

Contribution of Extracellular Vesicles and Molecular Chaperones in Age-Related Neurodegenerative Disorders of the CNS
 , ,
, , 
 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
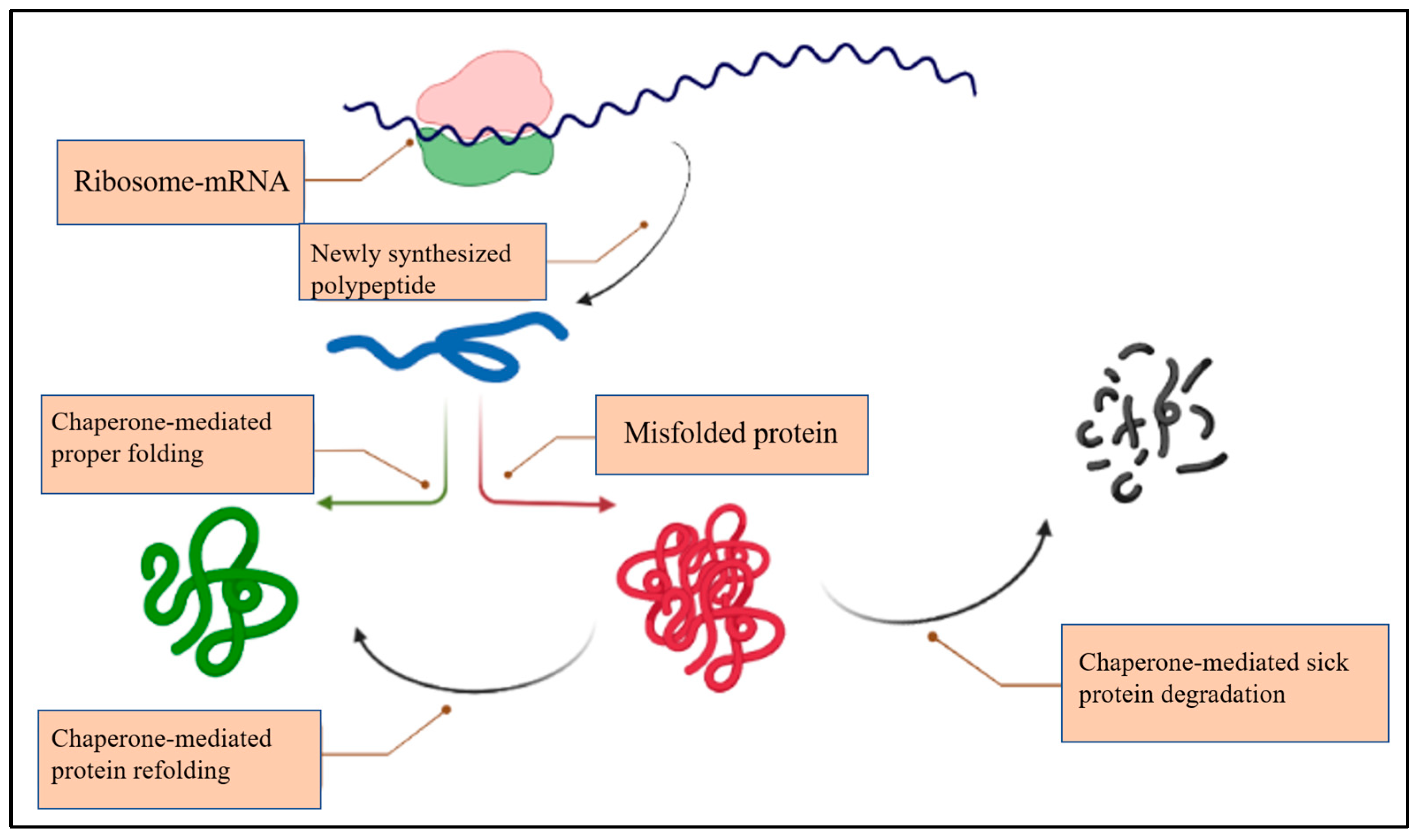
2. The Chaperone System (CS)
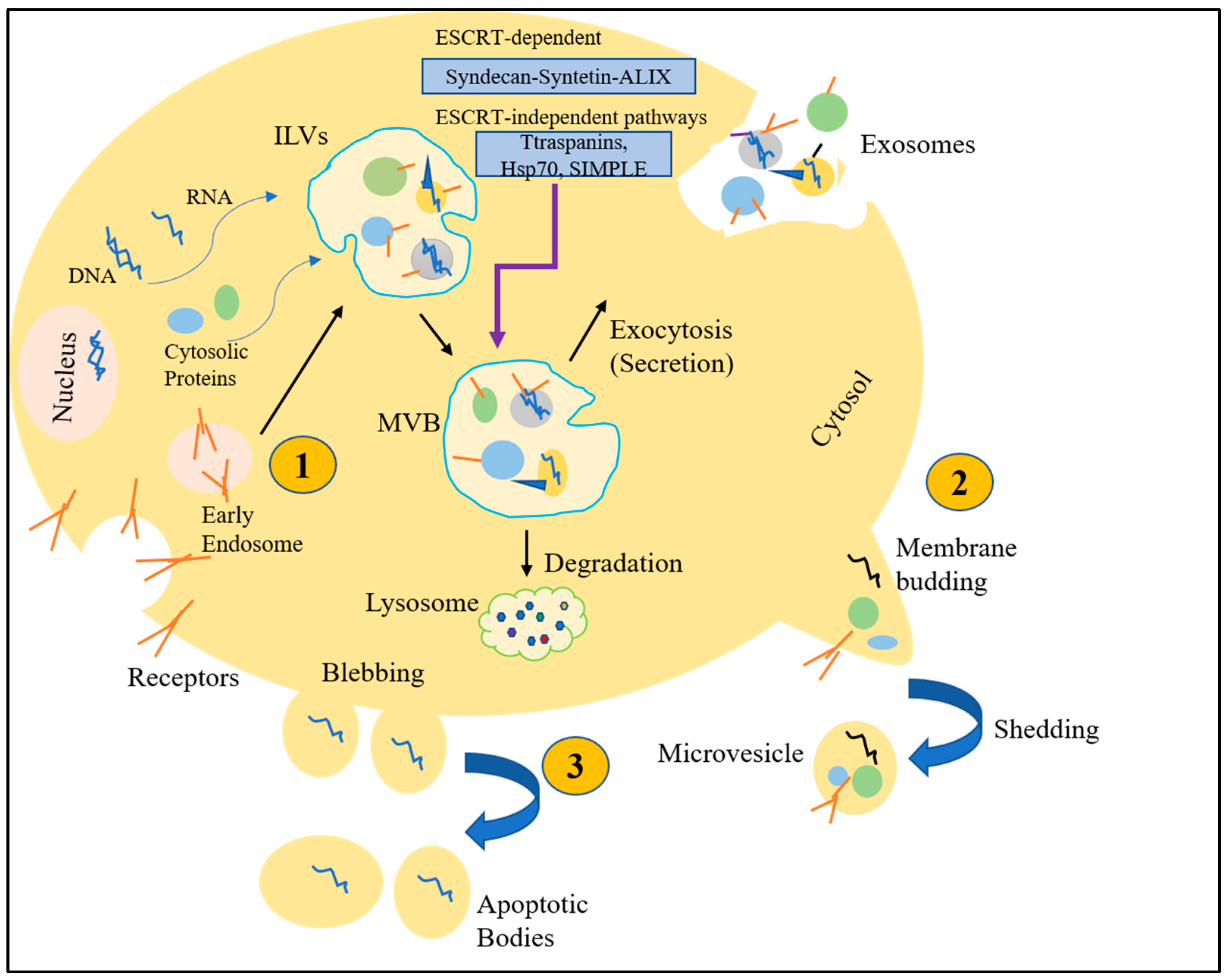
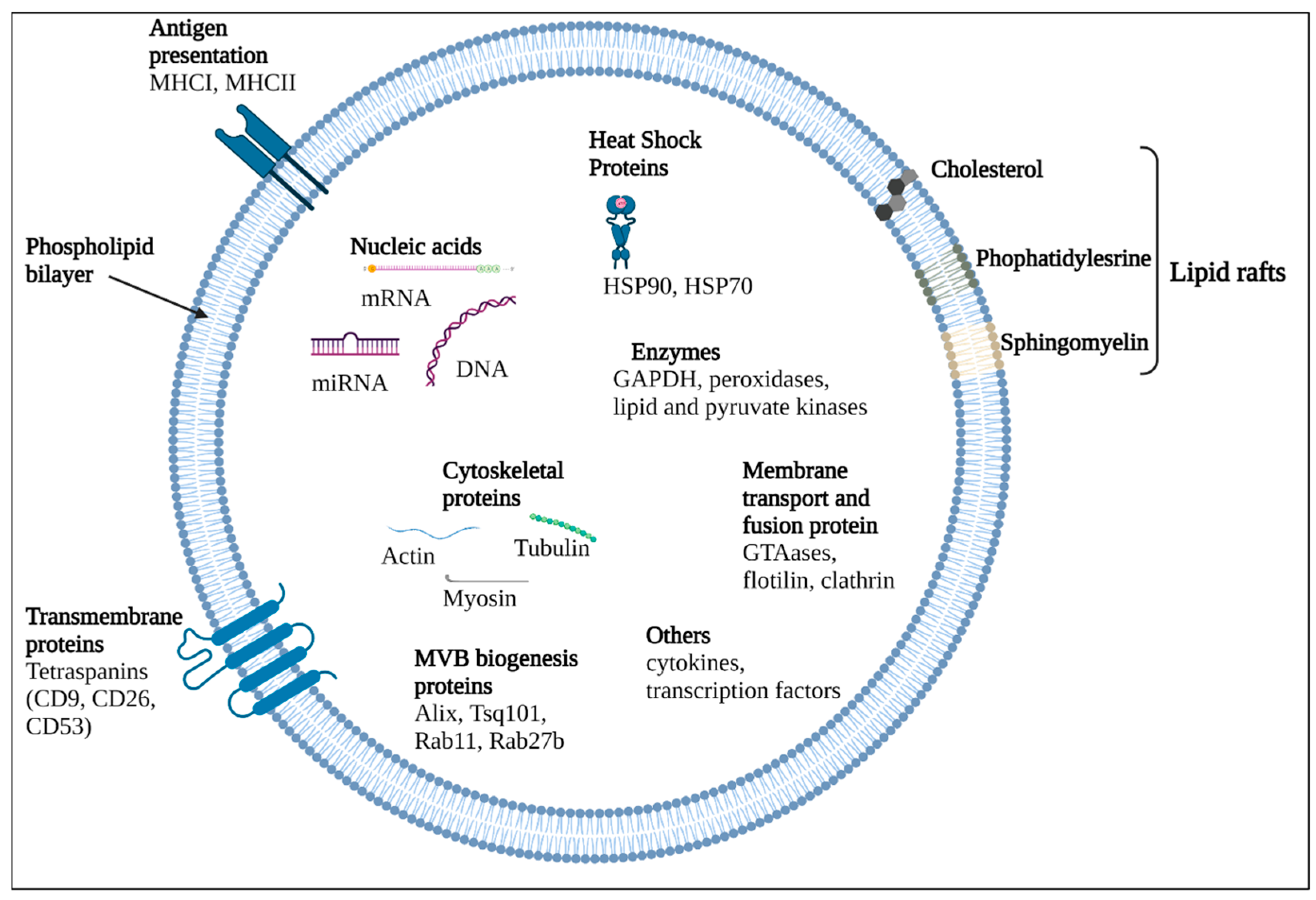
3. Extracellular Vesicles (EVs)
4. EVs in/of CNS
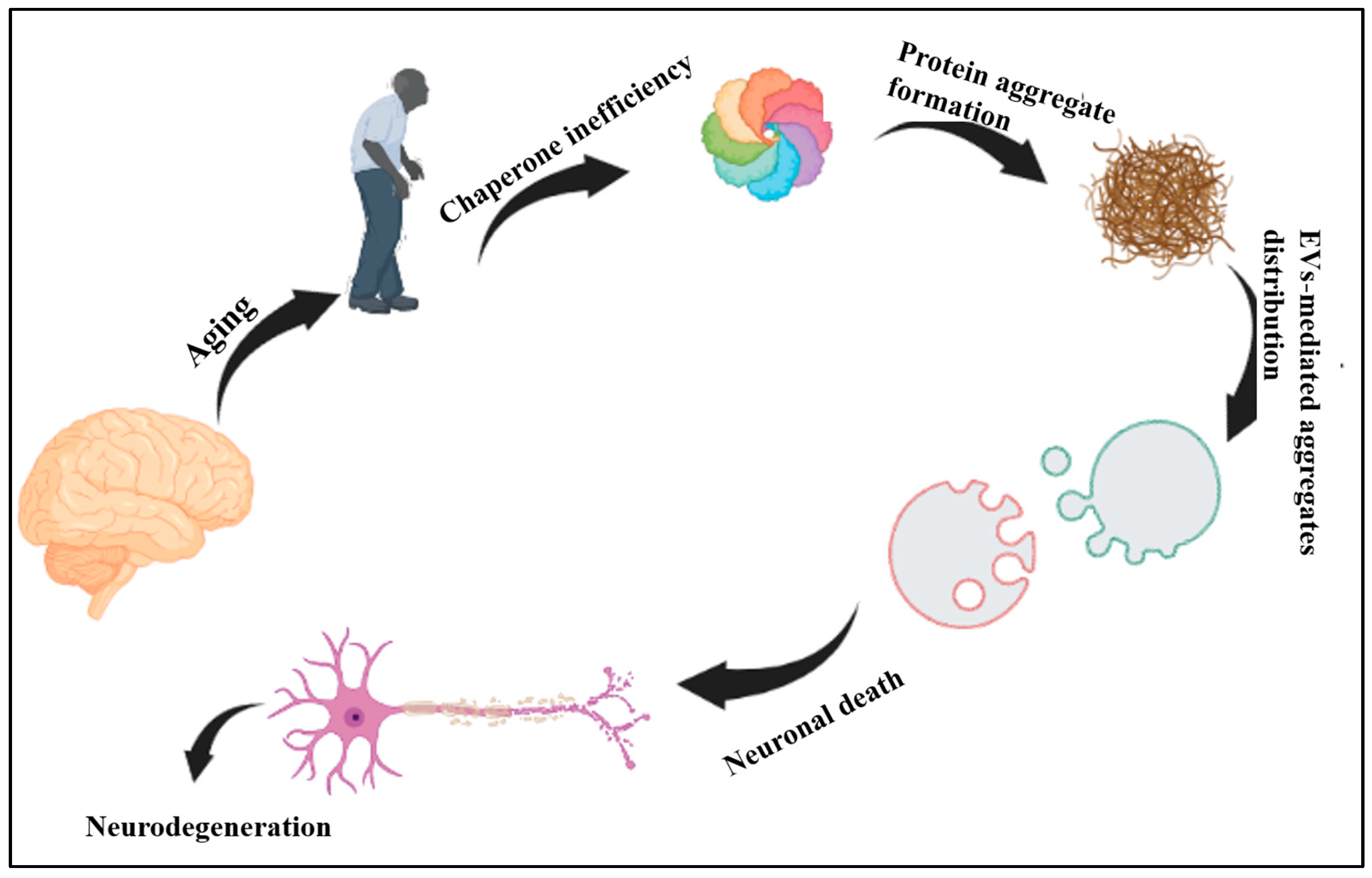
5. Ageing, CS and EVs
6. Alzheimer’s Disease, CS and EVs
7. Parkinson Disease, CS and EVs
8. Prion Disease, CS and EVs
9. Huntington’s Disease, CS and EVs
10. Therapeutic Applications of EVs and Molecular Chaperones in CNS Disorders
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer Disease |
| ALIX | ALG-2-interacting protein X |
| ALP | Autophagy Lysosomal Pathway |
| APP | Amyloid Precursor Protein |
| ARF6 | ADP-Ribosylation Factor 6 |
| ARRDC1 | Arrestin 1 Domain–Containing Protein 1 |
| Aβ | Amyloid β |
| BACE1 | Beta-Secretase 1 |
| BBB | Blood-Brain Barrier |
| BDNF | Brain Derived Neurotrophic Factor |
| BDNF | Brain-Derived Neurotrophic Factor |
| CCT | Chaperonin containing tailless complex polypeptide |
| CHMP4C | Caveolin-1 and charged MVP Protein 4C |
| circRNAs | Circular RNA |
| CJD | Creutzfeldt-Jakob Disease |
| CNS | Central nervous system |
| CS | Chaperone System |
| CSF | Cerebrospinal Fluid |
| DNAJB1 | DnaJ Heat Shock Protein Family (Hsp40) Member B1 |
| DNAJC5 | DnaJ Heat Shock Protein Family (Hsp40) Member C5 |
| ERK | Extracellular signal-Regulated Kinase |
| ESCRT | Endosomal-Sorting Complex Required for Transport |
| EV | Extracellular vesicle |
| FFI | Fatal Familial Insomnia |
| GDNF | Glial cell line-Derived Neurotrophic Factor |
| GPI | Glycosylphosphatidylinositol |
| GSLs | Glycosphingolipids |
| GSS | Gerstmann–Sträussler–Scheinker disease |
| HD | Huntington Disease |
| Hsc70 | Heat shock cognate 71 kDa protein |
| hsiRNAs | hydrophobically modified siRNAs |
| Hsp | Heat shock protein |
| HSPA4 | Heat Shock Protein Family A (Hsp70) Member 4 |
| HSPBs | small Heat Shock Proteins family |
| HTT | huntingtin gene |
| IGF1R | Insulin-like Growth Factor1 Receptor |
| IL-1β | Interleukin 1β |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| ILVs | Intraluminal vesicles |
| LAMP2 | Lysosome-Associated Membrane Protein 2 |
| LB | Lewy Bodies |
| lncRNAs | long non-coding RNAs |
| MHC | Major Histocompatibility Complex |
| MLCK | Myosin Light Chain Kinase |
| MMPs | Matrix Metalloproteinases |
| MVBs | Multivesicular Bodies |
| MVs | Microvesicles |
| NDE | Neural Derived Exosomes |
| NDEVs | Neural-Derived Evs |
| nSMase2 | neutral Sphingomyelinase 2 |
| PARK9/ATP13A2 | P-type ATPase ion pump |
| PD | Parkinson Disease |
| PDCD6IP | Programmed Cell Death 6 Interacting Protein |
| PLD | Phospholipase D |
| PM | Plasma Membrane |
| poly Q | polyglutamine |
| PQC | Protein Quality Control |
| PrPC | prion protein |
| REST | repressor element 1-silencing transcription factor |
| RVG | Rabies Virus Glycoprotein |
| SASP | Senescence-Associated Secretory Phenotype |
| sHsps | small Hsps |
| SIMPLE | Small Integral Membrane Protein of the Lysosome/late Endosome |
| SNARE | Transmembrane protein complex soluble N–ethylmaleiamide-Sensitive factor Attachment protein Receptor |
| TSAP6 | Tumor Suppressor-Activated Pathway 6 |
| UPS | Ubiquitin-Proteasome System |
| VPS4 | Vacuolar Protein Sorting-associated protein 4 |
| vSNARE | vesiclular SNARE |
| VTA1 | Vesicle Trafficking 1 |
References
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Croese, T.; Furlan, R. Extracellular vesicles in neurodegenerative diseases. Mol. Asp. Med. 2018, 60, 52–61. [Google Scholar] [CrossRef]
- Scalia, F.; Vitale, A.M.; Santonocito, R.; de Macario, E.M.; Macario, A.J.; Cappello, F. The neurochaperonopathies: Anomalies of the chaperone system with pathogenic effects in neurodegenerative and neuromuscular disorders. Appl. Sci. 2021, 11, 898. [Google Scholar] [CrossRef]
- Antona, V.; Scalia, F.; Giorgio, E.; Radio, F.C.; Brusco, A.; Oliveri, M.; Corsello, G.; Lo Celso, F.; Vadalà, M.; de Macario, E.C.; et al. A novel cct5 missense variant associated with early onset motor neuropathy. Int. J. Mol. Sci. 2020, 21, 7631. [Google Scholar] [CrossRef]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef]
- Leija-Salazar, M.; Piette, C.; Proukakis, C. Somatic mutations in neurodegeneration. Neuropathol. Appl. Neurobiol. 2018, 44, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative diseases: An overview of environmental risk factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef]
- Krtolica, A.; Campisi, J. Cancer and aging: A model for the cancer promoting effects of the aging stroma. Int. J. Biochem. Cell Biol. 2002, 34, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.-S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. “Good” and “Bad” microglia in parkinson’s disease: An understanding of homeostatic mechanisms in immunomodulation. In Inflammation in Parkinson’s Disease; Springer: Berlin/Heidelberg, Germany, 2014; pp. 105–126. [Google Scholar]
- Glezer, I.; Simard, A.; Rivest, S. Neuroprotective role of the innate immune system by microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Forman, M.S.; Trojanowski, J.Q.; Lee, V.M. Neurodegenerative diseases: A decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004, 10, 1055–1063. [Google Scholar] [CrossRef]
- Williams, A.J.; Paulson, H.L. Polyglutamine neurodegeneration: Protein misfolding revisited. Trends Neurosci. 2008, 31, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Mojaverrostami, S.; Pasbakhsh, P.; Madadi, S.; Nekoonam, S.; Zarini, D.; Noori, L.; Shiri, E.; Salama, M.; Zibara, K.; Ragerdi Kashani, I. Calorie restriction promotes remyelination in a Cuprizone-Induced demyelination mouse model of multiple sclerosis. Metab. Brain Dis. 2020, 35, 1211–1224. [Google Scholar] [CrossRef]
- Tam, S.; Spiess, C.; Auyeung, W.; Joachimiak, L.; Chen, B.; Poirier, M.A.; Frydman, J. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 2009, 16, 1279–1285. [Google Scholar] [CrossRef]
- Tam, S.; Geller, R.; Spiess, C.; Frydman, J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 2006, 8, 1155–1162. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Budnik, V.; Ruiz-Cañada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef]
- Zappulli, V.; Friis, K.P.; Fitzpatrick, Z.; Maguire, C.A.; Breakefield, X.O. Extracellular vesicles and intercellular communication within the nervous system. J. Clin. Investig. 2016, 126, 1198–1207. [Google Scholar] [CrossRef]
- Gupta, A.; Pulliam, L. Exosomes as mediators of neuroinflammation. J. Neuroinflamm. 2014, 11, 68. [Google Scholar] [CrossRef]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Möbius, W.; Goebbels, S.; Nave, K.-A.; et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte–neuron communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Simons, M. Exosomes: Vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 2013, 352, 33–47. [Google Scholar] [CrossRef]
