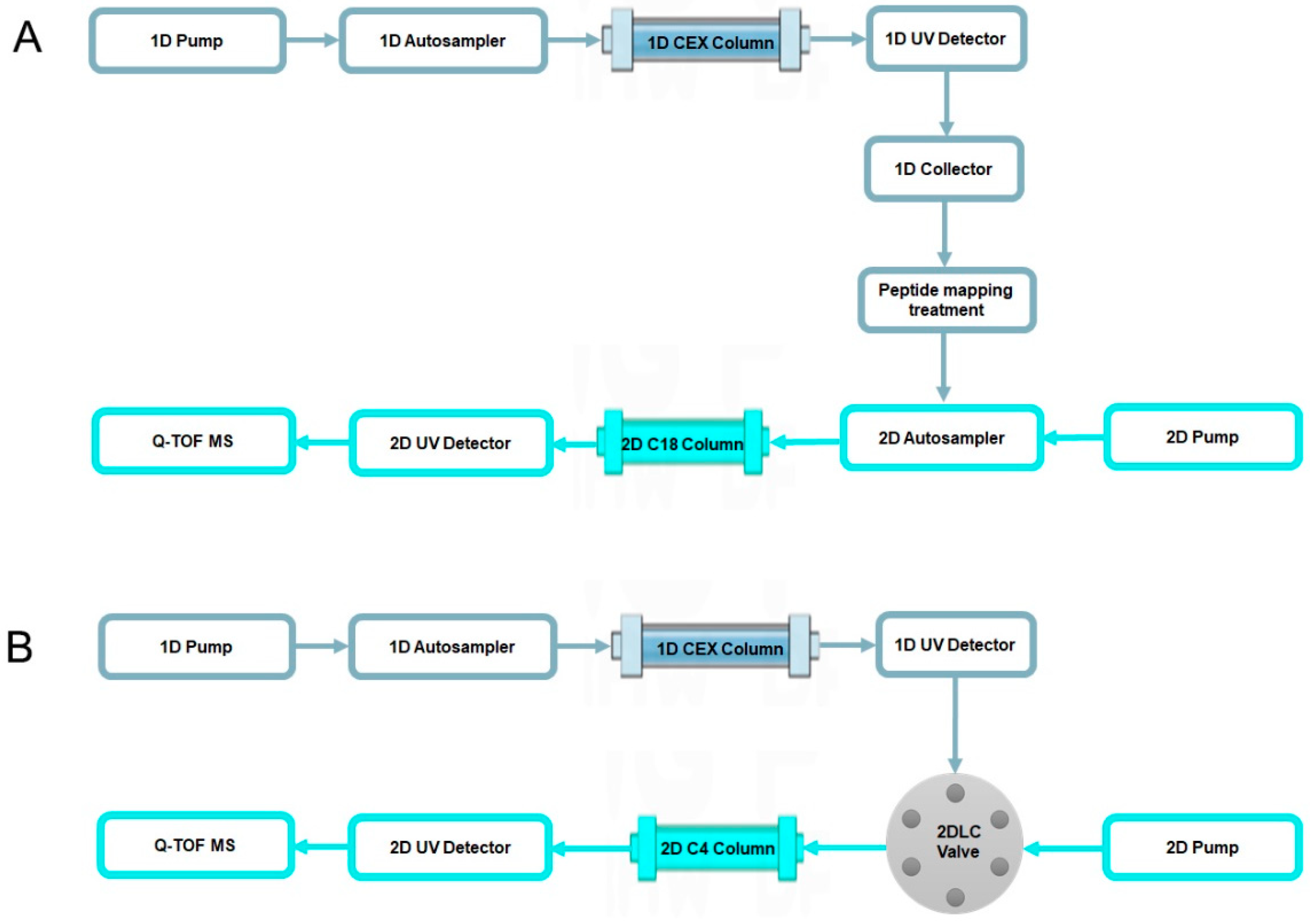
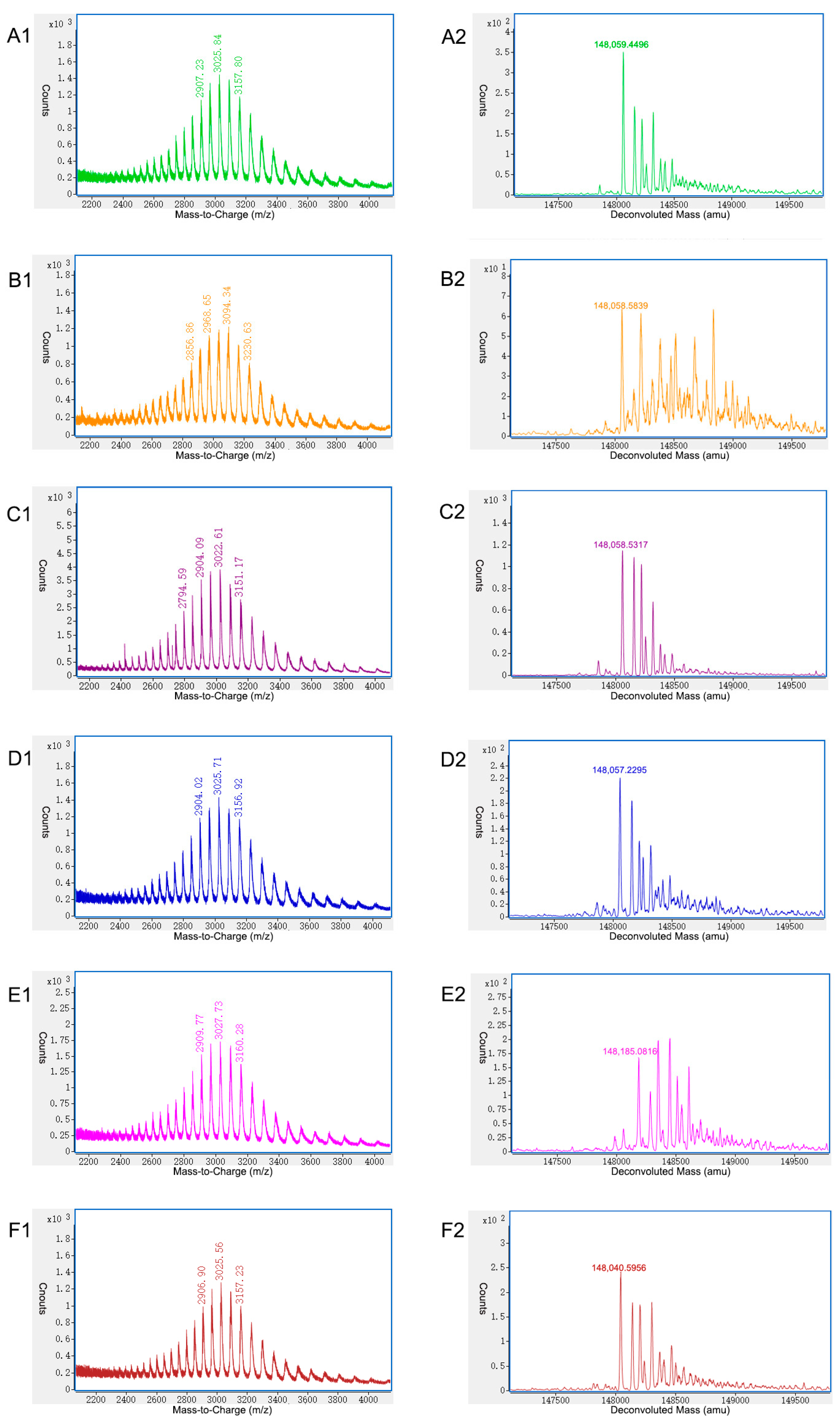
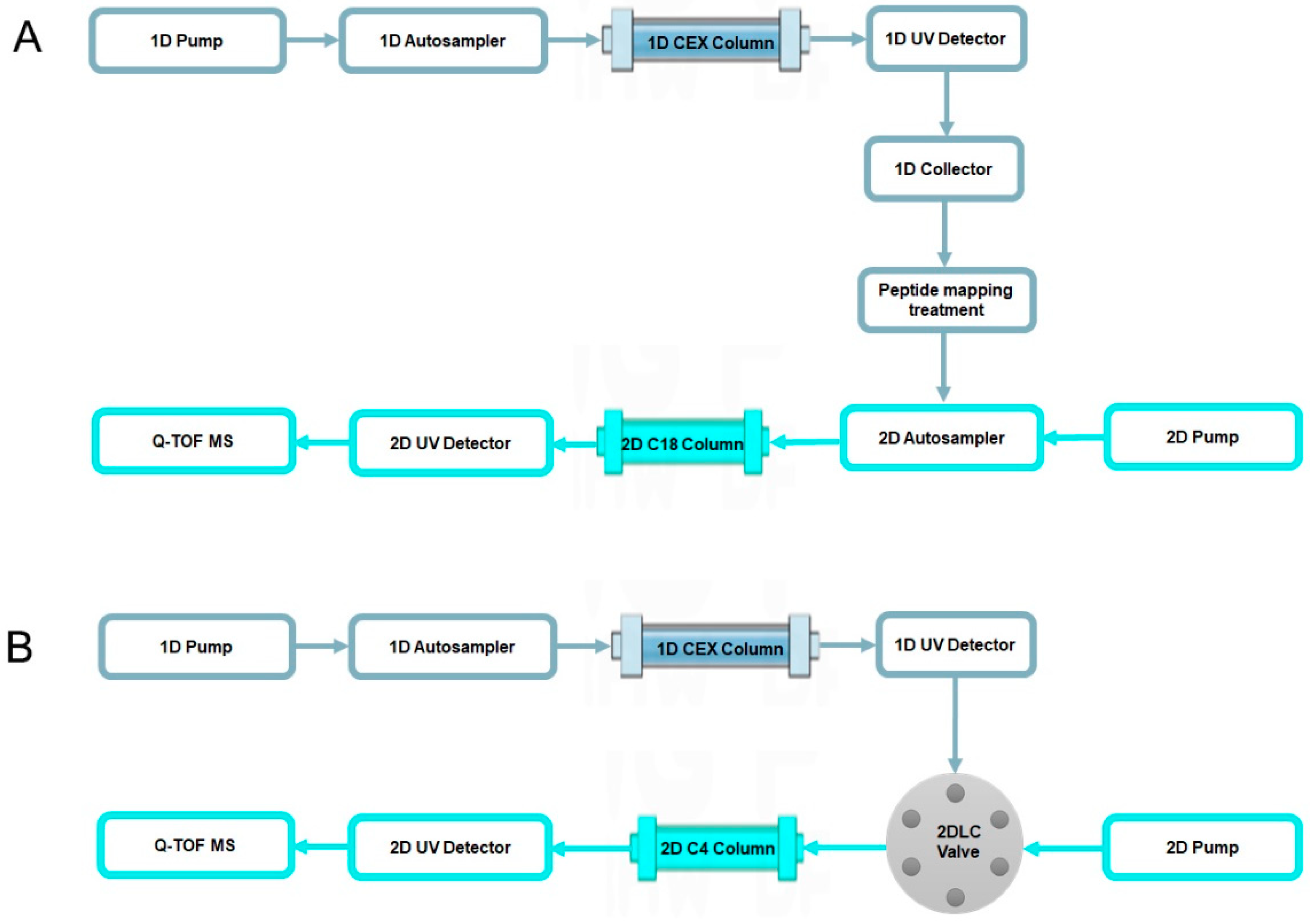
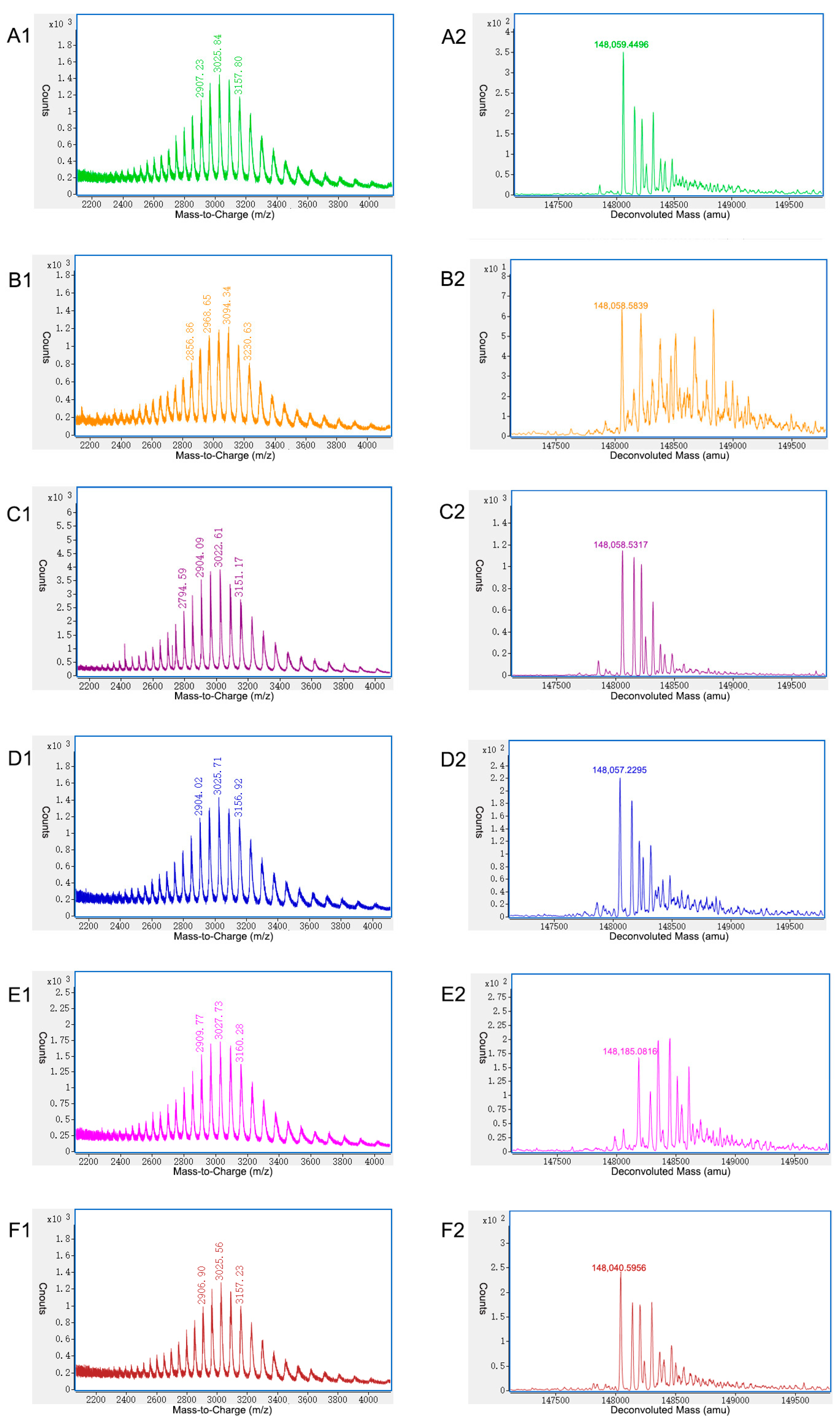
2.2. Characterization of Charge Variants at the Peptide Level by Off-Line 2D-LC-MS
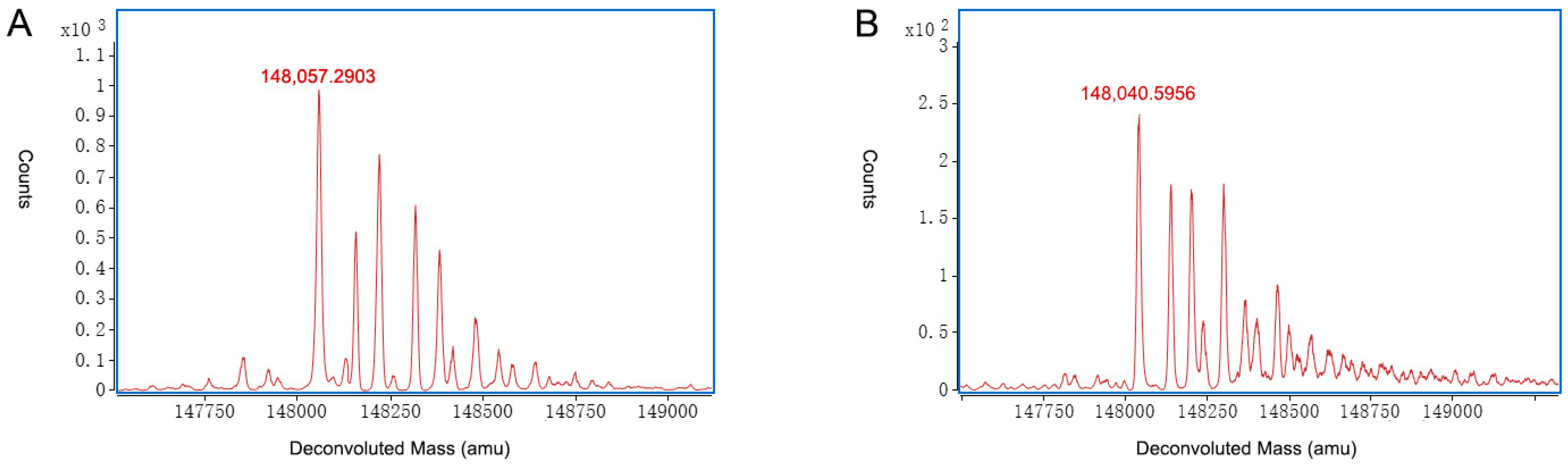
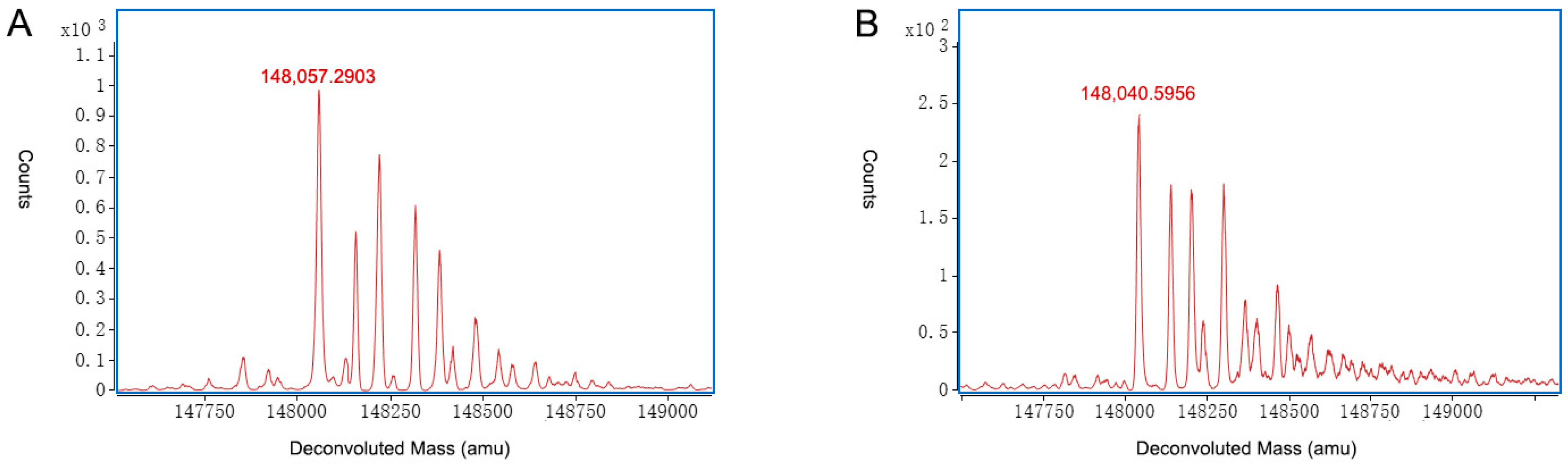
2.3. Characterization of Charge Variants at the Intact Protein Level by on-Line 2D-LC-MS
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Instrument
4.3. CEX Liquid Chromatography Analysis
4.4. Off-Line 2D-LC-MS Analysis
4.5. On-Line 2D-LC-MS Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Urquhart, L. Top companies and drugs by sales in 2022. Nat. Rev. Drug Discov. 2023, 22, 260. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to watch in 2023. MAbs 2023, 15, 2153410. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.P.; Grimaldi, C.; Grempler, R.; Hansel, S.; Kumar, S. Trends in industrialization of biotherapeutics: A survey of product characteristics of 89 antibody-based biotherapeutics. MAbs 2023, 15, 2191301. [Google Scholar] [CrossRef]
- Khetan, R.; Curtis, R.; Deane, C.M.; Hadsund, J.T.; Kar, U.; Krawczyk, K.; Kuroda, D.; Robinson, S.A.; Sormanni, P.; Tsumoto, K.; et al. Current advances in biopharmaceutical informatics: Guidelines, impact and challenges in the computational developability assessment of antibody therapeutics. MAbs 2022, 14, 2020082. [Google Scholar] [CrossRef] [PubMed]
- Farid, S.S.; Baron, M.; Stamatis, C.; Nie, W.; Coffman, J. Benchmarking biopharmaceutical process development and manufacturing cost contributions to R&D. MAbs 2020, 12, 1754999. [Google Scholar] [CrossRef]
