
The uPA/uPAR System Orchestrates the Inflammatory Response, Vascular Homeostasis, and Immune System in Fibrosis Progression


Abstract
1. Introduction
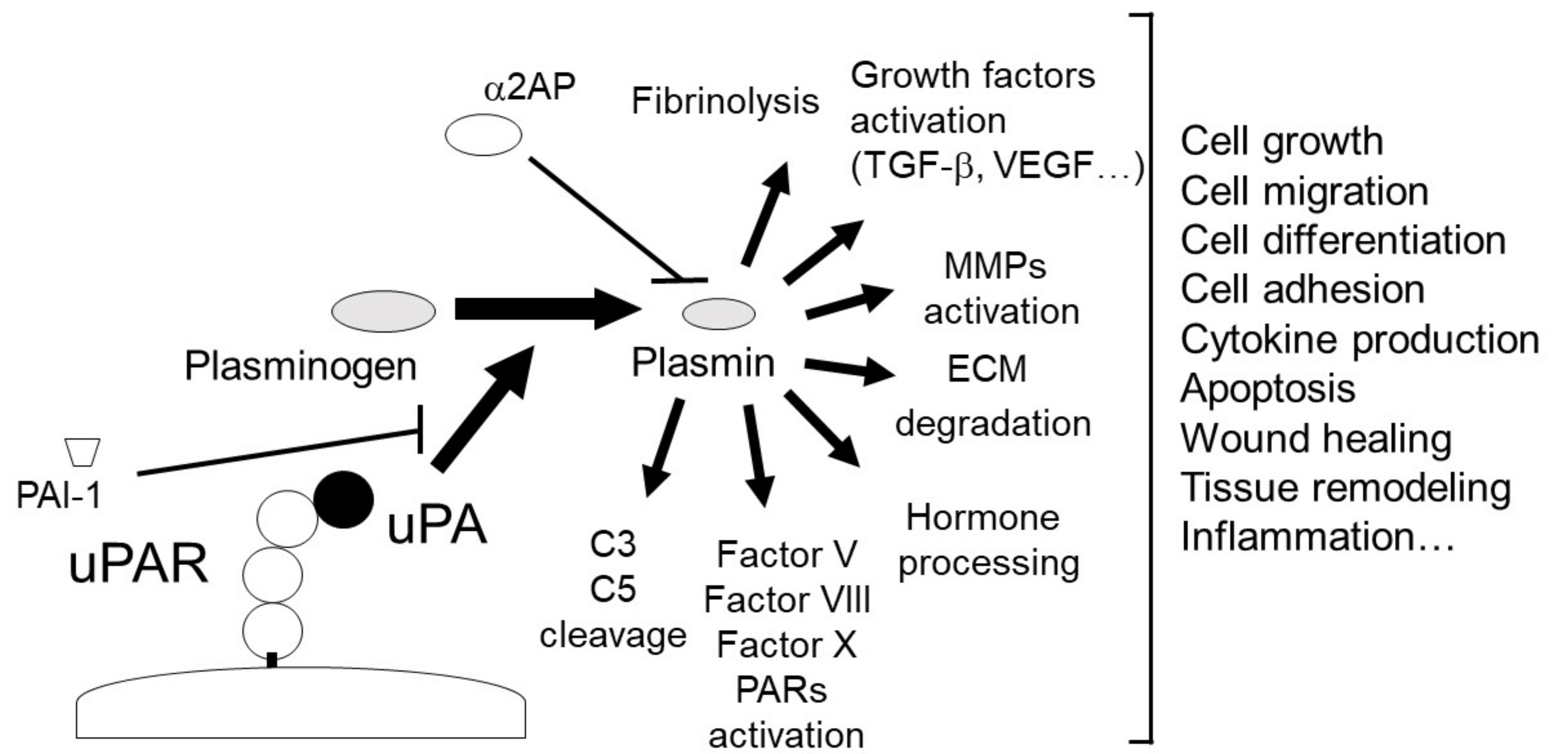
2. The uPA and uPAR System
2.1. uPA
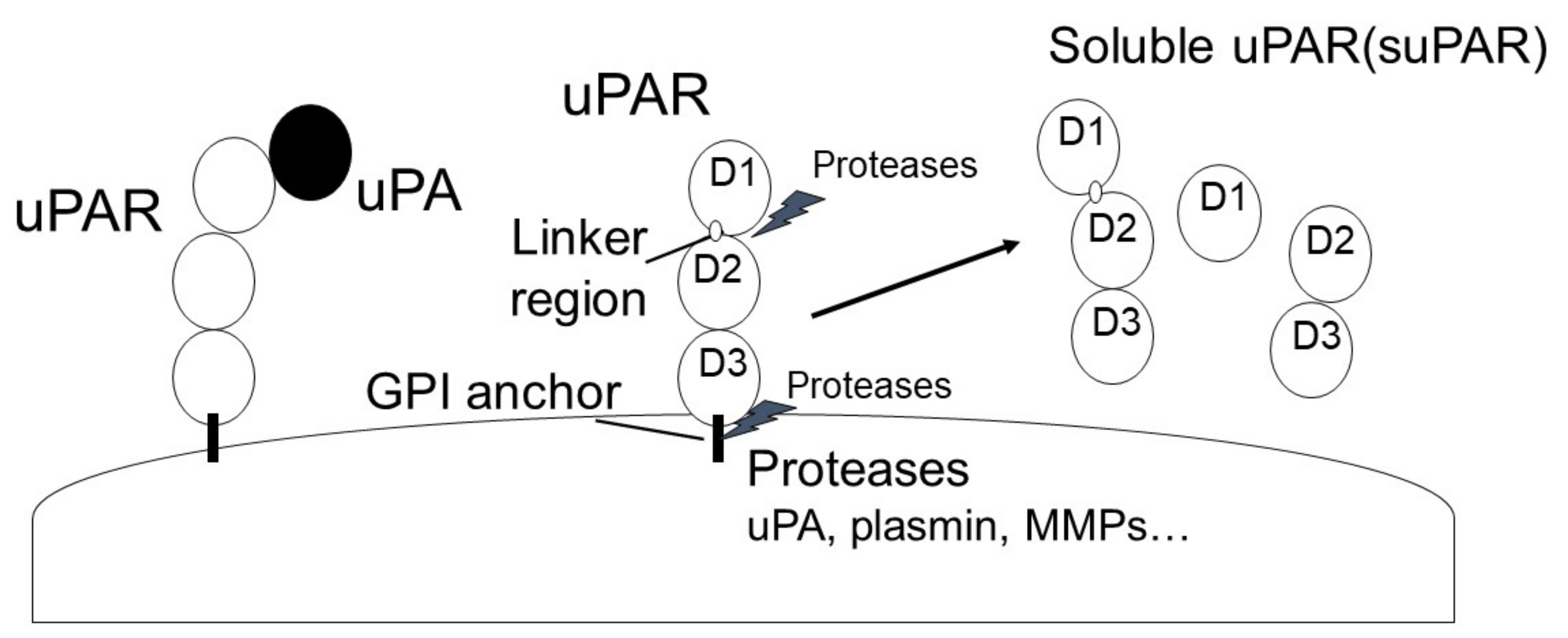
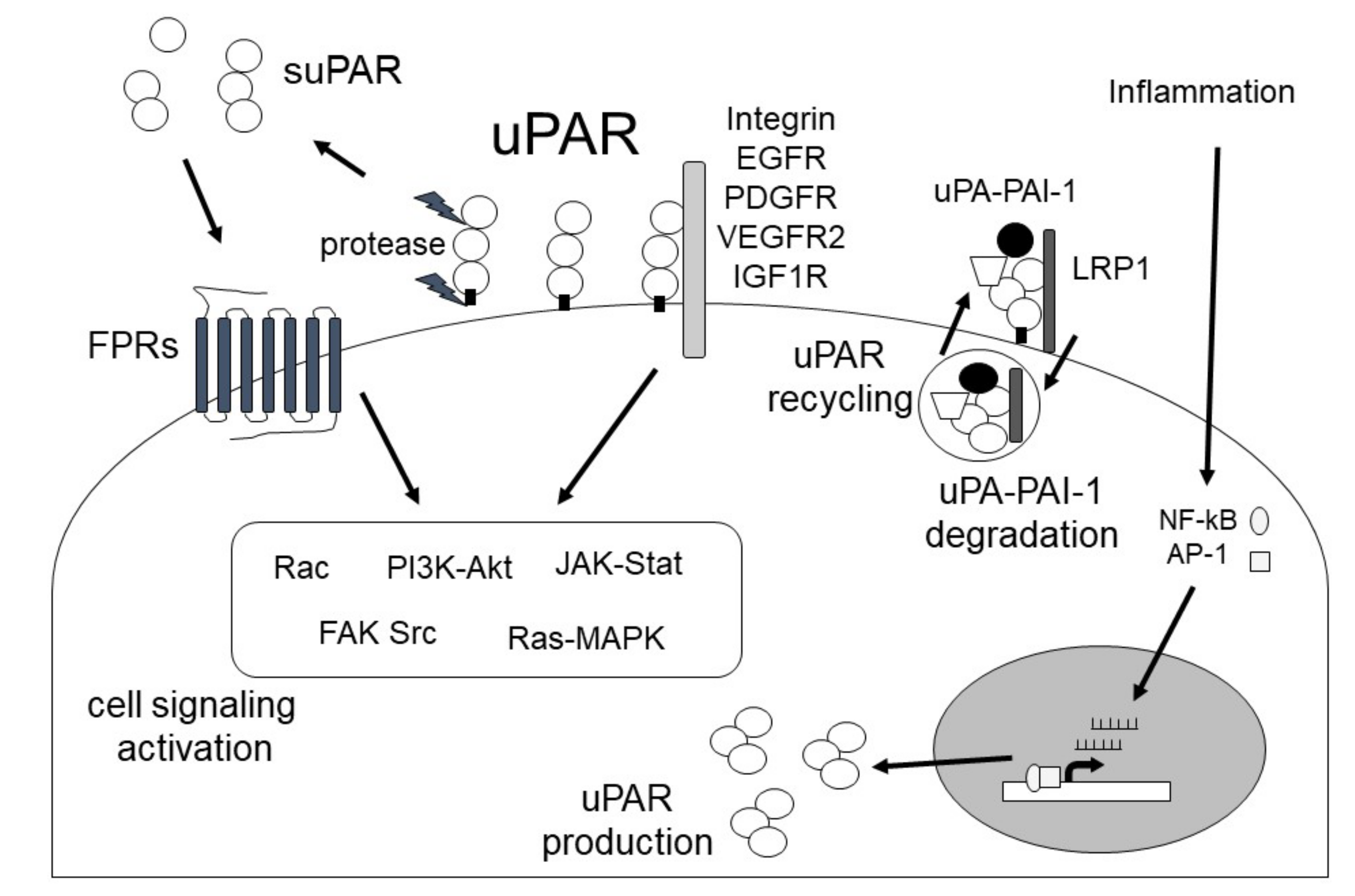
2.2. uPAR
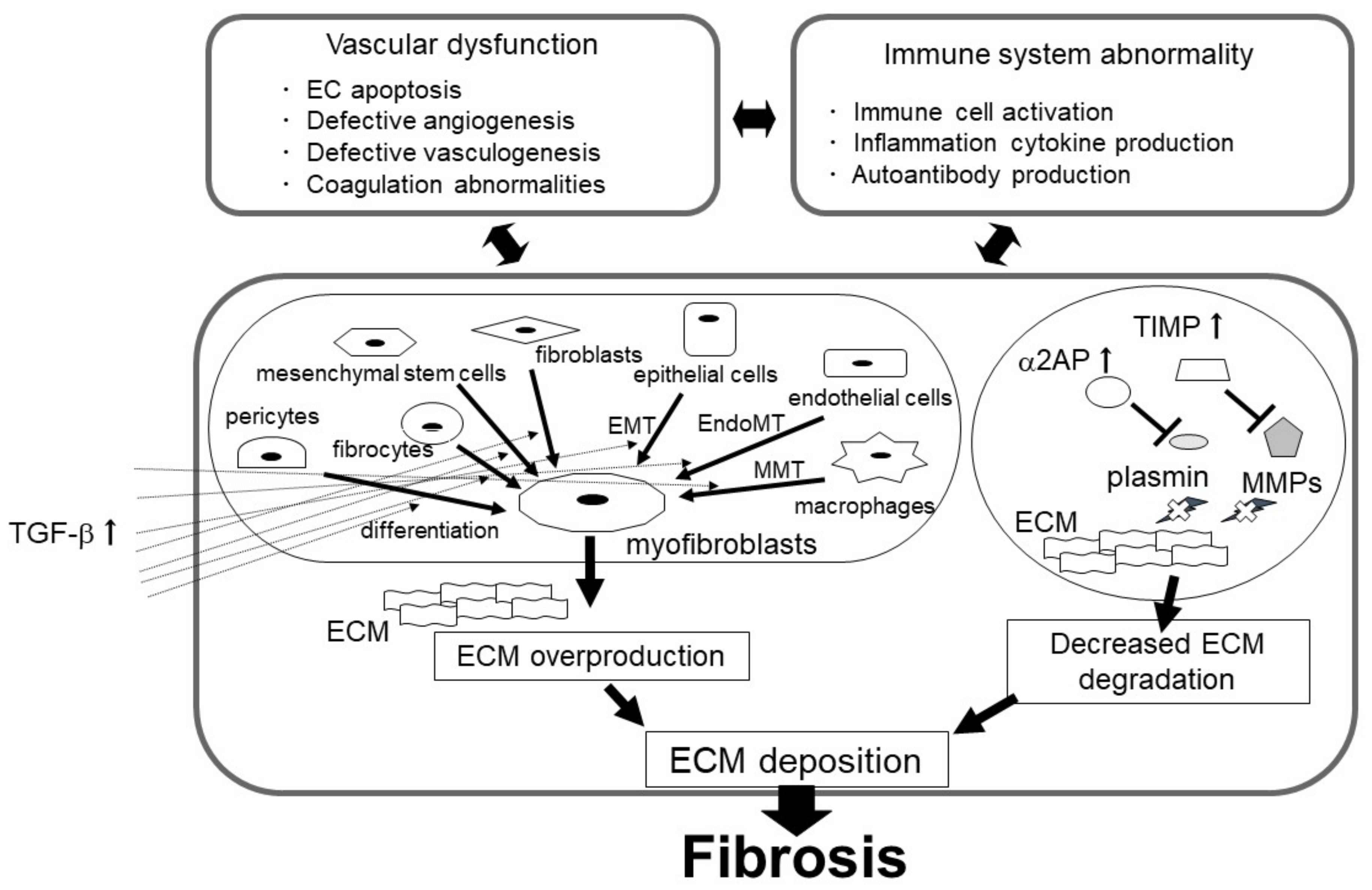
3. The Role of the uPA/uPAR System in Fibrosis
3.1. Myofibroblasts in Fibrosis
3.1.1. The uPA/uPAR System and Myofibroblasts
3.1.2. uPAR-Binding Protein and Myofibroblasts
3.1.3. Other Fibrinolytic Factors and Myofibroblasts
3.2. Suppression of ECM Depredating Protease in Fibrosis
4. Vascular Endothelial Dysfunction in Fibrosis
4.1. The Role of the uPA/uPAR System in EC Functions
4.2. The Role of the uPA/uPAR System in Angiogenesis
4.3. The Role of the uPA/uPAR System in Coagulation
4.4. The Role of the uPA/uPAR System in Vascular Tone Alteration and Hypertension
5. Immune Abnormalities and Inflammation in Fibrosis
6. The Role of the uPA/uPAR System in Inflammation and the Immune System
7. Conclusions and Therapeutic Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| α2AP | α2-antiplasmin |
| AMPK | AMP-activated protein kinase |
| ATF | amino-terminal fragment |
| CKD | chronic kidney disease |
| CSE | cigarette smoke extract |
| CTGF | connective tissue growth factor |
| DC | dendritic cell |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial-to-mesenchymal transition |
| EndoMT | endothelial-to-mesenchymal transition |
| eNOS | endothelial NOS |
| ET-1 | endothelin-1 |
| ERK | extracellular-signal-regulated kinase |
| FRP | formyl peptide receptor |
| FSGS | focal segmental glomerulosclerosis |
| GSK-3β | glycogen synthase kinase-3β |
| HGF | hepatocyte growth factor |
| HMGB1 | high mobility group box 1 |
| IGF1R | insulin-like growth factor 1 receptor |
| IFN | interferon |
| iNOS | inducible NOS |
| JAK | janus kinase |
| JNK | c-Jun N-terminal kinase |
| LRP | low-density lipoprotein receptor |
| MMT | macrophage-to-myofibroblast transition |
| MMP | matrix metalloproteinase |
| MSC | mesenchymal stem cells |
| NO | nitric oxide |
| NOS | NO synthase |
| PAH | pulmonary arterial hypertension |
| PAI-1 | plasminogen activator inhibitor-1 |
| PAR | protease-activated receptor |
| PDGF | platelet-derived growth factor |
| PDGFR | platelet-derived growth factor receptors |
| PI3K | phosphatidylinositol 3-kinase |
| Plg | plasminogen |
| PGE2 | prostaglandin E2 |
| PGI2 | prostacyclin |
| SLE | systemic lupus erythematosus |
| SSc | systemic sclerosis |
| STAT | signal transducer and activator of transcription protein |
| suPAR | soluble uPAR |
| TGF-β | transforming growth factor-β |
| TIMPs | tissue inhibitors of MMPs |
| TNF-α | tumor necrosis factor-α |
| uPA | urokinase plasminogen activator |
| uPAR | urokinase plasminogen activator receptor |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor |
References
- Wynn, T.; Ramalingam, T. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, X.; Yu, X.; Lan, H.-Y. Smad3 Signatures in Renal Inflammation and Fibrosis. Int. J. Biol. Sci. 2022, 18, 2795–2806. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Zapater, E.; Signes-Costa, J.; Montero, P.; Roger, I. Lung Fibrosis and Fibrosis in the Lungs: Is It All about Myofibroblasts? Biomedicines 2022, 10, 1423. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Shu, E. α2-Antiplasmin as a Potential Therapeutic Target for Systemic Sclerosis. Life 2022, 12, 396. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Xu, Z.; Yan, X. The role of the macrophage-to-myofibroblast transition in renal fibrosis. Front. Immunol. 2022, 13, 934377. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Salton, F.; Ruaro, B.; Confalonieri, P.; Confalonieri, M. Epithelial-Mesenchymal Transition: A Major Pathogenic Driver in Idio-pathic Pulmonary Fibrosis? Medicina 2020, 56, 608. [Google Scholar] [CrossRef]
- Confalonieri, P.; Volpe, M.C.; Jacob, J.; Maiocchi, S.; Salton, F.; Ruaro, B.; Confalonieri, M.; Braga, L. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells 2022, 11, 2095. [Google Scholar] [CrossRef]
- Egea-Zorrilla, A.; Vera, L.; Saez, B.; Pardo-Saganta, A. Promises and Challenges of Cell-Based Therapies to Promote Lung Re-generation in Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 2595. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, J.; Sun, H.; Zhang, Y.; Zou, D. New insights into fibrosis from the ECM degradation perspective: The macro-phage-MMP-ECM interaction. Cell Biosci. 2022, 12, 117. [Google Scholar] [CrossRef]
- Kanno, Y. The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis. Int. J. Mol. Sci. 2019, 20, 619. [Google Scholar] [CrossRef]
- Chuliá-Peris, L.; Carreres-Rey, C.; Gabasa, M.; Alcaraz, J.; Carretero, J.; Pereda, J. Matrix Metalloproteinases and Their Inhibitors in Pulmonary Fibrosis: EMMPRIN/CD147 Comes into Play. Int. J. Mol. Sci. 2022, 23, 6894. [Google Scholar] [CrossRef]
- Kanno, Y.; Kaneiwa, A.; Minamida, M.; Kanno, M.; Tomogane, K.; Takeuchi, K.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. The Absence of uPAR Is Associated with the Progression of Dermal Fibrosis. J. Investig. Dermatol. 2008, 128, 2792–2797. [Google Scholar] [CrossRef]
- Waszczykowska, A.; Podgórski, M.; Waszczykowski, M.; Gerlicz-Kowalczuk, Z.; Jurowski, P. Matrix Metalloproteinases MMP-2 and MMP-9, Their Inhibitors TIMP-1 and TIMP-2, Vascular Endothelial Growth Factor and sVEGFR-2 as Predictive Markers of Ischemic Retinopathy in Patients with Systemic Sclerosis-Case Series Report. Int. J. Mol. Sci. 2020, 21, 8703. [Google Scholar] [CrossRef]
- Serratì, S.C.M.; Margheri, F.; Guiducci, S.; Del Rosso, A.; Pucci, M.; Fibbi, G.; Bazzichi, L.; Bombardieri, S.; Matucci-Cerinic, M.; Del Rosso, M. Systemic sclerosis fibroblasts inhibit in vitro angiogenesis by MMP-12-dependent cleavage of the endothelial cell uro-kinase receptor. J. Pathol. 2006, 210, 240–248. [Google Scholar] [CrossRef]