- Vella, L.J.; Sharples, R.A.; Nisbet, R.M.; Cappai, R.; Hill, A.F. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur. Biophys. J. 2008, 37, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Kalani, A.; Tyagi, A.; Tyagi, N. Exosomes: Mediators of neurodegeneration, neuroprotection and therapeutics. Mol. Neurobiol. 2014, 49, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Leonardi, T.; Huang, B.; Iraci, N.; Vega, B.; Pluchino, S. Extracellular vesicles and their synthetic analogues in aging and age-associated brain diseases. Biogerontology 2015, 16, 147–185. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef]
- Mounier, N.; Arrigo, A.-P. Actin cytoskeleton and small heat shock proteins: How do they interact? Cell Stress Chaperones 2002, 7, 167. [Google Scholar] [CrossRef]
- Tiroli-Cepeda, A.O.; Ramos, C.H.I. An overview of the role of molecular chaperones in protein homeostasis. Protein Pept. Lett. 2011, 18, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.; de Macario, E.C. Molecular chaperones: Multiple functions, pathologies, and potential applications. Front. Biosci.-Landmark 2007, 12, 2588–2600. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Willison, K.R. The substrate specificity of eukaryotic cytosolic chaperonin CCT. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170192. [Google Scholar] [CrossRef]
- Vilasi, S.; Bulone, D.; Caruso Bavisotto, C.; Campanella, C.; Marino Gammazza, A.; San Biagio, P.L.; Cappello, F.; de Macario, E.C.; Macario, A.J.L. Chaperonin of group I: Oligomeric spectrum and biochemical and biological implications. Front. Mol. Biosci. 2018, 4, 99. [Google Scholar] [CrossRef]
- Jin, M.; Liu, C.; Han, W.; Cong, Y. TRiC/CCT chaperonin: Structure and function. In Macromolecular Protein Complexes II: Structure and Function; Springer: Berlin/Heidelberg, Germany, 2019; pp. 625–654. [Google Scholar]
- Scalia, F.; Gammazza, A.M.; de Macario, E.C.; Macario, A.J.; Cappello, F. Myelin pathology: Involvement of molecular chaperones and the promise of chaperonotherapy. Brain Sci. 2019, 9, 297. [Google Scholar] [CrossRef]
- Scalia, F.; Barone, R.; Rappa, F.; Gammazza, A.M.; Celso, F.L.; Bosco, G.L.; Barone, G.; Antona, V.; Vadalà, M.; Vitale, A.M.; et al. Muscle Histopathological Abnormalities in a Patient With a CCT5 Mutation Predicted to Affect the Apical Domain of the Chaperonin Subunit. Front. Mol. Biosci. 2022, 9, 887336. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.; De Macario, E.C. Chaperonopathies by defect, excess, or mistake. Ann. N. Y. Acad. Sci. 2007, 1113, 178–191. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavík, J.; Machala, M.; Zimmermann, P. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Conlon, M.M.; Rider, M.A.; Brownstein, N.C.; Meckes, D.G., Jr. Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 2016, 5, 31295. [Google Scholar] [CrossRef]
- Zhu, H.; Guariglia, S.; Yu, R.Y.; Li, W.; Brancho, D.; Peinado, H.; Lyden, D.; Salzer, J.; Bennett, C.; Chow, C.W. Mutation of SIMPLE in Charcot–Marie–Tooth 1C alters production of exosomes. Mol. Biol. Cell 2013, 24, 1619–1637. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neuro-Oncol. 2013, 113, 1–11. [Google Scholar] [CrossRef]
- Shi, M.; Sheng, L.; Stewart, T.; Zabetian, C.P.; Zhang, J. New windows into the brain: Central nervous system-derived extracellular vesicles in blood. Prog. Neurobiol. 2019, 175, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Gerst, J. SNAREs and SNARE regulators in membrane fusion and exocytosis. Cell. Mol. Life Sci. CMLS 1999, 55, 707–734. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Magini, A.; Buratta, S.; Brozzi, A.; Sagini, K.; Polchi, A.; Tancini, B.; Emiliani, C. Signaling pathways in exosomes biogenesis, secretion and fate. Genes 2013, 4, 152–170. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef]
- Rajendran, L.; Bali, J.; Barr, M.M.; Krämer-Albers, E.-M.; Picou, F.; Raposo, G.; van der Vos, K.E.; van Niel, G.; Wang, J.; Breakfield, X.O. Emerging roles of extracellular vesicles in the nervous system. J. Neurosci. 2014, 34, 15482–15489. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.; Bandari, S.K.; Liu, J.; Mobley, J.A.; Brown, E.E.; Sanderson, R.D. Fibronectin on the surface of myeloma cell-derived exosomes mediates exosome-cell interactions. J. Biol. Chem. 2016, 291, 1652–1663. [Google Scholar] [CrossRef]
- Stoeck, A.; Keller, S.; Riedle, S.; Sanderson, M.P.; Runz, S.; Le Naour, F.; Gutwein, P.; Ludwig, A.; Rubinstein, E.; Altevogt, P. A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem. J. 2006, 393, 609–618. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Raposo, G.; Stahl, P.D. Extracellular vesicles: A new communication paradigm? Nat. Rev. Mol. Cell Biol. 2019, 20, 509–510. [Google Scholar] [CrossRef] [PubMed]
- Cappello, F.; Logozzi, M.; Campanella, C.; Bavisotto, C.C.; Marcilla, A.; Properzi, F.; Fais, S. Exosome levels in human body fluids: A tumor marker by themselves? Eur. J. Pharm. Sci. 2017, 96, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Noori, L.; Arabzadeh, S.; Mohamadi, Y.; Mojaverrostami, S.; Mokhtari, T.; Akbari, M.; Hassanzadeh, G. Intrathecal administration of the extracellular vesicles derived from human Wharton’s jelly stem cells inhibit inflammation and attenuate the activity of inflammasome complexes after spinal cord injury in rats. Neurosci. Res. 2021, 170, 87–98. [Google Scholar] [CrossRef]
- Jan, A.T.; Malik, M.A.; Rahman, S.; Yeo, H.R.; Lee, E.J.; Abdullah, T.S.; Choi, I. Perspective insights of exosomes in neurodegenerative diseases: A critical appraisal. Front. Aging Neurosci. 2017, 9, 317. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes–vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Mahdavipour, M.; Hassanzadeh, G.; Seifali, E.; Mortezaee, K.; Aligholi, H.; Shekari, F.; Sarkoohi, P.; Zeraatpishe, Z.; Nazari, A.; Movassaghi, S.; et al. Effects of neural stem cell-derived extracellular vesicles on neuronal protection and functional recovery in the rat model of middle cerebral artery occlusion. Cell Biochem. Funct. 2020, 38, 373–383. [Google Scholar] [CrossRef]
- Qu, Y.; Franchi, L.; Nunez, G.; Dubyak, G.R. Nonclassical IL-1β secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J. Immunol. 2007, 179, 1913–1925. [Google Scholar] [CrossRef]
- Phoonsawat, W.; Aoki-Yoshida, A.; Tsuruta, T.; Sonoyama, K. Adiponectin is partially associated with exosomes in mouse serum. Biochem. Biophys. Res. Commun. 2014, 448, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, R.; Kemper, S.; Charrier, A.; Brigstock, D.R. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: Role of exosomes in horizontal transfer of Twist1. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 309, G491–G499. [Google Scholar] [CrossRef]