- Azevedo Reis Teixeira, A.; Erasmus, M.F.; D’Angelo, S.; Naranjo, L.; Ferrara, F.; Leal-Lopes, C.; Durrant, O.; Galmiche, C.; Morelli, A.; Scott-Tucker, A.; et al. Drug-like antibodies with high affinity, diversity and developability directly from next-generation antibody libraries. MAbs 2021, 13, 1980942. [Google Scholar] [CrossRef] [PubMed]
- Wolf Perez, A.M.; Sormanni, P.; Andersen, J.S.; Sakhnini, L.I.; Rodriguez-Leon, I.; Bjelke, J.R.; Gajhede, A.J.; De Maria, L.; Otzen, D.E.; Vendruscolo, M.; et al. In vitro and in silico assessment of the developability of a designed monoclonal antibody library. MAbs 2019, 11, 388–400. [Google Scholar] [CrossRef]
- Beck, A.; Nowak, C.; Meshulam, D.; Reynolds, K.; Chen, D.; Pacardo, D.B.; Nicholls, S.B.; Carven, G.J.; Gu, Z.; Fang, J.; et al. Risk-Based Control Strategies of Recombinant Monoclonal Antibody Charge Variants. Antibodies 2022, 11, 73. [Google Scholar] [CrossRef]
- King, C.; Patel, R.; Ponniah, G.; Nowak, C.; Neill, A.; Gu, Z.; Liu, H. Characterization of recombinant monoclonal antibody variants detected by hydrophobic interaction chromatography and imaged capillary isoelectric focusing electrophoresis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1085, 96–103. [Google Scholar] [CrossRef]
- Xiao, Z.; Yin, X.; Han, L.; Sun, B.; Shen, Z.; Liu, W.; Yu, F. A comprehensive approach for evaluating charge heterogeneity in biosimilars. Eur. J. Pharm. Sci. 2018, 115, 19–24. [Google Scholar] [CrossRef]
- Hosken, B.D.; Li, C.; Mullappally, B.; Co, C.; Zhang, B. Isolation and Characterization of Monoclonal Antibody Charge Variants by Free Flow Isoelectric Focusing. Anal. Chem. 2016, 88, 5662–5669. [Google Scholar] [CrossRef]