- Placido, L.; Romero, Y.; Maldonado, M.; Toscano-Marquez, F.; Ramírez, R.; Calyeca, J.; Mora, A.L.; Selman, M.; Pardo, A. Loss of MT1-MMP in Alveolar Epithelial Cells Exacerbates Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 2923. [Google Scholar] [CrossRef]
- Kalafatis, D.; Löfdahl, A.; Näsman, P.; Dellgren, G.; Wheelock, M.; Rendin, L.E.; Sköld, M.; Westergren-Thorsson, G. Distal Lung Microenvironment Triggers Release of Mediators Recognized as Potential Systemic Biomarkers for Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 13421. [Google Scholar] [CrossRef]
- Menou, A.; Duitman, J.; Crestani, B. The impaired proteases and anti-proteases balance in Idiopathic Pulmonary Fibrosis. Matrix Biol. 2018, 68-69, 382–403. [Google Scholar] [CrossRef]
- Niwa, H.; Kanno, Y.; Shu, E.; Seishima, M. Decrease in matrix metalloproteinase-3 activity in systemic sclerosis fibroblasts causes α2-antiplasmin and extracellular matrix deposition, and contributes to fibrosis development. Mol. Med. Rep. 2020, 22, 3001–3007. [Google Scholar] [CrossRef]
- Kanno, Y.; Kawashita, E.; Minamida, M.; Kaneiwa, A.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. Alpha2-antiplasmin is associated with the pro-gression of fibrosis. Am. J. Pathol. 2010, 176, 238–245. [Google Scholar] [CrossRef]
- Kanno, Y.; Kawashita, E.; Kokado, A.; Kuretake, H.; Ikeda, K.; Okada, K.; Seishima, M.; Ueshima, S.; Matsuo, O.; Matsuno, H. α2AP mediated myofibroblast formation and the development of renal fibrosis in unilateral ureteral obstruction. Sci. Rep. 2014, 4, srep05967. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Shu, E.; Kanoh, H.; Seishima, M. The Antifibrotic Effect of α2AP Neutralization in Systemic Sclerosis Dermal Fibroblasts and Mouse Models of Systemic Sclerosis. J. Investig. Dermatol. 2015, 136, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Miyashita, M.; Seishima, M.; Matsuo, O. α2AP is associated with the development of lupus nephritis through the regulation of plasmin inhibition and inflammatory responses. Immun. Inflamm. Dis. 2020, 8, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Kavian, N.; Batteux, F. Macro- and microvascular disease in systemic sclerosis. Vasc. Pharmacol. 2015, 71, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Braaten, J.V.; Handt, S.; Jerome, W.G.; Kirkpatrick, J.; Lewis, J.C.; Hantgan, R.R. Regulation of fibrinolysis by platelet-released plasminogen activator inhibitor 1: Light scattering and ultrastructural examination of lysis of a model platelet-fibrin thrombus. Blood 1993, 81, 1290-1299. [Google Scholar] [CrossRef]
- Kanno, Y.; Ishisaki, A.; Kawashita, E.; Chosa, N.; Nakajima, K.; Nishihara, T.; Toyoshima, K.; Okada, K.; Ueshima, S.; Matsushita, K. Plasminogen/Plasmin Modulates Bone Metabolism by Regulating the Osteoblast and Osteoclast Function. J. Biol. Chem. 2011, 286, 8952–8960. [Google Scholar] [CrossRef]
