
NTPDase1/CD39 Ectonucleotidase Is Necessary for Normal Arterial Diameter Adaptation to Flow

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Impact of CD39 Deletion on Hemodynamic Parameters
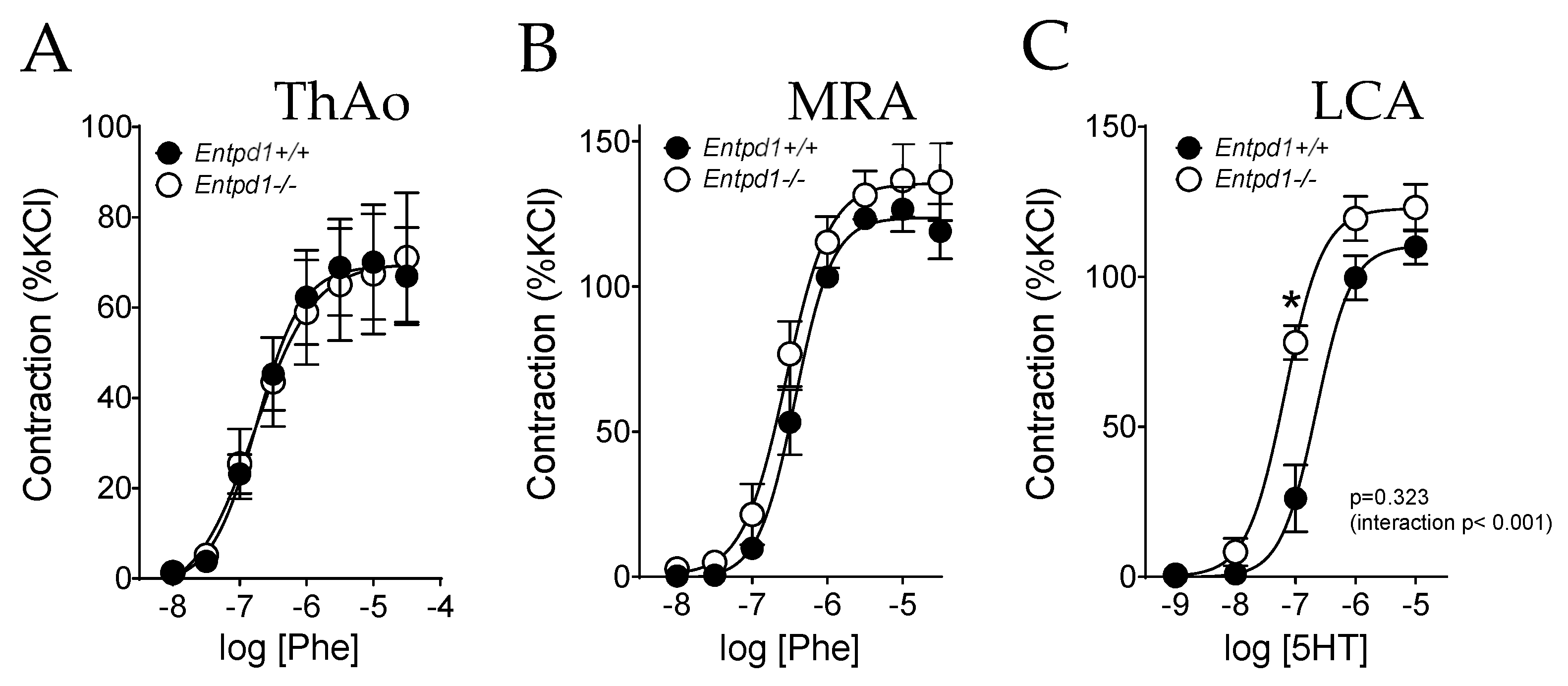
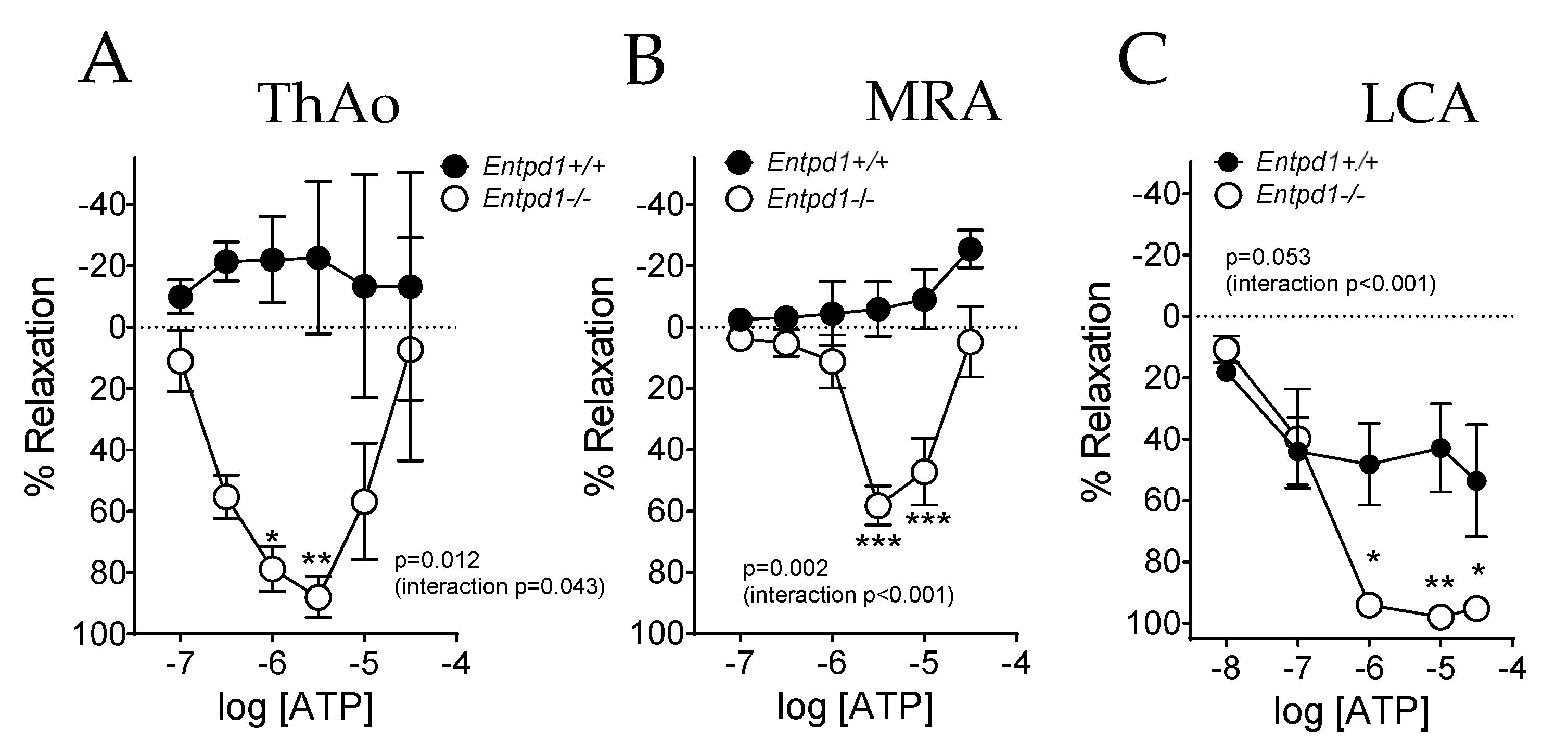
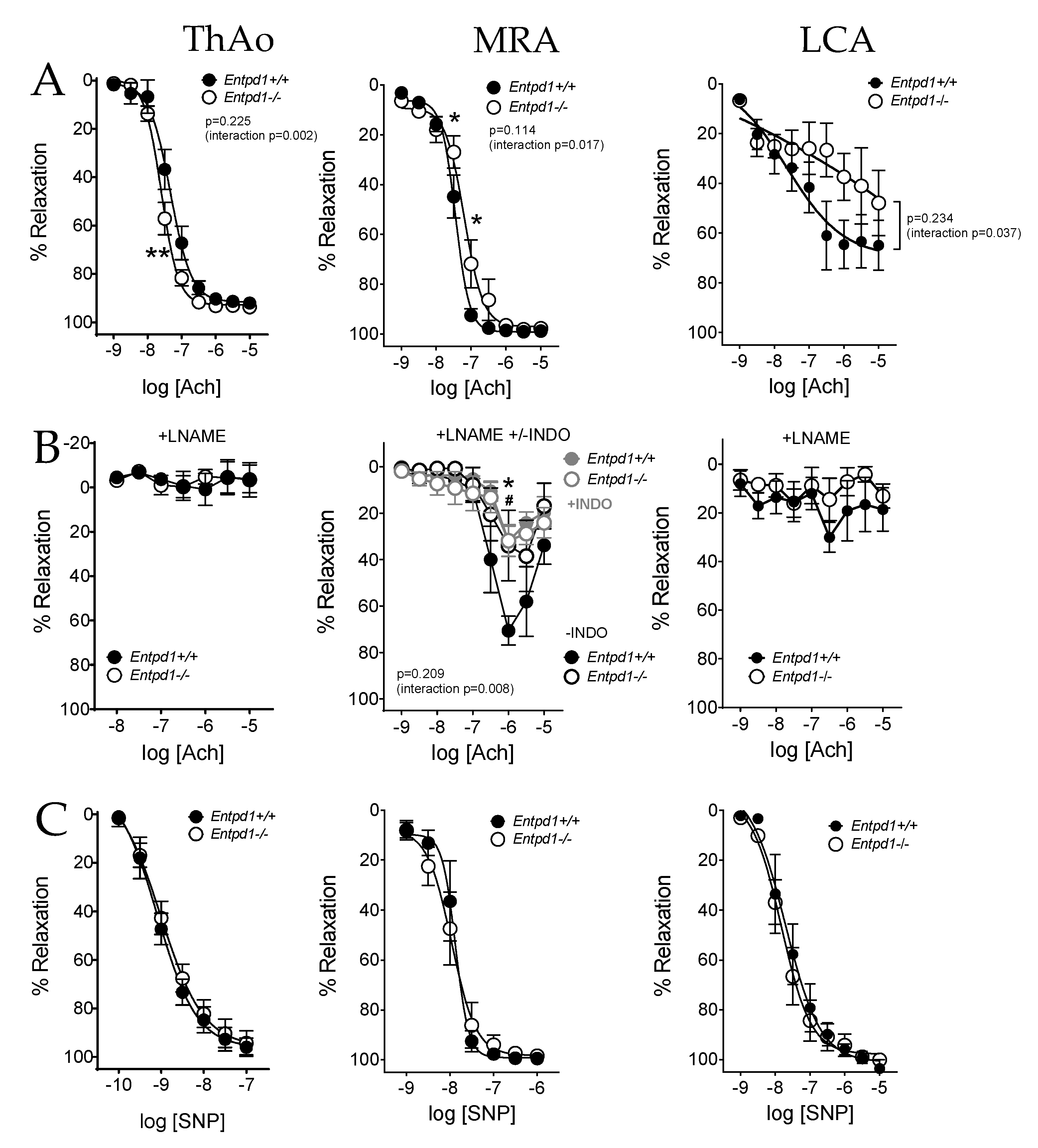
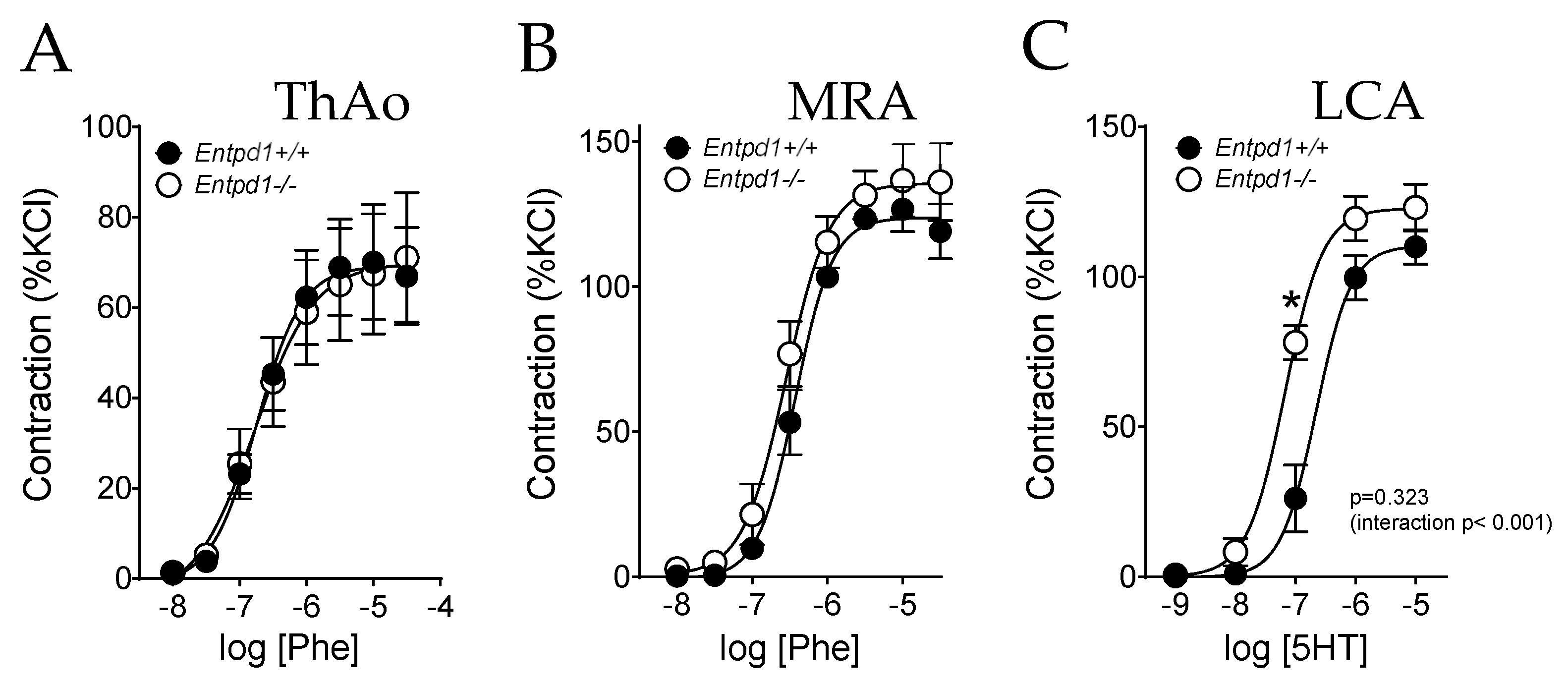
2.2. Impact of CD39 Deletion on Vasoreactivity of Conductance and Resistance Arteries
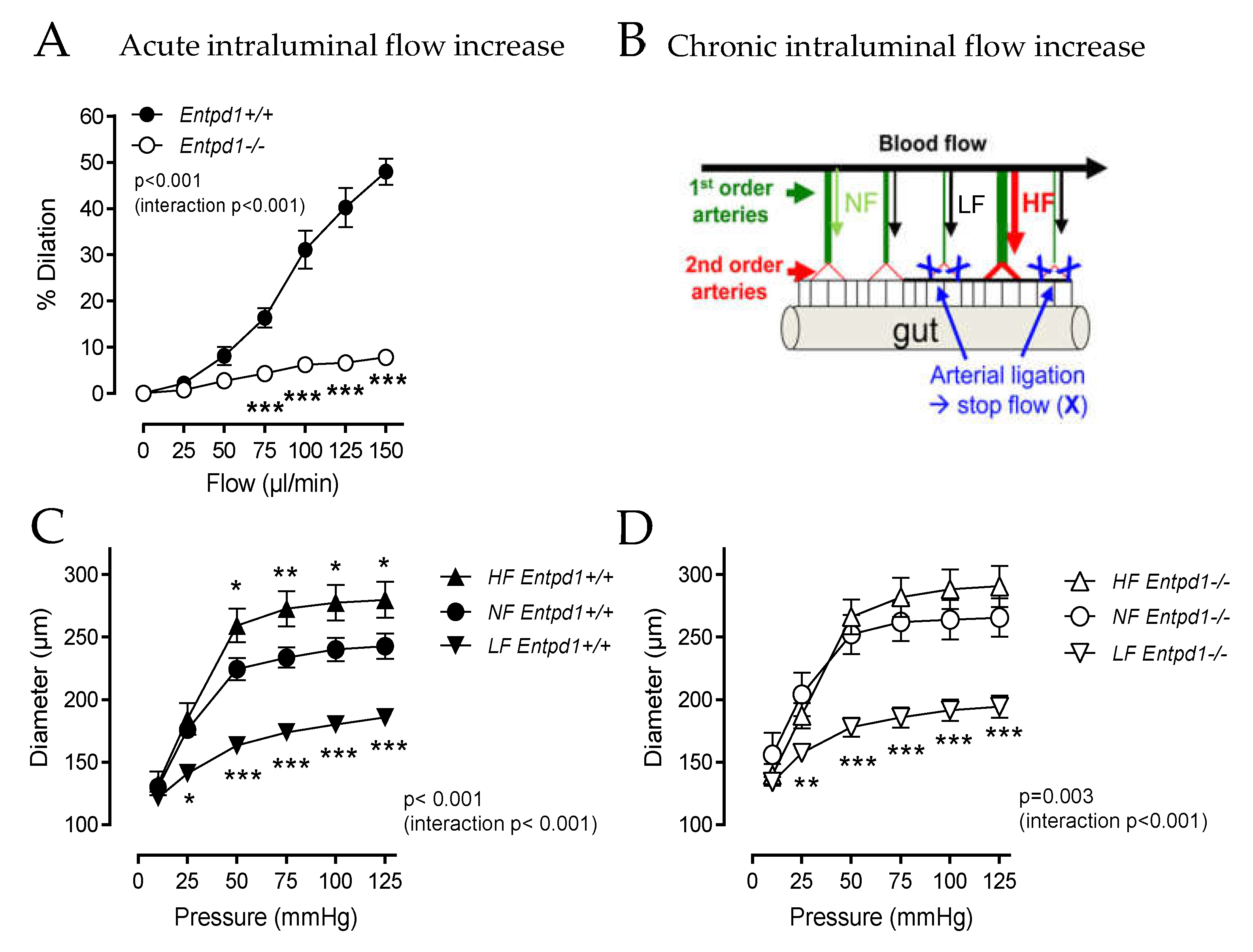
2.3. Impact of Entpd1 Deletion on Microvascular Response to Mechanical Stimuli
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Evaluation of Vascular Reactivity Using Wire Myography
4.3. Evaluation of Flow-Mediated Dilation Using Pressure-Myography
4.4. In Vivo Flow-Dependent Arterial Remodeling
4.5. Ex Vivo Working Heart Perfused in Semi-Recirculating Mode
4.6. In Vivo Telemetry
4.7. Millar Catheterization
4.8. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Celermajer, D.S.; Sorensen, K.E.; Gooch, V.M.; Spiegelhalter, D.J.; Miller, O.I.; Sullivan, I.D.; Lloyd, J.K.; Deanfield, J.E. Non-Invasive Detection of Endothelial Dysfunction in Children and Adults at Risk of Atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, D.H.J.; Black, M.A.; Pyke, K.E.; Padilla, J.; Atkinson, G.; Harris, R.A.; Parker, B.; Widlansky, M.E.; Tschakovsky, M.E.; Green, D.J. Assessment of Flow-Mediated Dilation in Humans: A Methodological and Physiological Guideline. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2–H12. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Kwon, T.-G.; Lennon, R.J.; Lerman, L.O.; Lerman, A. Prognostic Value of Flow-Mediated Vasodilation in Brachial Artery and Fingertip Artery for Cardiovascular Events: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2015, 4, e002270. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F. Hemodynamic Shear Stress and the Endothelium in Cardiovascular Pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial Function and Dysfunction: Testing and Clinical Relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic Signaling during Inflammation. N. Engl. J. Med. 2013, 368, 1260. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef]
- Cabou, C.; Martinez, L.O. The Interplay of Endothelial P2Y Receptors in Cardiovascular Health: From Vascular Physiology to Pathology. Int. J. Mol. Sci. 2022, 23, 5883. [Google Scholar] [CrossRef]