- Ponniah, G.; Nowak, C.; Neill, A.; Liu, H. Characterization of charge variants of a monoclonal antibody using weak anion exchange chromatography at subunit levels. Anal. Biochem. 2017, 520, 49–57. [Google Scholar] [CrossRef]
- Jiang, G.; Yu, C.; Yadav, D.B.; Hu, Z.; Amurao, A.; Duenas, E.; Wong, M.; Iverson, M.; Zheng, K.; Lam, X.; et al. Evaluation of Heavy-Chain C-Terminal Deletion on Product Quality and Pharmacokinetics of Monoclonal Antibodies. J. Pharm. Sci. 2016, 105, 2066–2072. [Google Scholar] [CrossRef] [PubMed]
- Camperi, J.; Grunert, I.; Heinrich, K.; Winter, M.; Ozipek, S.; Hoelterhoff, S.; Weindl, T.; Mayr, K.; Bulau, P.; Meier, M.; et al. Inter-laboratory study to evaluate the performance of automated online characterization of antibody charge variants by multi-dimensional LC-MS/MS. Talanta 2021, 234, 122628. [Google Scholar] [CrossRef] [PubMed]
- Jaag, S.; Shirokikh, M.; Lammerhofer, M. Charge variant analysis of protein-based biopharmaceuticals using two-dimensional liquid chromatography hyphenated to mass spectrometry. J. Chromatogr. A 2021, 1636, 461786. [Google Scholar] [CrossRef] [PubMed]
- Goyon, A.; Dai, L.; Chen, T.; Wei, B.; Yang, F.; Andersen, N.; Kopf, R.; Leiss, M.; Molhoj, M.; Guillarme, D.; et al. From proof of concept to the routine use of an automated and robust multi-dimensional liquid chromatography mass spectrometry workflow applied for the charge variant characterization of therapeutic antibodies. J. Chromatogr. A 2020, 1615, 460740. [Google Scholar] [CrossRef]
- Bathke, A.; Klemm, D.; Gstóttner, C.; Bell, C.; Kopf, R. Rapid online reduction and characterization of protein modifications using fully automated two-dimensional high performance liquid chromatography-mass spectrometry. LC-GC Eur. 2018, 31, 10–21. [Google Scholar]