- Ismail, A.; Shaker, B.; Bajou, K. The Plasminogen-Activator Plasmin System in Physiological and Pathophysiological Angio-genesis. Int. J. Mol. Sci. 2021, 23, 337. [Google Scholar] [CrossRef]
- Draxler, D.F.; Sashindranath, M.; Medcalf, R.L. Plasmin: A modulator of immune function. Semin. Thromb. Hemost. 2017, 43, 143–153. [Google Scholar] [CrossRef]
- Kanno, Y.; Sakai, A.; Miyashita, M.; Tsuchida, K.; Matsuo, O. Plasminogen deficiency is associated with improved glucose tolerance, and lower DPP-4 activity. Diabetes Res. Clin. Pract. 2016, 120, 190–193. [Google Scholar] [CrossRef]
- Law, R.H.; Abu-Ssaydeh, D.; Whisstock, J.C. New insights into the structure and function of the plasminogen/plasmin system. Curr. Opin. Struct. Biol. 2013, 23, 836–841. [Google Scholar] [CrossRef]
- Yang, D.; Yang, H.; Luiselli, G.; Ogagan, C.; Dai, H.; Chiu, L.; Carroll, R.S.; Johnson, M.D. Increased plasmin-mediated proteolysis of L1CAM in a mouse model of idiopathic normal pressure hydrocephalus. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef]
- Kanno, Y.; Ishisaki, A.; Kawashita, E.; Kuretake, H.; Ikeda, K.; Matsuo, O. uPA Attenuated LPS-induced Inflammatory Osteoclas-togenesis through the Plasmin/PAR-1/Ca2+/CaMKK/AMPK Axis. Int. J. Biol. Sci. 2016, 12, 63–71. [Google Scholar] [CrossRef]
- Kanno, Y.; Kuroki, A.; Minamida, M.; Kaneiwa, A.; Okada, K.; Tomogane, K.; Takeuchi, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. The absence of uPAR attenuates insulin-induced vascular smooth muscle cell migration and proliferation. Thromb. Res. 2008, 123, 336–341. [Google Scholar] [CrossRef]
- Kanno, Y.; Matsuno, H.; Kawashita, E.; Okada, K.; Suga, H.; Ueshima, S.; Matsuo, O. Urokinase-type plasminogen activator receptor is associated with the development of adipose tissue. Thromb. Haemost. 2010, 104, 1124–1132. [Google Scholar] [CrossRef]
- Del Rosso, M.; Margheri, F.; Serratì, S.; Chillà, A.; Laurenzana, A.; Fibbi, G. The urokinase receptor system, a key regulator at the intersection between inflammation, immunity, and coagulation. Curr. Pharm. Des. 2011, 17, 1924–1943. [Google Scholar] [CrossRef]
- Kanno, Y.; Ishisaki, A.; Miyashita, M.; Matsuo, O. The blocking of uPAR suppresses lipopolysaccharide-induced inflammatory osteoclastogenesis and the resultant bone loss through attenuation of integrin β3/Akt pathway. Immun. Inflamm. Dis. 2016, 4, 338–349. [Google Scholar] [CrossRef]
- Kanno, Y.; Maruyama, C.; Matsuda, A.; Ishisaki, A. uPA-derived peptide, Å6 is involved in the suppression of lipopolysaccar-ide-promoted inflammatory osteoclastogenesis and the resultant bone loss. Immun. Inflamm. Dis. 2017, 5, 289–299. [Google Scholar] [CrossRef]