- Bodin, P.; Bailey, D.; Burnstock, G. Increased Flow-Induced ATP Release from Isolated Vascular Endothelial Cells but Not Smooth Muscle Cells. Br. J. Pharmacol. 1991, 103, 1203–1205. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sokabe, T.; Matsumoto, T.; Yoshimura, K.; Shibata, M.; Ohura, N.; Fukuda, T.; Sato, T.; Sekine, K.; Kato, S.; et al. Impaired Flow-Dependent Control of Vascular Tone and Remodeling in P2X4-Deficient Mice. Nat. Med. 2006, 12, 133–137. [Google Scholar] [CrossRef]
- Wang, S.; Iring, A.; Strilic, B.; Albarrán Juárez, J.; Kaur, H.; Troidl, K.; Tonack, S.; Burbiel, J.C.; Müller, C.E.; Fleming, I.; et al. P2Y2 and Gq/G11 Control Blood Pressure by Mediating Endothelial Mechanotransduction. J. Clin. Investig. 2015, 125, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chennupati, R.; Kaur, H.; Iring, A.; Wettschureck, N.; Offermanns, S. Endothelial Cation Channel PIEZO1 Controls Blood Pressure by Mediating Flow-Induced ATP Release. J. Clin. Investig. 2016, 126, 4527–4536. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Cho, J.M.; Park, S.-K.; Ruan, T.; Li, Y.; Mueller, R.; Bean, T.; Reese, V.; Richardson, R.S.; Cai, J.; et al. Endothelial Cell Autophagy Maintains Shear Stress-Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.M.; Park, S.-K.; Kwon, O.S.; Taylor La Salle, D.; Cerbie, J.; Fermoyle, C.C.; Morgan, D.; Nelson, A.; Bledsoe, A.; Bharath, L.P.; et al. Activating P2Y1 Receptors Improves Function in Arteries with Repressed Autophagy. Cardiovasc. Res. 2023, 119, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Enjyoji, K.; Sévigny, J.; Lin, Y.; Frenette, P.S.; Christie, P.D.; Esch, J.S., 2nd; Imai, M.; Edelberg, J.M.; Rayburn, H.; Lech, M.; et al. Targeted Disruption of Cd39/ATP Diphosphohydrolase Results in Disordered Hemostasis and Thromboregulation. Nat. Med. 1999, 5, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, D.J.; Broekman, M.J.; Peschon, J.J.; Stocking, K.L.; Fujita, T.; Ramasamy, R.; Connolly, E.S., Jr.; Huang, J.; Kiss, S.; Zhang, Y.; et al. Elucidation of the Thromboregulatory Role of CD39/Ectoapyrase in the Ischemic Brain. J. Clin. Investig. 2002, 109, 1031–1040. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated Adenine Nucleotide Phosphohydrolysis and Nucleoside Signaling in Posthypoxic Endothelium: Role of Ectonucleotidases and Adenosine A2B Receptors. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Grenz, A.; Zhang, H.; Hermes, M.; Eckle, T.; Klingel, K.; Huang, D.Y.; Muller, C.E.; Robson, S.C.; Osswald, H.; Eltzschig, H.K. Contribution of E-NTPDase1 (CD39) to Renal Protection from Ischemia-Reperfusion Injury. Faseb. J. 2007, 21, 2863–2873. [Google Scholar] [CrossRef]