- Oezipek, S.; Hoelterhoff, S.; Breuer, S.; Bell, C.; Bathke, A. mD-UPLC-MS/MS: Next Generation of mAb Characterization by Multidimensional Ultraperformance Liquid Chromatography-Mass Spectrometry and Parallel On-Column LysC and Trypsin Digestion. Anal. Chem. 2022, 94, 8136–8145. [Google Scholar] [CrossRef]
- Liu, Y.D.; Cadang, L.; Bol, K.; Pan, X.; Tschudi, K.; Jazayri, M.; Camperi, J.; Michels, D.; Stults, J.; Harris, R.J.; et al. Challenges and Strategies for a Thorough Characterization of Antibody Acidic Charge Variants. Bioengineering 2022, 9, 641. [Google Scholar] [CrossRef]
- Schmid, I.; Bonnington, L.; Gerl, M.; Bomans, K.; Thaller, A.L.; Wagner, K.; Schlothauer, T.; Falkenstein, R.; Zimmermann, B.; Kopitz, J.; et al. Assessment of susceptible chemical modification sites of trastuzumab and endogenous human immunoglobulins at physiological conditions. Commun. Biol. 2018, 1, 28. [Google Scholar] [CrossRef]
- Spanov, B.; Olaleye, O.; Lingg, N.; Bentlage, A.E.H.; Govorukhina, N.; Hermans, J.; van de Merbel, N.; Vidarsson, G.; Jungbauer, A.; Bischoff, R. Change of charge variant composition of trastuzumab upon stressing at physiological conditions. J. Chromatogr. A 2021, 1655, 462506. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Joshi, S.; Guttman, A.; Rathore, A.S. N-Glycosylation of monoclonal antibody therapeutics: A comprehensive review on significance and characterization. Anal. Chim. Acta 2022, 1209, 339828. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Pan, H.; Flynn, G.C. C-terminal lysine processing of human immunoglobulin G2 heavy chain in vivo. Biotechnol. Bioeng. 