- Napolitano, F.; Montuori, N. The Role of the Plasminogen Activation System in Angioedema: Novel Insights on the Patho-genesis. J. Clin. Med. 2021, 10, 518. [Google Scholar] [CrossRef]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Navaratna, D.; Menicucci, G.; Maestas, J.; Srinivasan, R.; McGuire, P.; Das, A. A peptide inhibitor of the urokinase/urokinase receptor system inhibits alteration of the blood-retinal barrier in diabetes. FASEB J. 2008, 22, 3310–3317. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.P.; Patecki, M.; Tkachuk, S.; Kiyan, Y.; Haller, H.; Dumler, I. Urokinase receptor mediates osteoclastogenesis via M-CSF release from osteoblasts and the c-Fms/PI3K/Akt/NF-κB pathway in osteoclasts. J. Bone Miner. Res. 2015, 30, 379–388. [Google Scholar]
- Tomogane, K.; Kanno, Y.; Kawashita, E.; Okada, K.; Takeuchi, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. The Absence of Urokinase-type Plasminogen Activator Receptor Plays a Role in the Insulin-independent Glucose Metabolism. J. Cardiovasc. Pharmacol. 2011, 57, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Masucci, M.T.; Minopoli, M.; Di Carluccio, G.; Motti, M.L.; Carriero, M.V. Therapeutic Strategies Targeting Urokinase and Its Receptor in Cancer. Cancers 2022, 14, 498. [Google Scholar] [CrossRef]
- Yepes, M.; Woo, Y.; Martin-Jimenez, C. Plasminogen Activators in Neurovascular and Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 4380. [Google Scholar] [CrossRef]
- Kumar, A.A.; Buckley, B.J.; Ranson, M. The Urokinase Plasminogen Activation System in Pancreatic Cancer: Prospective Diagnostic and Therapeutic Targets. Biomolecules 2022, 12, 152. [Google Scholar] [CrossRef]
- Dinesh, P.; Rasool, M. uPA/uPAR signaling in rheumatoid arthritis: Shedding light on its mechanism of action. Pharmacol. Res. 2018, 134, 31–39. [Google Scholar] [CrossRef]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arter. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef]
- Alfano, D.; Franco, P.; Stoppelli, M.P. Modulation of Cellular Function by the Urokinase Receptor Signalling: A Mechanistic View. Front. Cell Dev. Biol. 2022, 10, 818616. [Google Scholar] [CrossRef]
- Enocsson, H.; Sjöwall, C.; Wetterö, J. Soluble urokinase plasminogen activator receptor--a valuable biomarker in systemic lupus erythematosus? Clin. Chim. Acta 2015, 444, 234–241. [Google Scholar] [CrossRef]
- Czekay, R.-P.; Kuemmel, T.A.; Orlando, R.A.; Farquhar, M.G. Direct Binding of Occupied Urokinase Receptor (uPAR) to LDL Receptor-related Protein Is Required for Endocytosis of uPAR and Regulation of Cell Surface Urokinase Activity. Mol. Biol. Cell 2001, 12, 1467–1479. [Google Scholar] [CrossRef]
- Chavakis, T.; Kanse, S.M.; Yutzy, B.; Lijnen, H.R.; Preissner, K.T. Vitronectin concentrates proteolytic activity on the cell surface and extracellular matrix by trapping soluble urokinase receptor-urokinase complexes. Blood 1998, 91, 2305–2312. [Google Scholar] [CrossRef]