- Kohler, D.; Eckle, T.; Faigle, M.; Grenz, A.; Mittelbronn, M.; Laucher, S.; Hart, M.L.; Robson, S.C.; Muller, C.E.; Eltzschig, H.K. CD39/Ectonucleoside Triphosphate Diphosphohydrolase 1 Provides Myocardial Protection during Cardiac Ischemia/Reperfusion Injury. Circulation 2007, 116, 1784–1794. [Google Scholar] [CrossRef]
- Kanthi, Y.; Hyman, M.C.; Liao, H.; Baek, A.E.; Visovatti, S.H.; Sutton, N.R.; Goonewardena, S.N.; Neral, M.K.; Jo, H.; Pinsky, D.J. Flow-Dependent Expression of Ectonucleotide Tri(Di)Phosphohydrolase-1 and Suppression of Atherosclerosis. J. Clin. Investig. 2015, 125, 3027–3036. [Google Scholar] [CrossRef]
- Kauffenstein, G.; Furstenau, C.R.; D’Orleans-Juste, P.; Sevigny, J. The Ecto-Nucleotidase NTPDase1 Differentially Regulates P2Y1 and P2Y2 Receptor-Dependent Vasorelaxation. Br. J. Pharmacol. 2010, 159, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Machida, T.; Heerdt, P.M.; Reid, A.C.; Schafer, U.; Silver, R.B.; Broekman, M.J.; Marcus, A.J.; Levi, R. Ectonucleoside Triphosphate Diphosphohydrolase 1/CD39, Localized in Neurons of Human and Porcine Heart, Modulates ATP-Induced Norepinephrine Exocytosis. J. Pharmacol. Exp. Ther. 2005, 313, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, U.; Machida, T.; Broekman, M.J.; Marcus, A.J.; Levi, R. Targeted Deletion of Ectonucleoside Triphosphate Diphosphohydrolase 1/CD39 Leads to Desensitization of Pre- and Postsynaptic Purinergic P2 Receptors. J. Pharmacol. Exp. Ther. 2007, 322, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Kauffenstein, G.; Laher, I.; Matrougui, K.; Guerineau, N.C.; Henrion, D. Emerging Role of G Protein-Coupled Receptors in Microvascular Myogenic Tone. Cardiovasc. Res. 2012, 95, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Mazza, L.F.; Yalavarthi, S.; Sule, G.; Ali, R.A.; Hodgin, J.B.; Kanthi, Y.; Pinsky, D.J. Ectonucleotidase-Mediated Suppression of Lupus Autoimmunity and Vascular Dysfunction. Front. Immunol. 2018, 9, 1322. [Google Scholar] [CrossRef] [PubMed]
- Henrion, D.; Terzi, F.; Matrougui, K.; Duriez, M.; Boulanger, C.M.; Colucci-Guyon, E.; Babinet, C.; Briand, P.; Friedlander, G.; Poitevin, P.; et al. Impaired Flow-Induced Dilation in Mesenteric Resistance Arteries from Mice Lacking Vimentin. J. Clin. Investig. 1997, 100, 2909–2914. [Google Scholar] [CrossRef] [PubMed]
- Campinho, P.; Vilfan, A.; Vermot, J. Blood Flow Forces in Shaping the Vascular System: A Focus on Endothelial Cell Behavior. Front. Physiol. 2020, 11, 552. [Google Scholar] [CrossRef]
- Billaud, M.; Lohman, A.W.; Straub, A.C.; Looft-Wilson, R.; Johnstone, S.R.; Araj, C.A.; Best, A.K.; Chekeni, F.B.; Ravichandran, K.S.; Penuela, S.; et al. Pannexin1 Regulates Alpha1-Adrenergic Receptor- Mediated Vasoconstriction. Circ. Res. 2011, 109, 80–85. [Google Scholar] [CrossRef]