- Pastuzyn, E.D.; Day, C.E.; Kearns, R.B.; Kyrke-Smith, M.; Taibi, A.V.; McCormick, J.; Yoder, N.; Belnap, D.M.; Erlendsson, S.; Morado, D.R.; et al. The neuronal gene arc encodes a repurposed retrotransposon gag protein that mediates intercellular RNA transfer. Cell 2018, 172, 275–288.e18. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Gray, E.; Heman-Ackah, S.M.; Mäger, I.; Talbot, K.; Andaloussi, S.E.; Wood, M.J.; Turner, M.R. Extracellular vesicles in neurodegenerative disease—Pathogenesis to biomarkers. Nat. Rev. Neurol. 2016, 12, 346–357. [Google Scholar] [CrossRef]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of extracellular vesicles across the blood-brain barrier: Brain pharmacokinetics and effects of inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, L.; Ma, F.; Wong, C.; Guo, X.; Chacko, J.; Farhoodi, H.P.; Zhang, S.X.; Zimak, J.; Ségaliny, A.; et al. Elucidation of exosome migration across the blood-brain barrier model in vitro. Cell. Mol. Bioeng. 2016, 9, 509–529. [Google Scholar] [CrossRef]
- Benz, F.; Liebner, S. Structure and Function of the Blood–Brain Barrier (BBB); Springer: Cham, Switzerland, 2020. [Google Scholar]
- Wurdinger, T.; Gatson, N.N.; Balaj, L.; Kaur, B.; Breakefield, X.O.; Pegtel, D.M. Extracellular vesicles and their convergence with viral pathways. Adv. Virol. 2012, 2012, 767694. [Google Scholar] [CrossRef]
- Villegas, J.; Broadwell, R. Transcytosis of protein through the mammalian cerebral epithelium and endothelium. II. Adsorptive transcytosis of WGA-HRP and the blood-brain and brain-blood barriers. J. Neurocytol. 1993, 22, 67–80. [Google Scholar] [CrossRef]
- Banks, W.A.; Freed, E.O.; Wolf, K.M.; Robinson, S.M.; Franko, M.; Kumar, V.B. Transport of human immunodeficiency virus type 1 pseudoviruses across the blood-brain barrier: Role of envelope proteins and adsorptive endocytosis. J. Virol. 2001, 75, 4681–4691. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Akerstrom, V. HIV-1 protein gp120 crosses the blood-brain barrier: Role of adsorptive endocytosis. Life Sci. 1997, 61, PL119–PL125. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Neuroimmune axes of the blood–brain barriers and blood–brain interfaces: Bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Seifali, E.; Hassanzadeh, G.; Mahdavipour, M.; Mortezaee, K.; Moini, A.; Satarian, L.; Shekari, F.; Nazari, A.; Movassaghi, S.; Akbari, M. Extracellular Vesicles Derived from Human Umbilical Cord Perivascular Cells Improve Functional Recovery in Brain Ischemic Rat via the Inhibition of Apoptosis. Iran. Biomed. J. 2020, 24, 347. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Park, S.-H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and-aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef]
- De Toda, I.M.; Maté, I.; Vida, C.; Cruces, J.; De la Fuente, M. Immune function parameters as markers of biological age and predictors of longevity. Aging (Albany NY) 2016, 8, 3110. [Google Scholar] [CrossRef] [PubMed]
- Ambra, R.; Mocchegiani, E.; Giacconi, R.; Canali, R.; Rinna, A.; Malavolta, M.; Virgili, F. Characterization of the hsp70 response in lymphoblasts from aged and centenarian subjects and differential effects of in vitro zinc supplementation. Exp. Gerontol. 2004, 39, 1475–1484. [Google Scholar] [CrossRef]
- Khodakarami, A. Investigating the Role of the CCT Chaperonin in Stem Cell Identity and Aging; Universität zu Köln: Cologne, Germany, 2017. [Google Scholar]
- Llewellyn, J.; Mallikarjun, V.; Appleton, E.; Osipova, M.; Gilbert, H.T.; Richardson, S.M.; Hubbard, S.J.; Swift, J. Senescence inhibits the chaperone response to thermal stress. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gidalevitz, T.; Ben-Zvi, A.; Ho, K.H.; Brignull, H.R.; Morimoto, R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 2006, 311, 1471–1474. [Google Scholar] [CrossRef]
- Csermely, P.; Sőti, C.; Kalmar, E.; Papp, E.; Pato, B.; Vermes, A.; Sreedhar, A.S. Molecular chaperones, evolution and medicine. J. Mol. Struct. THEOCHEM 2003, 666, 373–380. [Google Scholar] [CrossRef]
- Nardai, G.; Végh, E.M.; Prohászka, Z.; Csermely, P. Chaperone-related immune dysfunction: An emergent property of distorted chaperone networks. Trends Immunol. 2006, 27, 74–79. [Google Scholar] [CrossRef]
- Arslan, M.A.; Csermely, P.; Sőti, C. Protein homeostasis and molecular chaperones in aging. Biogerontology 2006, 7, 383–389. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.; Desprez, P.; Krtolica, A.; Campisi, J.J. ARoPMD. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Ungerleider, K.; Beck, J.; Lissa, D.; Turnquist, C.; Horikawa, I.; Harris, B.T.; Harris, C.C. Astrocyte senescence and SASP in neurodegeneration: Tau joins the loop. Cell Cycle 2021, 20, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Hong, L.; Shen, H.; Du, Q.; Ye, Q.; Chen, X.; Zhang, J. Estradiol-induced senescence of hypothalamic astrocytes contributes to aging-related reproductive function declines in female mice. Aging Albany NY 2020, 12, 6089. [Google Scholar] [CrossRef]
- Urbanelli, L.; Buratta, S.; Sagini, K.; Tancini, B.; Emiliani, C. Extracellular vesicles as new players in cellular senescence. Int. J. Mol. Sci. 2016, 17, 1408. [Google Scholar] [CrossRef]
- Soria, F.N.; Pampliega, O.; Bourdenx, M.; Meissner, W.G.; Bezard, E.; Dehay, B. Exosomes, an unmasked culprit in neurodegenerative diseases. Front. Neurosci. 2017, 11, 26. [Google Scholar] [CrossRef]
- Misawa, T.; Tanaka, Y.; Okada, R.; Takahashi, A. Biology of extracellular vesicles secreted from senescent cells as senescence-associated secretory phenotype factors. Geriatr. Gerontol. Int. 2020, 20, 539–546. [Google Scholar] [CrossRef]
- Song, Z.; Xu, Y.; Deng, W.; Zhang, L.; Zhu, H.; Yu, P.; Qu, Y.; Zhao, W.; Han, Y.; Qin, C. Brain derived exosomes are a double-edged sword in Alzheimer’s disease. Front. Mol. Neurosci. 2020, 13, 79. [Google Scholar] [CrossRef]
- Lim, Y.-J.; Lee, S.-J. Are exosomes the vehicle for protein aggregate propagation in neurodegenerative diseases? Acta Neuropathol. Commun. 2017, 5, 64. [Google Scholar] [CrossRef]
- Lee, B.-R.; Kim, J.-H.; Choi, E.-S.; Cho, J.-H.; Kim, E. Effect of young exosomes injected in aged mice. Int. J. Nanomed. 2018, 13, 5335. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016, 30, 4141–4148. [Google Scholar] [CrossRef]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef]
- Yu, X.; Riley, T.; Levine, A.J. The regulation of the endosomal compartment by p53 the tumor suppressor gene. FEBS J. 2009, 276, 2201–2212. [Google Scholar] [CrossRef]
- Feng, Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, a001057. [Google Scholar] [CrossRef]