- Selleri, C.; Montuori, N.; Ricci, P.; Visconte, V.; Carriero, M.V.; Sidenius, N.; Serio, B.; Blasi, F.; Rotoli, B.; Rossi, G. Involvement of the urokinase-type plasminogen activator receptor in hematopoietic stem cell mobilization. Blood 2005, 105, 2198–2205. [Google Scholar] [CrossRef]
- Li Santi, A.; Napolitano, F.; Montuori, N.; Ragno, P. The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 22084111. [Google Scholar] [CrossRef]
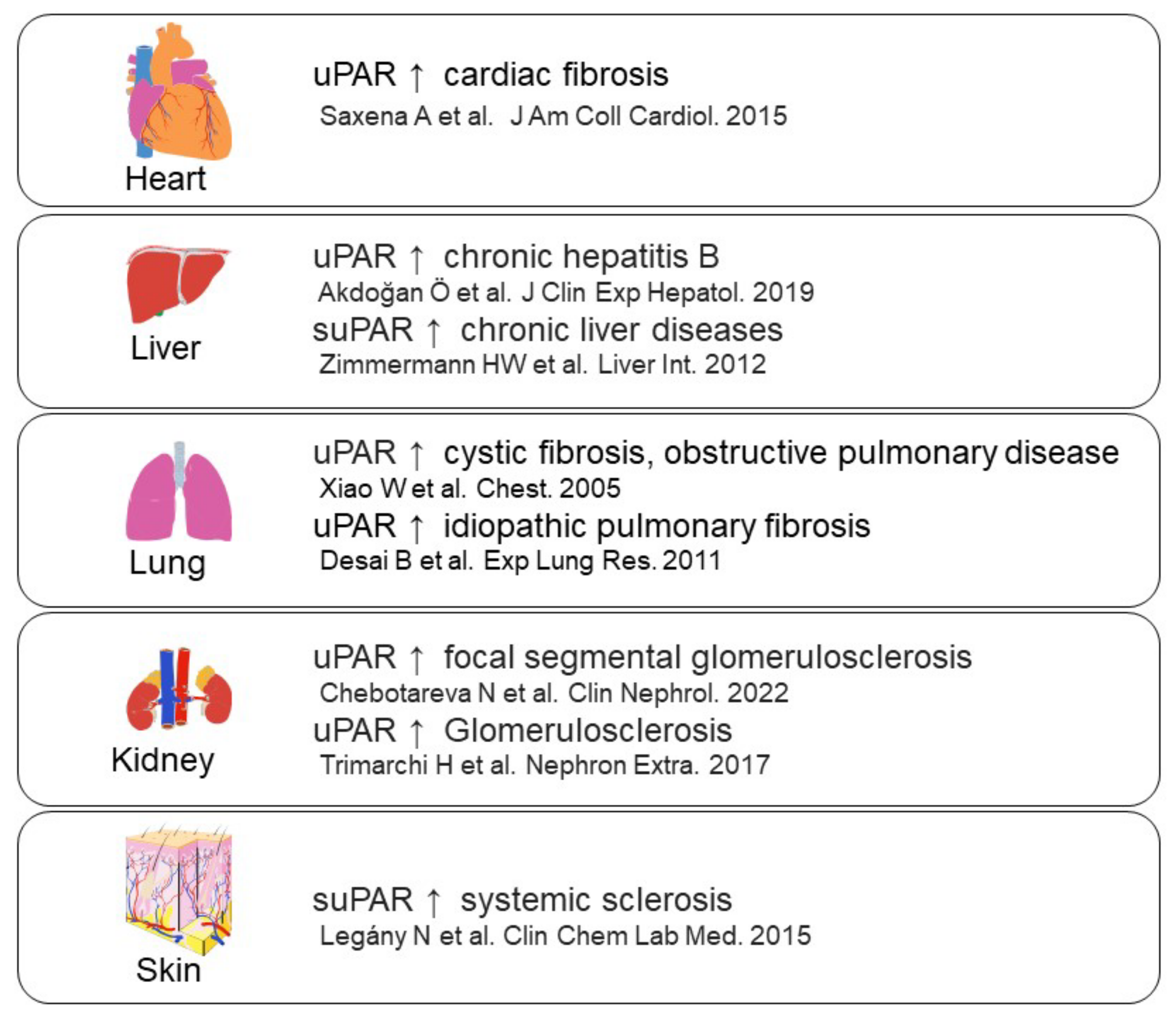
- Saxena, A.; Izmirly, P.M.; Han, S.W.; Briassouli, P.; Rivera, T.L.; Zhong, H.; Friedman, D.M.; Clancy, R.M.; Buyon, J.P. Serum Biomarkers of Inflammation, Fibrosis, and Cardiac Function in Facilitating Diagnosis, Prognosis, and Treatment of Anti-SSA/Ro-Associated Cardiac Neonatal Lupus. J. Am. Coll. Cardiol. 2015, 66, 930–939. [Google Scholar] [CrossRef]
- Akdoğan, J.P.; Yücel, A.A.; Sargin, Z.G.; Sönmez, C.; Yilmaz, G.E.; Özenirler, S. Evaluation of Plasma Urokinase-Type Plasminogen Activator Receptor (UPAR) in Patients with Chronic Hepatitis B, C and Non-Alcoholic Fatty Liver Disease (NAFLD) as Serological Fibrosis Marker. J. Clin. Exp. Hepatol. 2019, 9, 29–33. [Google Scholar] [CrossRef]
- Zimmermann, H.W.; Koch, A.; Seidler, S.; Trautwein, C.; Tacke, F. Circulating soluble urokinase plasminogen activator is elevated in patients with chronic liver disease, discriminates stage and aetiology of cirrhosis and predicts prognosis. Liver Int. 2012, 32, 500–509. [Google Scholar] [CrossRef]
- Xiao, W.; Hsu, Y.-P.; Ishizaka, A.; Kirikae, T.; Moss, R.B. Sputum Cathelicidin, Urokinase Plasminogen Activation System Components, and Cytokines Discriminate Cystic Fibrosis, COPD, and Asthma Inflammation. Chest 2005, 128, 2316–2326. [Google Scholar] [CrossRef]
- Desai, B.; Mattson, J.; Paintal, H.; Nathan, M.; Shen, F.; Beaumont, M.; Malinao, M.C.; Li, Y.; Canfield, J.; Basham, B. Differential expression of monocyte/macrophage-selective markers in human idiopathic pulmonary fibrosis. Exp. Lung Res. 2011, 37, 227–238. [Google Scholar] [CrossRef]
- Chebotareva, N.; Vinogradov, A.; Cao, V.; Gindis, A.; Berns, A.; Alentov, I.; Sergeeva, N. Serum levels of plasminogen activator urokinase receptor and cardiotrophin-like cytokine factor 1 in patients with nephrotic syndrome. Clin. Nephrol. 2022, 97, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, H.; Canzonieri, R.; Schiel, A.; Costales-Collaguazo, C.; Stern, A.; Paulero, M.; Rengel, T.; Andrews, J.; Iotti, A.; Forrester, M. In IgA Nephropathy, Glomerulo-sclerosis Is Associated with Increased Urinary CD80 Excretion and Urokinase-Type Plasminogen Activator Receptor-Positive Podocyturia. Nephron Extra 2017, 7, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Legány, N.; Toldi, G.; Distler, J.H.; Beyer, C.; Szalay, B.; Kovács, L.; Vásárhelyi, B.; Balog, A. Increased plasma soluble urokinase plasminogen activator receptor levels in systemic sclerosis: Possible association with microvascular abnormalities and extent of fibrosis. Clin. Chem. Lab. Med. 2015, 53, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Gilbane, A.J.; Denton, C.P.; Holmes, A.M. Scleroderma pathogenesis: A pivotal role for fibroblasts as effector cells. Thromb. Haemost. 