- Murali, S.; Zhang, M.; Nurse, C.A. Evidence That 5-HT Stimulates Intracellular Ca2+ Signalling and Activates Pannexin-1 Currents in Type II Cells of the Rat Carotid Body. J. Physiol. 2017, 595, 4261–4277. [Google Scholar] [CrossRef]
- Goepfert, C.; Sundberg, C.; Sévigny, J.; Enjyoji, K.; Hoshi, T.; Csizmadia, E.; Robson, S. Disordered Cellular Migration and Angiogenesis in Cd39-Null Mice. Circulation 2001, 104, 3109–3115. [Google Scholar] [CrossRef]
- Guns, P.J.; Van Assche, T.; Fransen, P.; Robaye, B.; Boeynaems, J.M.; Bult, H. Endothelium-Dependent Relaxation Evoked by ATP and UTP in the Aorta of P2Y2-Deficient Mice. Br. J. Pharmacol. 2006, 147, 569–574. [Google Scholar] [CrossRef]
- Chen, X.; Qian, S.; Hoggatt, A.; Tang, H.; Hacker, T.A.; Obukhov, A.G.; Herring, P.B.; Seye, C.I. Endothelial Cell-Specific Deletion of P2Y2 Receptor Promotes Plaque Stability in Atherosclerosis-Susceptible ApoE-Null Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 75–83. [Google Scholar] [CrossRef]
- Lustig, K.D.; Erb, L.; Landis, D.M.; Hicks-Taylor, C.S.; Zhang, X.; Sportiello, M.G.; Weisman, G.A. Mechanisms by Which Extracellular ATP and UTP Stimulate the Release of Prostacyclin from Bovine Pulmonary Artery Endothelial Cells. Biochim. Biophys. Acta 1992, 1134, 61–72. [Google Scholar] [CrossRef]
- Favre, J.; Musette, P.; Douin-Echinard, V.; Laude, K.; Henry, J.-P.; Arnal, J.-F.; Thuillez, C.; Richard, V. Toll-like Receptors 2-Deficient Mice Are Protected against Postischemic Coronary Endothelial Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Chachisvilis, M.; Zhang, Y.-L.; Frangos, J.A. G Protein-Coupled Receptors Sense Fluid Shear Stress in Endothelial Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15463–15468. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Imamura, H.; Ando, J. Shear Stress Augments Mitochondrial ATP Generation That Triggers ATP Release and Ca2+ Signaling in Vascular Endothelial Cells. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1477–H1485. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mather, S.; Huang, Y.; Garland, C.J.; Yao, X. Extracellular ATP Facilitates Flow-Induced Vasodilatation in Rat Small Mesenteric Arteries. Am. J. Physiol. 2004, 286, H1688–H1695. [Google Scholar] [CrossRef] [PubMed]