- Saksena, S.; Sun, J.; Chu, T.; Emr, S.D. ESCRTing proteins in the endocytic pathway. Trends Biochem. Sci. 2007, 32, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Lespagnol, A.; Duflaut, D.; Beekman, C.; Blanc, L.; Fiucci, G.; Marine, J.-C.; Vidal, M.; Amson, R.; Telerman, A. Exosome secretion, including the DNA damage-induced p53-dependent secretory pathway, is severely compromised in TSAP6/Steap3-null mice. Cell Death Differ. 2008, 15, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Weiner-Gorzel, K.; Dempsey, E.; Milewska, M.; McGoldrick, A.; Toh, V.; Walsh, A.; Lindsay, S.; Gubbins, L.; Cannon, A.; Sharpe, D.; et al. Overexpression of the microRNA miR-433 promotes resistance to paclitaxel through the induction of cellular senescence in ovarian cancer cells. Cancer Med. 2015, 4, 745–758. [Google Scholar] [CrossRef]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging 2009, 1, 402. [Google Scholar] [CrossRef]
- Serpente, M.; Fenoglio, C.; D’Anca, M.; Arcaro, M.; Sorrentino, F.; Visconte, C.; Arighi, A.; Fumagalli, G.G.; Porretti, L.; Cattaneo, A.; et al. MiRNA profiling in plasma neural-derived small extracellular vesicles from patients with Alzheimer’s disease. Cells 2020, 9, 1443. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Lowenstein, C.J. MiR-34, SIRT1, and p53: The feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, M.; Chen, B.; Calin, G.A. Exosomal lncRNAs as new players in cell-to-cell communication. Transl. Cancer Res. 2018, 7 (Suppl. 2), S243. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, D.P.; Bitar, M.; Jacobs, F.M.; Barry, G. Long non-coding RNAs in neuronal aging. Non-Coding RNA 2018, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Grammatikakis, I.; Panda, A.C.; Abdelmohsen, K.; Gorospe, M. Long noncoding RNAs (lncRNAs) and the molecular hallmarks of aging. Aging Albany NY 2014, 6, 992. [Google Scholar] [CrossRef] [PubMed]
- Massone, S.; Vassallo, I.; Fiorino, G.; Castelnuovo, M.; Barbieri, F.; Borghi, R.; Tabaton, M.; Robello, M.; Gatta, E.; Russo, C.; et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol. Dis. 2011, 41, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, K.M.; Noh, J.H.; Yoon, J.-H.; Abdelmohsen, K.; Gorospe, M. Long noncoding RNAs in diseases of aging. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2016, 1859, 209–221. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Q.; Chen, J.; Hu, J.; Wang, S.; Sun, Y. Role of long non-coding RNA HULC in cell proliferation, apoptosis and tumor metastasis of gastric cancer: A clinical and in vitro investigation. Oncol. Rep. 2014, 31, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Panda, A.C.; Kang, M.-J.; Guo, R.; Kim, J.; Grammatikakis, I.; Yoon, J.H.; Dudekula, D.B.; Noh, J.H.; Yang, X.; et al. 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res. 2014, 42, 10099–10111. [Google Scholar] [CrossRef]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 Suppresses Target Mrna. Mol. Cell 2012, 47, 648–655, Erratum in Mol. Cell 2013, 50, 303. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The cellular phase of Alzheimer’s disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef]
- Chaari, A. Molecular chaperones biochemistry and role in neurodegenerative diseases. Int. J. Biol. Macromol. 2019, 131, 396–411. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.; Liang, Y.; Chow, T.C.; Tang, H.; Wu, S.L.; Wai, M.S.; Yew, D.T. Mutated tau, amyloid and neuroinflammation in Alzheimer disease—A brief review. Prog. Histochem. Cytochem. 2016, 51, 1–8. [Google Scholar] [CrossRef]
- Querfurth, H.; LaFerla, F. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 1844–1845. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, S.; Velázquez-Delgado, E.M.; Abbruzzese, G.; Hardy, J.A. Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J. Mol. Biol. 2011, 406, 75–91. [Google Scholar] [CrossRef]
- Janel, N.; Alexopoulos, P.; Badel, A.; Lamari, F.; Camproux, A.; Lagarde, J.; Simon, S.; Fraudet-Tarisse, C.; Lamourette, P.; Arbones, M.; et al. Combined assessment of DYRK1A, BDNF and homocysteine levels as diagnostic marker for Alzheimer’s disease. Transl. Psychiatry 2017, 7, e1154. [Google Scholar] [CrossRef] [PubMed]
- Leshchyns’ka, I.; Sytnyk, V. Synaptic cell adhesion molecules in Alzheimer’s disease. Neural. Plast. 2016, 2016, 6427537. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Schenk, D. Alzheimer’s disease: Molecular understanding predicts amyloid-based therapeutics. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 545–584. [Google Scholar] [CrossRef]
- Wyttenbach, A. Role of heat shock proteins during polyglutamine neurodegeneration. J. Mol. Neurosci. 2004, 23, 69–95. [Google Scholar] [CrossRef]
- Shimura, H.; Schwartz, D.; Gygi, S.P.; Kosik, K.S. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004, 279, 4869–4876. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. BioMed Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef] [PubMed]
- Radke, E.G.; Braun, J.M.; Nachman, R.M.; Cooper, G.S. Phthalate exposure and neurodevelopment: A systematic review and meta-analysis of human epidemiological evidence. Environ. Int. 2020, 137, 105408. [Google Scholar] [CrossRef]
- Tittelmeier, J.; Nachman, E.; Nussbaum-Krammer, C. Molecular chaperones: A double-edged sword in neurodegenerative diseases. Front. Aging Neurosci. 2020, 12, 581374. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xu, B.; Chen, J.; Sui, Y.; Ren, L.; Li, J.; Zhang, H.; Guo, L.; Sun, X. Micro-RNA-137 inhibits tau hyperphosphorylation in Alzheimer’s disease and targets the CACNA1C gene in transgenic mice and human neuroblastoma SH-SY5Y cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 5635. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of tau aggregates and neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Matts, R.L.; Brandt, G.E.; Lu, Y.; Dixit, A.; Mollapour, M.; Wang, S.; Donnelly, A.C.; Neckers, L.; Verkhivker, G.; Blagg, B.S.J. A systematic protocol for the characterization of Hsp90 modulators. Bioorg. Med. Chem. 2011, 19, 684–692. [Google Scholar] [CrossRef]
- Sőti, C.; Csermely, P. Molecular chaperones and the aging process. Biogerontology 2000, 1, 225–233. [Google Scholar] [CrossRef]
- Sherman, M.Y.; Goldberg, A.L. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001, 29, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Imarisio, S.; Menzies, F.M.; Jimenez-Sanchez, M.; Siddiqi, F.H.; Wu, X.; Renna, M.; O’Kane, C.J.; Crowther, D.C.; Rubinsztein, D.C. CCT complex restricts neuropathogenic protein aggregation via autophagy. Nat. Commun. 2016, 7, 13821. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [PubMed]
- Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A.F. Inhibition of γ-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008, 22, 1469–1478. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gelé, P.; Drobeck, H.; Bégard, S.; Galas, M.C.; Delacourte, A.; Beauvillain, J.-C.; Buée, L.; et al. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J. Biol. Chem. 2007, 282, 18197–18205. [Google Scholar] [CrossRef]