2013, 15, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Hasanzadeh, A.; Rafiei, A.; Kazemi, M.; Beiromvand, M.; Bahreini, A.; Khanahmad, H. The Role of Tissue Inhibitor of Metallopro-teinase-1 and 2 in Echinococcus granulosus senso lato-Induced Human Hepatic Fibrosis. Acta Parasitol. 2022, 67, 851–857. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2019, 16, 11–31. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Tschumperlin, D.J.; Kim, K.K.; Osterholzer, J.J.; Subbotina, N.; Ajayi, I.O.; Teitz-Tennenbaum, S.; Virk, A.; Dotson, M.; Liu, F. Urokinase Plasminogen Activator Overex-pression Reverses Established Lung Fibrosis. Thromb Haemost. 2019, 119, 1968–1980. [Google Scholar]
- Sun, C.; Li, D.G.; Chen, Y.W.; Chen, Y.W.; Wang, B.C.; Sun, Q.L.; Lu, H.M. Transplantation of urokinase-type plasminogen activator gene-modified bone marrow-derived liver stem cells reduces liver fibrosis in rats. J. Gene Med. 2008, 10, 855–866. [Google Scholar] [CrossRef]
- Dergilev, K.; Beloglazova, I.; Tsokolaeva, Z.; Vasilets, Y.; Parfenova, E. Deficiency of Urokinase-Type Plasminogen Activator Receptor Is Associated with the Development of Perivascular Fibrosis in Mouse Heart. Bull. Exp. Biol. Med. 2022, 173, 5–9. [Google Scholar] [CrossRef]
- Manetti, M.; Rosa, I.; Milia, A.F.; Guiducci, S.; Carmeliet, P.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Inactivation of urokinase-type plasminogen activator receptor (uPAR) gene induces dermal and pulmonary fibrosis and peripheral microvasculopathy in mice: A new model of experimental scleroderma? Ann. Rheum. Dis. 2014, 73, 1700–1709. [Google Scholar] [CrossRef]
- Zhang, G.; Kim, H.; Cai, X.; López-Guisa, J.M.; Alpers, C.E.; Liu, Y.; Carmeliet, P.; Eddy, A.A. Urokinase Receptor Deficiency Accelerates Renal Fibrosis in Obstructive Nephropathy. J. Am. Soc. Nephrol. 2003, 14, 1254–1271. [Google Scholar] [CrossRef]
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.K.; Saleem, M. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 2011, 17, 952–960. [Google Scholar] [CrossRef]
- Wei, C.; Möller, C.C.; Altintas, M.M.; Li, J.; Schwarz, K.; Zacchigna, S.; Xie, L.; Henger, A.; Schmid, H.; Rastaldi, M.P. Modification of kidney barrier function by the urokinase receptor. Nat. Med. 2007, 14, 55–63. [Google Scholar] [CrossRef]
- Wei, C.; Li, J.; Adair, B.D.; Zhu, K.; Cai, J.; Merchant, M.; Samelko, B.; Liao, Z.; Koh, K.H.; Tardi, N.J. uPAR isoform 2 forms a dimer and induces severe kidney disease in mice. J. Clin. Investig. 2019, 129, 1946–1959. [Google Scholar] [CrossRef]
- Dal Monte, M.; Cammalleri, M.; Pecci, V.; Carmosino, M.; Procino, G.; Pini, A.; De Rosa, M.; Pavone, V.; Svelto, M.; Bagnoli, P. Inhibiting the urokinase-type plasminogen activator receptor system recovers STZ-induced diabetic nephropathy. J. Cell. Mol. Med. 2019, 23, 1034–1049. [Google Scholar] [CrossRef]