- Kauffenstein, G.; Pelletier, J.; Lavoie, E.G.; Kukulski, F.; Martin-Satue, M.; Dufresne, S.S.; Frenette, J.; Ribas Furstenau, C.; Sereda, M.J.; Toutain, B.; et al. Nucleoside Triphosphate Diphosphohydrolase-1 Ectonucleotidase Is Required for Normal Vas Deferens Contraction and Male Fertility through Maintaining P2X1 Receptor Function. J. Biol. Chem. 2014, 289, 28629–28639. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.; Bodin, P.; Burnstock, G. Effect of Shear Stress on the Release of Soluble Ecto-Enzymes ATPase and 5’-Nucleotidase along with Endogenous ATP from Vascular Endothelial Cells. Br. J. Pharmacol. 2000, 129, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Kroll, R.G.; Powell, C.; Chen, J.; Snider, N.T.; St. Hilaire, C.; Reddy, A.; Kim, J.; Pinsky, D.J.; Murthy, V.L.; Sutton, N.R. Circulating Ectonucleotidases Signal Impaired Myocardial Perfusion at Rest and Stress. J. Am. Heart Assoc. 2023, 12, e027920. [Google Scholar] [CrossRef] [PubMed]
- Loufrani, L.; Henrion, D. Role of the Cytoskeleton in Flow (Shear Stress)-Induced Dilation and Remodeling in Resistance Arteries. Med. Biol. Eng. Comput. 2008, 46, 451–460. [Google Scholar] [CrossRef]
- Dumont, O.; Pinaud, F.; Guihot, A.L.; Baufreton, C.; Loufrani, L.; Henrion, D. Alteration in Flow (Shear Stress)-Induced Remodelling in Rat Resistance Arteries with Aging: Improvement by a Treatment with Hydralazine. Cardiovasc. Res. 2008, 77, 600–608. [Google Scholar] [CrossRef]
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, Immunity, and Hypertension. Hypertension 2011, 57, 132–140. [Google Scholar] [CrossRef]
- Guckelberger, O.; Sun, X.F.; Sévigny, J.; Imai, M.; Kaczmarek, E.; Enjyoji, K.; Kruskal, J.B.; Robson, S.C. Beneficial Effects of CD39/Ecto-Nucleoside Triphosphate Diphosphohydrolase-1 in Murine Intestinal Ischemia-Reperfusion Injury. Thromb. Haemost. 2004, 91, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Kauffenstein, G.; Tamareille, S.; Prunier, F.; Roy, C.; Ayer, A.; Toutain, B.; Billaud, M.; Isakson, B.E.; Grimaud, L.; Loufrani, L.; et al. Central Role of P2Y6 UDP Receptor in Arteriolar Myogenic Tone. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Haanes, K.A.; Spray, S.; Syberg, S.; Jørgensen, N.R.; Robaye, B.; Boeynaems, J.-M.; Edvinsson, L. New Insights on Pyrimidine Signalling within the Arterial Vasculature—Different Roles for P2Y2 and P2Y6 Receptors in Large and Small Coronary Arteries of the Mouse. J. Mol. Cell. Cardiol. 2016, 93, 1–11. [Google Scholar] [CrossRef]
- Sutton, N.R.; Hayasaki, T.; Hyman, M.C.; Anyanwu, A.C.; Liao, H.; Petrovic-Djergovic, D.; Badri, L.; Baek, A.E.; Walker, N.; Fukase, K.; et al. Ectonucleotidase CD39-Driven Control of Postinfarction Myocardial Repair and Rupture. JCI Insight 2017, 2, e89504. [Google Scholar] [CrossRef] [PubMed]