2011, 108, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Raoufi, F.; Bailly, M.A.; Fayadat-Dilman, L.; Tomazela, D. Hyphenation of strong cation exchange chromatography to native mass spectrometry for high throughput online characterization of charge heterogeneity of therapeutic monoclonal antibodies. MAbs 2020, 12, 1763762. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, M.; Harmes, D.C.; Stoll, D.R.; Staples, G.O.; Fekete, S.; Guillarme, D.; Beck, A. Comparison of originator and biosimilar therapeutic monoclonal antibodies using comprehensive two-dimensional liquid chromatography coupled with time-of-flight mass spectrometry. MAbs 2016, 8, 1224–1234. [Google Scholar] [CrossRef]
- Spanov, B.; Olaleye, O.; Mesurado, T.; Govorukhina, N.; Jungbauer, A.; van de Merbel, N.C.; Lingg, N.; Bischoff, R. Pertuzumab Charge Variant Analysis and Complementarity-Determining Region Stability Assessment to Deamidation. Anal. Chem. 2023, 95, 3951–3958. [Google Scholar] [CrossRef]
- Dulce, R.A.; Schulman, I.H.; Hare, J.M. S-glutathionylation: A redox-sensitive switch participating in nitroso-redox balance. Circ. Res. 2011, 108, 531–533. [Google Scholar] [CrossRef]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef]
- Liu, H.; Gaza-Bulseco, G.; Faldu, D.; Chumsae, C.; Sun, J. Heterogeneity of monoclonal antibodies. J. Pharm. Sci. 2008, 97, 2426–2447. [Google Scholar] [CrossRef]
- Hintersteiner, B.; Lingg, N.; Zhang, P.; Woen, S.; Hoi, K.M.; Stranner, S.; Wiederkum, S.; Mutschlechner, O.; Schuster, M.; Loibner, H.; et al. Charge heterogeneity: Basic antibody charge variants with increased binding to Fc receptors. MAbs 2016, 8, 1548–1560. [Google Scholar] [CrossRef] [PubMed]