- Perez-Gonzalez, R.; Gauthier, S.A.; Kumar, A.; Levy, E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 2012, 287, 43108–43115. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Yamamoto, N.; Yanagisawa, K. Accelerated release of exosome-associated GM1 ganglioside (GM1) by endocytic pathway abnormality: Another putative pathway for GM1-induced amyloid fibril formation. J. Neurochem. 2008, 105, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dinkins, M.; He, Q.; Zhu, G.; Poirier, C.; Campbell, A.; Mayer-Proschel, m.; Bieberich, E. Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4): Potential mechanism of apoptosis induction in Alzheimer disease (AD). J. Biol. Chem. 2012, 287, 21384–21395. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef]
- Agosta, F.; Dalla Libera, D.; Spinelli, E.G.; Finardi, A.; Canu, E.; Bergami, A.; Chiavetto, L.B.; Baronio, M.; Comi, G.; Martino, G.; et al. Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and A lzheimer disease. Ann. Neurol. 2014, 76, 813–825. [Google Scholar] [CrossRef]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef]
- Ghidoni, R.; Paterlini, A.; Albertini, V.; Glionna, M.; Monti, E.; Schiaffonati, L.; Benussi, L.; Levy, E.; Binetti, G. Cystatin C is released in association with exosomes: A new tool of neuronal communication which is unbalanced in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Sundelöf, J.; Ärnlöv, J.; Ingelsson, E.; Sundström, J.; Basu, S.; Zethelius, B.; Larsson, A.; Irizarry, M.C.; Giedraitis, V.; Rönnemaa, E.; et al. Serum cystatin C and the risk of Alzheimer disease in elderly men. Neurology 2008, 71, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Sakai, S.; Mitsutake, S.; Okada, M.; Tahara, H.; Furukawa, J.-I.; Fujitani, N.; Shinohara, Y.; Igarashi, Y. Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 2014, 289, 24488–24498. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Sun, H.; Usuki, S.; Sakai, S.; Hanamatsu, H.; Mioka, T.; Kimura, N.; Okada, M.; Tahara, H.; Furukawa, J.-I.; et al. A potential function for neuronal exosomes: Sequestering intracerebral amyloid-β peptide. FEBS Lett. 2015, 589, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, N.C.; Alvarez, V.E.; Lee, N.C.Y.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimer’s Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef]
- Tang, Z.; Ioja, E.; Bereczki, E.; Hultenby, K.; Li, C.; Guan, Z.; Winblad, B.; Pei, J.J. mTor mediates tau localization and secretion: Implication for Alzheimer’s disease. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 1646–1657. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584. [Google Scholar] [CrossRef]
- Shi, M.; Kovac, A.; Korff, A.; Cook, T.J.; Ginghina, C.; Bullock, K.M.; Yang, L.; Stewart, T.; Zheng, D.; Aro, P.; et al. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimer’s Dement. 2016, 12, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Saman, S.; Lee, N.C.; Inoyo, I.; Jin, J.; Li, Z.; Doyle, T.; McKee, N.C.; Hall, G.F. Proteins recruited to exosomes by tau overexpression implicate novel cellular mechanisms linking tau secretion with Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 40, S47–S70. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Breydo, L.; Green, R.; Kis, V.; Puska, G.; Lőrincz, P.; Perju-Dumbrava, L.; Giera, R.; Piker, W.; Lutz, M.; et al. Intracellular processing of disease-associated α-synuclein in the human brain suggests prion-like cell-to-cell spread. Neurobiol. Dis. 2014, 69, 76–92. [Google Scholar] [CrossRef]
- Sot, B.; Rubio-Muñoz, A.; Leal-Quintero, A.; Martínez-Sabando, J.; Marcilla, M.; Roodveldt, C.; Valpuesta, J.M. The chaperonin CCT inhibits assembly of α-synuclein amyloid fibrils by a specific, conformation-dependent interaction. Sci. Rep. 2017, 7, 40859. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Uryu, K.; Richter-Landsberg, C.; Welch, W.; Sun, E.; Goldbaum, O.; Norris, E.H.; Pham, C.T.; Yazawa, I.; Hilburger, K.; Micsenyi, M.; et al. Convergence of heat shock protein 90 with ubiquitin in filamentous α-synuclein inclusions of α-synucleinopathies. Am. J. Pathol. 2006, 168, 947–961. [Google Scholar] [CrossRef]
- Xiong, R.; Zhou, W.; Siegel, D.; Kitson, R.R.; Freed, C.R.; Moody, C.J.; Ross, D. A novel Hsp90 inhibitor activates compensatory heat shock protein responses and autophagy and alleviates mutant A53T α-synuclein toxicity. Mol. Pharmacol. 2015, 88, 1045–1054. [Google Scholar] [CrossRef]
- St Martin, J.L.; Klucken, J.; Outeiro, T.F.; Nguyen, P.; Keller-McGandy, C.; Cantuti-Castelvetri, I.; Grammatopoulos, T.N.; Standaert, D.G.; Hyman, B.T.; Mclean, P.J. Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J. Neurochem. 2007, 100, 1449–1457. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Klucken, J.; Strathearn, K.E.; Liu, F.; Nguyen, P.; Rochet, J.-C.; Hyman, B.T. Small heat shock proteins protect against α-synuclein-induced toxicity and aggregation. Biochem. Biophys. Res. Commun. 2006, 351, 631–638. [Google Scholar] [CrossRef]
- Rekas, A.; Adda, C.G.; Aquilina, J.A.; Barnham, K.J.; Sunde, M.; Galatis, D.; Williamson, N.A.; Masters, C.L.; Anders, R.F.; Robinson, C.V.; et al. Interaction of the molecular chaperone αB-crystallin with α-synuclein: Effects on amyloid fibril formation and chaperone activity. J. Mol. Biol. 2004, 340, 1167–1183. [Google Scholar] [CrossRef]
- Dimant, H.; Ebrahimi-Fakhari, D.; McLean, P.J. Molecular chaperones and co-chaperones in Parkinson disease. Neurosci. 2012, 18, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; Mclean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef]
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-synuclein aggregation by exosomes. J. Biol. Chem. 2015, 290, 2969–2982. [Google Scholar] [CrossRef]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 regulates secretion of exosomes and α-synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef]
- Kong, S.M.; Chan, B.K.; Park, J.-S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.A.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Poehler, A.-M.; Xiang, W.; Spitzer, P.; May, V.E.L.; Meixner, H.; Rockenstein, E.; Chunta, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Xie, S.; Liu, F.; Simone, L.C.; Caplan, S.; Qin, X.; Naslavsky, N. Rapid degradation of the complement regulator, CD59, by a novel inhibitor. J. Biol. Chem. 2014, 289, 12109–12125. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, P.S.; Huang, Y.; Gysbers, A.; Cheng, D.; Gai, W.P.; Outeiro, T.F.; Halliday, J.M. LRRK2 interactions with α-synuclein in Parkinson’s disease brains and in cell models. J. Mol. Med. 2013, 91, 513–522. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef]