- Goodwill, A.G.; Dick, G.M.; Kiel, A.M.; Tune, J.D. Regulation of Coronary Blood Flow. Compr. Physiol. 2017, 7, 321–382. [Google Scholar] [CrossRef] [PubMed]
- Koszalka, P.; Ozuyaman, B.; Huo, Y.; Zernecke, A.; Flogel, U.; Braun, N.; Buchheiser, A.; Decking, U.K.; Smith, M.L.; Sevigny, J.; et al. Targeted Disruption of Cd73/Ecto-5’-Nucleotidase Alters Thromboregulation and Augments Vascular Inflammatory Response. Circ. Res. 2004, 95, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Korchazhkina, O.; Wright, G.; Exley, C. Intravascular ATP and Coronary Vasodilation in the Isolated Working Rat Heart. Br. J. Pharmacol. 1999, 127, 701–708. [Google Scholar] [CrossRef]
- Roy, C.; Tabiasco, J.; Caillon, A.; Delneste, Y.; Merot, J.; Favre, J.; Guihot, A.L.; Martin, L.; Nascimento, D.C.; Ryffel, B.; et al. Loss of Vascular Expression of Nucleoside Triphosphate Diphosphohydrolase-1/CD39 in Hypertension. Purinergic Signal. 2017, 14, 73–82. [Google Scholar] [CrossRef]
- Zhao, T.V.; Li, Y.; Liu, X.; Xia, S.; Shi, P.; Li, L.; Chen, Z.; Yin, C.; Eriguchi, M.; Chen, Y.; et al. ATP Release Drives Heightened Immune Responses Associated with Hypertension. Sci. Immunol. 2019, 4, eaau6426. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sokabe, T.; Ohura, N.; Nakatsuka, H.; Kamiya, A.; Ando, J. Endogenously Released ATP Mediates Shear Stress-Induced Ca2+ Influx into Pulmonary Artery Endothelial Cells. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H793–H803. [Google Scholar] [CrossRef]
- De Giorgi, M.; Enjyoji, K.; Jiang, G.; Csizmadia, E.; Mitsuhashi, S.; Gumina, R.J.; Smolenski, R.T.; Robson, S.C. Complete Deletion of Cd39 Is Atheroprotective in Apolipoprotein E-Deficient Mice. J. Lipid. Res. 2017, 58, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Visovatti, S.H.; Hyman, M.C.; Goonewardena, S.N.; Anyanwu, A.C.; Kanthi, Y.; Robichaud, P.; Wang, J.; Petrovic-Djergovic, D.; Rattan, R.; Burant, C.F.; et al. Purinergic Dysregulation in Pulmonary Hypertension. Am. J. Physiology. Heart Circ. Physiol. 2016, 311, H286–H298. [Google Scholar] [CrossRef] [PubMed]
- Koziak, K.; Bojakowska, M.; Robson, S.C.; Bojakowski, K.; Soin, J.; Csizmadia, E.; Religa, P.; Gaciong, Z.; Kaczmarek, E. Overexpression of CD39/Nucleoside Triphosphate Diphosphohydrolase-1 Decreases Smooth Muscle Cell Proliferation and Prevents Neointima Formation after Angioplasty. J. Thromb. Haemost. 