- Teshima, G.; Li, M.X.; Danishmand, R.; Obi, C.; To, R.; Huang, C.; Kung, J.; Lahidji, V.; Freeberg, J.; Thorner, L.; et al. Separation of oxidized variants of a monoclonal antibody by anion-exchange. J. Chromatogr. A 2011, 1218, 2091–2097. [Google Scholar] [CrossRef] [PubMed]
- Chumsae, C.; Gaza-Bulseco, G.; Sun, J.; Liu, H. Comparison of methionine oxidation in thermal stability and chemically stressed samples of a fully human monoclonal antibody. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 850, 285–294. [Google Scholar] [CrossRef]
- Fujii, N.; Takata, T.; Fujii, N.; Aki, K. Isomerization of aspartyl residues in crystallins and its influence upon cataract. Biochim. Biophys. Acta 2016, 1860, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Sreedhara, A.; Cordoba, A.; Zhu, Q.; Kwong, J.; Liu, J. Characterization of the isomerization products of aspartate residues at two different sites in a monoclonal antibody. Pharm. Res. 2012, 29, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.A.; Borchardt, R.T.; Eigenbrot, C.; Shia, S.; Wang, Y.J.; Shire, S.J.; Liu, J.L. Aspartate isomerization in the complementarity-determining regions of two closely related monoclonal antibodies. Biochemistry 2007, 46, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.C.; Chelius, D.; Xiao, G.; Khor, H.K.; Coulibaly, S.; Bondarenko, P.V. Accumulation of succinimide in a recombinant monoclonal antibody in mildly acidic buffers under elevated temperatures. Pharm. Res. 2007, 24, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Valliere-Douglass, J.; Jones, L.; Shpektor, D.; Kodama, P.; Wallace, A.; Balland, A.; Bailey, R.; Zhang, Y. Separation and characterization of an IgG2 antibody containing a cyclic imide in CDR1 of light chain by hydrophobic interaction chromatography and mass spectrometry. Anal. Chem. 2008, 80, 3168–3174. [Google Scholar] [CrossRef]