- Stuendl, A.; Kunadt, M.; Kruse, N.; Bartels, C.; Moebius, W.; Danzer, K.M.; Mollenhauer, B.; Schneider, A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 2016, 139, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Prabhakar, S.; Balaj, L.; Lai, C.P.; Cerione, R.A.; Breakefield, X.O. Delivery of therapeutic proteins via extracellular vesicles: Review and potential treatments for Parkinson’s disease, glioma, and schwannoma. Cell. Mol. Neurobiol. 2016, 36, 417–427. [Google Scholar] [CrossRef]
- Garbayo, E.; Ansorena, E.; Lana, H.; del Mar Carmona-Abellan, M.; Marcilla, I.; Lanciego, J.L.; Luquin, M.R.; Blanco-Prieto, M.J. Brain delivery of microencapsulated GDNF induces functional and structural recovery in parkinsonian monkeys. Biomaterials 2016, 110, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef]
- Ugalde, C.L.; Finkelstein, D.I.; Lawson, V.A.; Hill, A.F. Pathogenic mechanisms of prion protein, amyloid-β and α-synuclein misfolding: The prion concept and neurotoxicity of protein oligomers. J. Neurochem. 2016, 139, 162–180. [Google Scholar] [CrossRef]
- Tagliapietra, M.; Zanusso, G.; Fiorini, M.; Bonetto, N.; Zarantonello, G.; Zambon, A.; Ermani, M.; Monaco, S.; Manara, R.; Cagnin, A. Accuracy of diagnostic criteria for sporadic Creutzfeldt-Jakob disease among rapidly progressive dementia. J. Alzheimer’s Dis. 2013, 34, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Harischandra, D.S.; Kondru, N.; Martin, D.P.; Kanthasamy, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.G. Role of proteolytic activation of protein kinase Cδ in the pathogenesis of prion disease. Prion 2014, 8, 143–153. [Google Scholar] [CrossRef]
- Hetz, C.A.; Soto, C. Stressing out the ER: A role of the unfolded protein response in prion-related disorders. Curr. Mol. Med. 2006, 6, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Kovács, G.G.; Kurucz, I.; Budka, H.; Ádori, C.; Müller, F.; Ács, P.; Klöppel, S.; Schätzl, H.M.; Mayer, R.J.; László, L. Prominent stress response of Purkinje cells in Creutzfeldt–Jakob disease. Neurobiol. Dis. 2001, 8, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Kenward, N.; Hope, J.; Landon, M.; Mayer, R.J. Expression of polyubiquitin and heat-shock protein 70 genes increases in the later stages of disease progression in scrapie-infected mouse brain. J. Neurochem. 1994, 62, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.; Edskes, H.; Gorkovskiy, A.; Bezsonov, E.; Stroobant, E. Yeast and fungal prions: Amyloid-handling systems, amyloid structure, and prion biology. Adv. Genet. 2016, 93, 191–236. [Google Scholar]
- Wickner, R.B.; Bezsonov, E.E.; Son, M.; Ducatez, M.; DeWilde, M.; Edskes, H.K. Anti-prion systems in yeast and inositol polyphosphates. Biochemistry 2018, 57, 1285–1292. [Google Scholar] [CrossRef]
- Wang, S.-B.; Shi, Q.; Xu, Y.; Xie, W.-L.; Zhang, J.; Tian, C.; Guo, Y.; Wang, K.; Zhang, B.Y.; Chen, C.; et al. Protein disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutant-induced cytotoxicity. PLoS ONE 2012, 7, e38221. [Google Scholar] [CrossRef]
- Mays, C.E.; Armijo, E.; Morales, R.; Kramm, C.; Flores, A.; Tiwari, A.; Bian, J.; Telling, G.C.; Pandita, T.K.; Hunt, C.R.; et al. Prion disease is accelerated in mice lacking stress-induced heat shock protein 70 (HSP70). J. Biol. Chem. 2019, 294, 13619–13628. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.; Taraboulos, A.; Prusiner, S. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992, 267, 16188–16199. [Google Scholar] [CrossRef] [PubMed]
- Stöckel, J.; Hartl, F.U. Chaperonin-mediated de novo generation of prion protein aggregates. J. Mol. Biol. 2001, 313, 861–872. [Google Scholar] [CrossRef]
- Kiselev, G.G.; Naletova, I.N.; Sheval, E.V.; Stroylova, Y.Y.; Schmalhausen, E.V.; Haertlé, T.; Muronetz, V.I. Chaperonins induce an amyloid-like transformation of ovine prion protein: The fundamental difference in action between eukaryotic TRiC and bacterial GroEL. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2011, 1814, 1730–1738. [Google Scholar] [CrossRef]
- Kudryavtseva, S.; Stroylova, Y.; Kurochkina, L.; Muronetz, V. The chaperonin TRiC is blocked by native and glycated prion protein. Arch. Biochem. Biophys. 2020, 683, 108319. [Google Scholar] [CrossRef]
- Vella, L.J.; Greenwood, D.L.; Cappai, R.; Scheerlinck, J.-P.Y.; Hill, A.F. Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet. Immunol. Immunopathol. 2008, 124, 385–393. [Google Scholar] [CrossRef]
- Guo, B.B.; Bellingham, S.A.; Hill, A.F. Stimulating the release of exosomes increases the intercellular transfer of prions. J. Biol. Chem. 2016, 291, 5128–5137. [Google Scholar] [CrossRef]
- Vella, L.; Sharples, R.; Lawson, V.; Masters, C.; Cappai, R.; Hill, A. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2007, 211, 582–590. [Google Scholar] [CrossRef]
- Bellingham, S.A.; Coleman, B.M.; Hill, A.F. Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res. 2012, 40, 10937–10949. [Google Scholar] [CrossRef]
- Arellano-Anaya, Z.E.; Huor, A.; Leblanc, P.; Lehmann, S.; Provansal, M.; Raposo, G.; Andréoletti, O.; Vilette, D. Prion strains are differentially released through the exosomal pathway. Cell. Mol. Life Sci. 2015, 72, 1185–1196. [Google Scholar] [CrossRef]
- Apolinário, T.; Paiva, C.; Agostinho, L. Intermediate alleles of Huntington’s disease HTT gene in different populations worldwide: A systematic review. Genet. Mol. Res. 2017, 16, gmr16029648. [Google Scholar] [CrossRef]
- Wyant, K.J.; Ridder, A.J.; Dayalu, P. Huntington’s disease—Update on treatments. Curr. Neurol. Neurosci. Rep. 2017, 17, 33. [Google Scholar] [CrossRef]
- Zhang, X.; Abels, E.R.; Redzic, J.S.; Margulis, J.; Finkbeiner, S.; Breakefield, X.O. Potential transfer of polyglutamine and CAG-repeat RNA in extracellular vesicles in Huntington’s disease: Background and evaluation in cell culture. Cell. Mol. Neurobiol. 2016, 36, 459–470. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Breydo, L.; Redington, J.; Uversky, V. Effects of intrinsic and extrinsic factors on aggregation of physiologically important intrinsically disordered proteins. Int. Rev. Cell Mol. Biol. 2017, 329, 145–185. [Google Scholar]
- Wacker, J.L.; Huang, S.-Y.; Steele, A.D.; Aron, R.; Lotz, G.P.; Nguyen, Q.; Giorgini, F.; Roberson, E.D.; Lindquist, S.; Masliah, E.; et al. Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s disease. J. Neurosci. 2009, 29, 9104–9114. [Google Scholar] [CrossRef]
- Warrick, J.M.; Chan, H.; Gray-Board, G.L.; Chai, Y.; Paulson, H.L.; Bonini, N.M. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 1999, 23, 425–428. [Google Scholar] [CrossRef]
- Wacker, J.L.; Zareie, M.H.; Fong, H.; Sarikaya, M.; Muchowski, P.J. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 2004, 11, 1215–1222. [Google Scholar] [CrossRef]