2008, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Behdad, A.; Sun, X.; Khalpey, Z.; Enjyoji, K.; Wink, M.; Wu, Y.; Usheva, A.; Robson, S.C. Vascular Smooth Muscle Cell Expression of Ectonucleotidase CD39 (ENTPD1) Is Required for Neointimal Formation in Mice. Purinergic Signal 2009, 5, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, T.L.; Arnold, E.; Boerth, N.J.; Lincoln, T.M. Inhibition of Smooth Muscle Cell Growth by Nitric Oxide and Activation of cAMP-Dependent Protein Kinase by cGMP. Am. J. Physiol. 1994, 267, C1405–C1413. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.W.; Hoshi, T.; Wu, Y.; Sun, X.; Enjyoji, K.; Cszimadia, E.; Sundberg, C.; Robson, S.C. Disordered Purinergic Signaling Inhibits Pathological Angiogenesis in Cd39/Entpd1-Null Mice. Am. J. Pathol. 2007, 171, 1395–1404. [Google Scholar] [CrossRef]
- Enjyoji, K.; Kotani, K.; Thukral, C.; Blumel, B.; Sun, X.; Wu, Y.; Imai, M.; Friedman, D.; Csizmadia, E.; Bleibel, W.; et al. Deletion of Cd39/Entpd1 Results in Hepatic Insulin Resistance. Diabetes 2008, 57, 2311–2320. [Google Scholar] [CrossRef]
- Mulvany, M.J.; Halpern, W. Contractile Properties of Small Arterial Resistance Vessels in Spontaneously Hypertensive and Normotensive Rats. Circ. Res. 1977, 41, 19–26. [Google Scholar] [CrossRef]
- Unthank, J.L.; Fath, S.W.; Burkhart, H.M.; Miller, S.C.; Dalsing, M.C. Wall Remodeling during Luminal Expansion of Mesenteric Arterial Collaterals in the Rat. Circ. Res. 1996, 79, 1015–1023. [Google Scholar] [CrossRef]
- Loufrani, L.; Dubroca, C.; You, D.; Li, Z.; Levy, B.; Paulin, D.; Henrion, D. Absence of Dystrophin in Mice Reduces NO-Dependent Vascular Function and Vascular Density: Total Recovery after a Treatment with the Aminoglycoside Gentamicin. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 671–676. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Entpd1+/+ | Entpd1−/− | p Value |
|---|---|---|---|
| n | 8 | 9 | |
| MAP (mmHg) | 109 ± 2 | 107 ± 1 | 0.168 |
| SBP (mmHg) | 126 ± 2 | 121 ± 1 | * 0.018 |
| DBP (mmHg) | 90.0 ± 1.4 | 91.5 ± 1.4 | 0.471 |
| PP (mmHg) | 36.3 ± 0.9 | 29.7 ± 2.2 | * 0.019 |
| HR (beats/min) | 564 ± 10 | 534 ± 6 | * 0.022 |
| Activity (a.u.) | 4.77 ± 0.43 | 5.72 ± 0.87 | 0.365 |
| Variable | Entpd1+/+ | Entpd1−/− | p Value | ||
|---|---|---|---|---|---|
| n | 5 | 6 | |||
| MBP (mmHg) | 70.0 | ±2.9 | 79.3 | ±4.6 | 0.144 |
| SBP (mmHg) | 88.4 | ±2.1 | 94.2 | ±4.1 | 0.263 |
| DBP (mmHg) | 54.2 | ±3.9 | 67.2 | ±4.5 | 0.064 |
| PP (mmHg) | 34.2 | ±2.2 | 27.1 | ±0.9 | * 0.011 |
| Pmax (mmHg) | 93.8 | ±2.3 | 95.0 | ±2.6 | 0.740 |
| Pmin (mmHg) | 3.88 | ±0.71 | 5.50 | ±0.83 | 0.182 |
| Ped (mmHg) | 6.98 | ±0.71 | 9.17 | ±1.13 | 0.154 |
| CT (msec) | 11.6 | ±0.4 | 11.8 | ±0.4 | 0.693 |