- Gstottner, C.; Klemm, D.; Haberger, M.; Bathke, A.; Wegele, H.; Bell, C.; Kopf, R. Fast and Automated Characterization of Antibody Variants with 4D HPLC/MS. Anal. Chem. 2018, 90, 2119–2125. [Google Scholar] [CrossRef]
- Chen, Z.; Zeng, M.; Park, S.-J.; Balakrishnan, G.; Zhou, K.; Pan, D.; Das, T.K. Bridging size and charge variants of a therapeutic monoclonal antibody by two-dimensional liquid chromatography. J. Pharm. Biomed. Anal. 2020, 183, 113178. [Google Scholar] [CrossRef]
- Mouchahoir, T.; Schiel, J.E. Development of an LC-MS/MS peptide mapping protocol for the NISTmAb. Anal. Bioanal. Chem. 2018, 410, 2111–2126. [Google Scholar] [CrossRef] [PubMed]
- Sisodiya, S.M. Precision medicine and therapies of the future. Epilepsia 2020, 62, S90–S105. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.S.; Raju, R.; Kshirsagar, R.; Ivanov, A.R.; Gilbert, A.; Zang, L.; Karger, B.L. Multi-Omics Study on the Impact of Cysteine Feed Level on Cell Viability and mAb Production in a CHO Bioprocess. Biotechnol. J. 2018, 14, 1800352. [Google Scholar] [CrossRef] [PubMed]
- Prade, E.; Zeck, A.; Stiefel, F.; Unsoeld, A.; Mentrup, D.; Arango Gutierrez, E.; Gorr, I.H. Cysteine in cell culture media induces acidic IgG1 species by disrupting the disulfide bond network. Biotechnol. Bioeng. 2020, 118, 1091–1104. [Google Scholar] [CrossRef]
- Chevallier, V.; Zoller, M.; Kochanowski, N.; Andersen, M.R.; Workman, C.T.; Malphettes, L. Use of novel cystine analogs to decrease oxidative stress and control product quality. J. Biotechnol. 2021, 327, 1–8. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | P1 | P2 | P3 | P4 | P5 | P6 |
|---|---|---|---|---|---|---|