- Carmichael, J.; Chatellier, J.; Woolfson, A.; Milstein, C.; Fersht, A.R.; Rubinsztein, D.C. Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 9701–9705. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Galaz-Montoya, J.G.; Schmid, M.F.; Cong, Y.; Ma, B.; Spiess, C.; Frydman, J.; Ludtke, S.J.; Chiu, W. TRiC’s tricks inhibit huntingtin aggregation. Elife 2013, 2, e00710. [Google Scholar] [CrossRef]
- Yang, W.; Dunlap, J.R.; Andrews, R.B.; Wetzel, R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 2002, 11, 2905–2917. [Google Scholar] [CrossRef]
- Sontag, E.M.; Joachimiak, L.A.; Tan, Z.; Tomlinson, A.; Housman, D.E.; Glabe, C.G.; Potkin, S.G.; Frydman, J.; Thompson, L.M. Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates mutant Huntingtin cellular phenotypes. Proc. Natl. Acad. Sci. USA 2013, 110, 3077–3082. [Google Scholar] [CrossRef]
- Sergeeva, O.A.; Chen, B.; Haase-Pettingell, C.; Ludtke, S.J.; Chiu, W.; King, J.A. Human CCT4 and CCT5 chaperonin subunits expressed in Escherichia coli form biologically active homo-oligomers. J. Biol. Chem. 2013, 288, 17734–17744. [Google Scholar] [CrossRef]
- Nardai, G.; Csermely, P.; Söti, C. Chaperone function and chaperone overload in the aged. A preliminary analysis. Exp. Gerontol. 2002, 37, 1257–1262. [Google Scholar] [CrossRef]
- Barna, J.; Csermely, P.; Vellai, T. Roles of heat shock factor 1 beyond the heat shock response. Cell. Mol. Life Sci. 2018, 75, 2897–2916. [Google Scholar] [CrossRef]
- Didiot, M.-C.; Hall, L.M.; Coles, A.H.; Haraszti, R.A.; Godinho, B.M.; Chase, K.; Sapp, E.; Ly, S.; Alterman, J.F.; Hassler, M.R.; et al. Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol. Ther. 2016, 24, 1836–1847. [Google Scholar] [CrossRef]
- Lee, M.; Liu, T.; Im, W.; Kim, M. Exosomes from adipose-derived stem cells ameliorate phenotype of Huntington’s disease in vitro model. Eur. J. Neurosci. 2016, 44, 2114–2119. [Google Scholar] [CrossRef]
- Labbadia, J.; Novoselov, S.S.; Bett, J.S.; Weiss, A.; Paganetti, P.; Bates, G.P.; Cheetham, M.E. Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain 2012, 135, 1180–1196. [Google Scholar] [CrossRef]
- Lee, S.-T.; Im, W.; Ban, J.-J.; Lee, M.; Jung, K.-H.; Lee, S.K.; Chu, K.; Kim, M. Exosome-based delivery of miR-124 in a Huntington’s disease model. J. Mov. Disord. 2017, 10, 45. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Van den Broek, B.; Wuyts, C.; Irobi, J. Extracellular vesicle-associated small heat shock proteins as therapeutic agents in neurodegenerative diseases and beyond. Adv. Drug Deliv. Rev. 2021, 179, 114009. [Google Scholar] [CrossRef]
- Kakkar, V.; Månsson, C.; de Mattos, E.P.; Bergink, S.; van der Zwaag, M.; van Waarde, M.A.; Klusterhuis, N.J.; Melki, R.; van Cruchten, R.T.P.; Al-Karadaghi, S.; et al. The S/T-rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol. Cell 2016, 62, 272–283. [Google Scholar] [CrossRef]
- Bason, M.; Meister-Broekema, M.; Alberts, N.; Dijkers, P.; Bergink, S.; Sibon, O.C.; Kampinga, H.H. Astrocytic expression of the chaperone DNAJB6 results in non-cell autonomous protection in Huntington’s disease. Neurobiol. Dis. 2019, 124, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wang, Y.; Huang, Y.; Zhang, H.; Lu, H.; Zheng, J.C. Exosomal miRNAs in central nervous system diseases: Biomarkers, pathological mediators, protective factors and therapeutic agents. Prog. Neurobiol. 2019, 183, 101694. [Google Scholar] [CrossRef]
- Sterzenbach, U.; Putz, U.; Low, L.-H.; Silke, J.; Tan, S.-S.; Howitt, J. Engineered exosomes as vehicles for biologically active proteins. Mol. Ther. 2017, 25, 1269–1278. [Google Scholar] [CrossRef]
- Bruno, S.; Grange, C.; Deregibus, M.C.; Calogero, R.A.; Saviozzi, S.; Collino, F.; Morando, L.; Busca, A.; Falda, M.; Bussolati, B.; et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J. Am. Soc. Nephrol. 2009, 20, 1053–1067. [Google Scholar] [CrossRef]
- Andaloussi, S.E.; Mäger, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Kucia, M.; Jadczyk, T.; Greco, N.J.; Wojakowski, W.; Tendera, M.; Ratajczak, J. Pivotal role of paracrine effects in stem cell therapies in regenerative medicine: Can we translate stem cell-secreted paracrine factors and microvesicles into better therapeutic strategies? Leukemia 2012, 26, 1166–1173. [Google Scholar] [CrossRef]
- Yeo, R.W.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv. Drug. Deliv. Rev. 2013, 65, 336–341. [Google Scholar] [CrossRef]
- Takeuchi, T.; Suzuki, M.; Fujikake, N.; Popiel, H.A.; Kikuchi, H.; Futaki, S.; Wada, K.; Nagai, Y. Intercellular chaperone transmission via exosomes contributes to maintenance of protein homeostasis at the organismal level. Proc. Natl. Acad. Sci. USA 2015, 112, E2497–E2506. [Google Scholar] [CrossRef]
- Joshi, B. Extracellular Vesicles for Intracellular Drug Delivery; University of Groningen: Groningen, The Netherlands, 2021. [Google Scholar]
- Tran, P.H.; Xiang, D.; Tran, T.T.; Yin, W.; Zhang, Y.; Kong, L.; Chen, K.; Sun, M.; Li, Y.; Hou, Y.; et al. Exosomes and nanoengineering: A match made for precision therapeutics. Adv. Mater. 2020, 32, 1904040. [Google Scholar] [CrossRef]
- Rayner, K.; Sun, J.; Chen, Y.-X.; McNulty, M.; Simard, T.; Zhao, X.; Wells, D.J.; de Belleroche, J.; O’Brien, E.R. Heat shock protein 27 protects against atherogenesis via an estrogen-dependent mechanism: Role of selective estrogen receptor beta modulation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1751–1756. [Google Scholar] [CrossRef]
- De Maio, A.; Vazquez, D. Extracellular heat shock proteins: A new location, a new function. Shock Augusta GA 2013, 40, 239. [Google Scholar] [CrossRef] [PubMed]
- Binder, R.; Vatner, R.; Srivastava, P. The heat-shock protein receptors: Some answers and more questions. Tissue Antigens 2004, 64, 442–451. [Google Scholar] [CrossRef]
- Salari, S.; Seibert, T.; Chen, Y.-X.; Hu, T.; Shi, C.; Zhao, X.; Cuerrier, C.M.; Raizman, J.E.; O’Brien, E.R. Extracellular HSP27 acts as a signaling molecule to activate NF-κB in macrophages. Cell Stress Chaperones 2013, 18, 53–63. [Google Scholar] [CrossRef]
- Nafar, F.; Williams, J.B.; Mearow, K.M. Astrocytes release HspB1 in response to amyloid-β exposure in vitro. J. Alzheimer’s Dis. 2016, 49, 251–263. [Google Scholar] [CrossRef]
- Cox, D.; Whiten, D.R.; Brown, J.W.; Horrocks, M.H.; San Gil, R.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef]
- Meriin, A.; Sherman, M. Role of molecular chaperones in neurodegenerative disorders. Int. J. Hyperth. 2005, 21, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lechman, E.R.; Bianco, N.; Menon, R.; Keravala, A.; Nash, J.; Mi, Z.; Watkins, S.C.; Gambotto, A.; Robbins, P.D. Exosomes derived from IL-10-treated dendritic cells can suppress inflammation and collagen-induced arthritis. J. Immunol. 2005, 174, 6440–6448. [Google Scholar] [CrossRef] [PubMed]
- Elman-Shina, K.; Efrati, S. Ischemia as a common trigger for Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1012779. [Google Scholar] [CrossRef]