| RT (msec) | 43.7 | ±0.6 | 42.2 | ±0.5 | 0.063 |
| dP/dt max (mmHg/s) | 6698 | ±327 | 6762 | ±489 | 0.919 |
| dP/dt min (mmHg/s) | −5557 | ±126 | −5196 | ±318 | 0.353 |
| Tau (msec) | 7.60 | ±0.51 | 6.50 | ±0.34 | 0.098 |
| HR (beats/min) | 401 | ±19 | 460 | ±15 | * 0.035 |
| Variable | Entpd1+/+ | Entpd1−/− | p Value |
|---|---|---|---|
| n | 5 | 5 | |
| ThAo | |||
| Phe EC50 | 2.04 × 10−7 ± 3.89 × 10−8 | 1.90 × 10−7 ± 2.92 × 10−8 | 0.944 |
| Phe Emax | 70.73 ± 11.62 | 70.2 ± 13.94 | 0.532 |
| Ach EC50 | 4.98 × 10−8 ± 1.11 × 10−8 | 2.70 × 10−8 ± 4.07 × 10−9 | 0.222 |
| Ach Emax | 91.41 ± 1.42 | 92.8 ± 2.12 | 0.532 |
| MRA | |||
| Phe EC50 | 4.04 × 10−7 ± 5.53 × 10−8 | 3.24 × 10−7 ± 7.61 × 10−8 | 0.532 |
| Phe Emax | 127.06 ± 6.87 | 138.92 ± 12.89 | 0.532 |
| Ach EC50 | 3.65 × 10−8 ± 6.57 × 10−9 | 1.04 × 10−7 ± 5.09 × 10−8 | 0.095 |
| Ach Emax | 99.02 ± 0.57 | 98.39 ± 0.24 | 0.532 |
| LCA | |||
| 5HT EC50 | 2.61 × 10−7 ± 7.30 × 10−8 | 7.14 × 10−8 ± 5.39 × 10−8 | * 0.029 |
| 5HT Emax | 110.30 ± 5.86 | 123.65 ± 8.47 | 0.486 |
| Ach EC50 | 6.75 × 10−8 ± 5.13 × 10−8 | 3.10 × 10−7 ± 2.13 × 10−7 | 0.873 |
| Ach Emax | 67.84 ± 10.62 | 54.71 ± 13.89 | 0.524 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favre, J.; Roy, C.; Guihot, A.-L.; Drouin, A.; Laprise, M.; Gillis, M.-A.; Robson, S.C.; Thorin, E.; Sévigny, J.; Henrion, D.; et al. NTPDase1/CD39 Ectonucleotidase Is Necessary for Normal Arterial Diameter Adaptation to Flow. Int. J. Mol. Sci. 2023, 24, 15038. https://doi.org/10.3390/ijms242015038
Favre J, Roy C, Guihot A-L, Drouin A, Laprise M, Gillis M-A, Robson SC, Thorin E, Sévigny J, Henrion D, et al. NTPDase1/CD39 Ectonucleotidase Is Necessary for Normal Arterial Diameter Adaptation to Flow. International Journal of Molecular Sciences. 2023; 24(20):15038. https://doi.org/10.3390/ijms242015038
Chicago/Turabian StyleFavre, Julie, Charlotte Roy, Anne-Laure Guihot, Annick Drouin, Manon Laprise, Marc-Antoine Gillis, Simon C. Robson, Eric Thorin, Jean Sévigny, Daniel Henrion, and et al. 2023. "NTPDase1/CD39 Ectonucleotidase Is Necessary for Normal Arterial Diameter Adaptation to Flow" International Journal of Molecular Sciences 24, no. 20: 15038. https://doi.org/10.3390/ijms242015038
APA StyleFavre, J., Roy, C., Guihot, A.-L., Drouin, A., Laprise, M., Gillis, M.-A., Robson, S. C., Thorin, E., Sévigny, J., Henrion, D., & Kauffenstein, G. (2023). NTPDase1/CD39 Ectonucleotidase Is Necessary for Normal Arterial Diameter Adaptation to Flow. International Journal of Molecular Sciences, 24(20), 15038. https://doi.org/10.3390/ijms242015038