| HC N387 Deamidation | 98.41% | 6.84% | 5.48% | 13.76% | 9.08% | 4.00% |
| LC N30 Deamidation | 34.86% | 4.08% | 5.37% | 2.63% | 5.93% | 7.02% |
| HC C-terminal K Loss | 97.42% | 95.19% | 99.93% | 94.62% | 48.56% | 93.13% |
| HC M255 Oxidation | 2.61% | 8.13% | 4.51% | 11.88% | 8.70% | 6.27% |
| HC M431 Oxidation | 1.46% | 1.32% | 0.66% | 3.61% | 2.21% | 1.66% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, X.; He, B. Combination of On-Line and Off-Line Two-Dimensional Liquid Chromatography-Mass Spectrometry for Comprehensive Characterization of mAb Charge Variants and Precise Instructions for Rapid Process Development. Int. J. Mol. Sci. 2023, 24, 15184. https://doi.org/10.3390/ijms242015184
Jin X, He B. Combination of On-Line and Off-Line Two-Dimensional Liquid Chromatography-Mass Spectrometry for Comprehensive Characterization of mAb Charge Variants and Precise Instructions for Rapid Process Development. International Journal of Molecular Sciences. 2023; 24(20):15184. https://doi.org/10.3390/ijms242015184
Chicago/Turabian StyleJin, Xiaoqing, and Bingfang He. 2023. "Combination of On-Line and Off-Line Two-Dimensional Liquid Chromatography-Mass Spectrometry for Comprehensive Characterization of mAb Charge Variants and Precise Instructions for Rapid Process Development" International Journal of Molecular Sciences 24, no. 20: 15184. https://doi.org/10.3390/ijms242015184
APA StyleJin, X., & He, B. (2023). Combination of On-Line and Off-Line Two-Dimensional Liquid Chromatography-Mass Spectrometry for Comprehensive Characterization of mAb Charge Variants and Precise Instructions for Rapid Process Development. International Journal of Molecular Sciences, 24(20), 15184. https://doi.org/10.3390/ijms242015184