- Yang, J.; Cao, L.-L.; Wang, X.-P.; Guo, W.; Guo, R.-B.; Sun, Y.-Q.; Xue, T.-F.; Cai, Z.-Y.; Ji, J.; Cheng, H.; et al. Neuronal extracellular vesicle derived miR-98 prevents salvageable neurons from microglial phagocytosis in acute ischemic stroke. Cell Death Dis. 2021, 12, 23. [Google Scholar] [CrossRef]
- Li, Y.; Tang, Y.; Yang, G.-Y. Therapeutic application of exosomes in ischaemic stroke. Stroke Vasc. Neurol. 2021, 6, 483–495. [Google Scholar] [CrossRef]
- Xin, H.; Li, Y.; Chopp, M. Exosomes/miRNAs as mediating cell-based therapy of stroke. Front. Cell. Neurosci. 2014, 8, 377. [Google Scholar] [CrossRef]
- Cai, Z.-Y.; Xiao, M.; Quazi, S.H.; Ke, Z.-Y. Exosomes: A novel therapeutic target for Alzheimer’s disease? Neural Regen. Res. 2018, 13, 930. [Google Scholar] [CrossRef]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.-G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Lai, R.C.; Yeo, R.W.; Tan, K.H.; Lim, S.K. Exosomes for drug delivery-a novel application for the mesenchymal stem cell. Biotechnol. Adv. 2013, 31, 543–551. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Lee, Y.; Lakhal-Littleton, S.; Li, J.; Seow, Y.; Gardiner, C.; Alvarez-Erviti, L.; Sargent, I.; Wood, M.J.A. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat. Protoc. 2012, 7, 2112–2126. [Google Scholar] [CrossRef] [PubMed]
- Batagov, A.O.; Kuznetsov, V.A.; Kurochkin, I.V. Identification of nucleotide patterns enriched in secreted RNAs as putative cis-acting elements targeting them to exosome nano-vesicles. BMC Genom. 2011, 12 (Suppl. 3), S18. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 2013, 172, 229–238. [Google Scholar] [CrossRef]
- Chen, T.S.; Arslan, F.; Yin, Y.; Tan, S.S.; Lai, R.C.; Choo, A.B.; Padmanabhan, J.; Lee, C.N.; de Kleijn, D.P.V.; Lim, S.K. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J. Transl. Med. 2011, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhu, J.; Ma, Q.; Zhao, Y.; Wang, Y.; Hu, X.; Chen, J.; Zhu, W.; Han, Z.; Yu, H. Exosomes derived from human umbilical cord MSCs rejuvenate aged MSCs and enhance their functions for myocardial repair. Stem Cell Res. Ther. 2020, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Zhuang, X.; Wang, Q.; Jiang, H.; Deng, Z.B.; Wang, B.; Zhang, L.; Kakar, S.; Jun, Y.; Miller, D.; et al. Interspecies communication between plant and mouse gut host cells through edible plant derived exosome-like nanoparticles. Mol. Nutr. Food Res. 2014, 58, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Narbute, K.; Piļipenko, V.; Pupure, J.; Dzirkale, Z.; Jonavičė, U.; Tunaitis, V.; Kriaučiūnaitė, K.; Jarmalavičiūtė, A.; Jansone, B.; Kluša, V.; et al. Intranasal administration of extracellular vesicles derived from human teeth stem cells improves motor symptoms and normalizes tyrosine hydroxylase expression in the substantia nigra and striatum of the 6-hydroxydopamine-treated rats. Stem Cells Transl. Med. 2019, 8, 490–499. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Weight (KDa) | Classical Family |
|---|---|
| ≥200 | Sacsin |
| 100–199 | Hsp100–110 |
| 81–99 | Hsp90 |
| 65–80 | Hsp70/DnaK |
| 55–64 | Hsp60 (chaperonins Group I and II, e.g., Cpn60 and CCT) |
| 35–54 | Hsp40/DnaJ |
| ≤34 | sHsp (crystallins) |
| Exosomes | |
|---|---|
| Biogenesis |
|
| Microvesicles | |
| |
| Exosomes | |
| Secretion |
|
| Microvesicles | |
| |
| Exosomes and Microvesicles | |
| Uptake |
|
| First Author/Year | Target Group | CNS Disorder | Source of EVs | Effective Cargoes | Main Results |
|---|---|---|---|---|---|
| Zhang et al. (2020) [266] | Old people mesenchymal stem cells (OMSCs) | Aging | Umbilical mesenchymal stem cells (UMSCs) | miR-136 | Reduced senescence phenotype of OMSCs |
| Mu et al. (2014) [267] | Mice | Aging | Plants (ginger, grapefruit and carrot) | Proteins, lipids, microRNAs | Expression of heme oxygenase-1 (HO-1), IL-10 and Nrf2 |
| Narbute et al. (2019) [268] | Rats | Parkinson | Stem cells from the dental pulp of human exfoliated deciduous teeth (SHEDs) | Proteins, lipids and RNAs | Motor function improvement and normalization of tyrosine hydroxylase in striatum and substantia nigra |
| Haney et al. (2015) [193] | Mice | Parkinson | Raw 264.7 macrophages | Loaded catalase | Decreased oxidative stress and increase neuronal survival |
| Yuyama et al. (2014) [158] | Mice | Alzheimer | Neuroblastoma, Neuro2a (N2a) derived exsosomes | Abundant glycosphingolipids (GSLs) | Carrying Aβ on the exosome surface GSLs to deliver it to microglia |
| Alvarez-Erviti et al. (2011) [79] | Mice | Alzheimer | Dendritic cells (DC) | siRNA | BACE 1 gene knockdown in brain |
| Zhuang et al. (2011) [241] | Mice | LPS-induced brain inflammation | Tumor cell lines such as 4T1, CT26 | Exosomes loaded with curcumin and JSI-124 | Induction of apoptosis in microglia and inhibit brain inflammation |
| Takeuchi et al. (2015) [244] | Neuro2a cultured cells and eyes of Drosophila | Aggregated polyglutamine | Neuro2a cells | Hsp40, 70-rich exosomes | Elevated chaperones, Hsp40 and Hsp70, improves proteostasis both in cultured cells and in Drosophila |
| Bason et al. (2019) [236] | Mice | Huntington | Human DNAJB6 | DNAJB6-rich EVs | Suppression of PolyQ aggregation and related neurode- generation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noori, L.; Filip, K.; Nazmara, Z.; Mahakizadeh, S.; Hassanzadeh, G.; Caruso Bavisotto, C.; Bucchieri, F.; Marino Gammazza, A.; Cappello, F.; Wnuk, M.; et al. Contribution of Extracellular Vesicles and Molecular Chaperones in Age-Related Neurodegenerative Disorders of the CNS. Int. J. Mol. Sci. 2023, 24, 927. https://doi.org/10.3390/ijms24020927
Noori L, Filip K, Nazmara Z, Mahakizadeh S, Hassanzadeh G, Caruso Bavisotto C, Bucchieri F, Marino Gammazza A, Cappello F, Wnuk M, et al. Contribution of Extracellular Vesicles and Molecular Chaperones in Age-Related Neurodegenerative Disorders of the CNS. International Journal of Molecular Sciences. 2023; 24(2):927. https://doi.org/10.3390/ijms24020927
Chicago/Turabian StyleNoori, Leila, Kamila Filip, Zohreh Nazmara, Simin Mahakizadeh, Gholamreza Hassanzadeh, Celeste Caruso Bavisotto, Fabio Bucchieri, Antonella Marino Gammazza, Francesco Cappello, Maciej Wnuk, and et al. 2023. "Contribution of Extracellular Vesicles and Molecular Chaperones in Age-Related Neurodegenerative Disorders of the CNS" International Journal of Molecular Sciences 24, no. 2: 927. https://doi.org/10.3390/ijms24020927
APA StyleNoori, L., Filip, K., Nazmara, Z., Mahakizadeh, S., Hassanzadeh, G., Caruso Bavisotto, C., Bucchieri, F., Marino Gammazza, A., Cappello, F., Wnuk, M., & Scalia, F. (2023). Contribution of Extracellular Vesicles and Molecular Chaperones in Age-Related Neurodegenerative Disorders of the CNS. International Journal of Molecular Sciences, 24(2), 927. https://doi.org/10.3390/ijms24020927