- Schuliga, M.; Grainge, C.; Westall, G.; Knight, D. The fibrogenic actions of the coagulant and plasminogen activation systems in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2018, 97, 108–117. [Google Scholar] [CrossRef]
- Oh, H.; Park, H.E.; Song, M.S.; Kim, H.; Baek, J.-H. The Therapeutic Potential of Anticoagulation in Organ Fibrosis. Front. Med. 2022, 9, 866746. [Google Scholar] [CrossRef]
- Bauman, K.A.; Wettlaufer, S.H.; Okunishi, K.; Vannella, K.M.; Stoolman, J.S.; Huang, S.K.; Courey, A.J.; White, E.S.; Hogaboam, C.M.; Simon, R.H. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Investig. 2010, 120, 1950–1960. [Google Scholar] [CrossRef]
- Kanno, Y.; Hirade, K.; Ishisaki, A.; Nakajima, K.; Suga, H.; Into, T.; Matsushita, K.; Okada, K.; Matsuo, O.; Matsuno, H. Lack of alpha2-antiplasmin improves cutaneous wound healing via over-released vascular endothelial growth factor-induced angiogenesis in wound lesions. J. Thromb. Haemost. 2006, 4, 1602–1610. [Google Scholar] [CrossRef]
- Kanno, Y.; Kuroki, A.; Okada, K.; Tomogane, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. alpha2-Antiplasmin is involved in the production of transforming growth factor beta1 and fibrosis. J. Thromb. Haemost. 2007, 5, 2266–2273. [Google Scholar] [CrossRef]
- Beier, J.I.; Kaiser, J.P.; Guo, L.; Martínez-Maldonado, M.; Arteel, G.E. Plasminogen activator inhibitor-1 deficient mice are protected from angiotensin II-induced fibrosis. Arch. Biochem. Biophys. 2011, 510, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Chuang-Tsai, S.; Sisson, T.H.; Hattori, N.; Tsai, C.G.; Subbotina, N.M.; Hanson, K.E.; Simon, R.H. Reduction in Fibrotic Tissue Formation in Mice Genetically Deficient in Plasminogen Activator Inhibitor. Am. J. Pathol. 2003, 163, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Woods, E.L.; Dally, J.; Kong, D.; Steadman, R.; Moseley, R.; Midgley, A.C. Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis. Biomolecules 2021, 11, 1095. [Google Scholar] [CrossRef] [PubMed]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 8361945. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xia, H.; Yao, S. Regulatory T cells are a double-edged sword in pulmonary fibrosis. Int. Immunopharmacol. 2020, 84, 106443. [Google Scholar] [CrossRef]
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2015, 7, 8809–8822. [Google Scholar] [CrossRef]
- Derada Troletti, C.; Fontijn, R.D.; Gowing, E.; Charabati, M.; van Het Hof, B.; Didouh, I.; van der Pol, S.M.A.; Geerts, D.; Prat, A.; van Horssen, J. Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. 2019, 10, 45. [Google Scholar] [CrossRef]
- Wu, N.; Wang, Y.; Wang, K.; Zhong, B.; Liao, Y.; Liang, J.; Jiang, N. Cathepsin K regulates the tumor growth and metastasis by IL-17/CTSK/EMT axis and mediates M2 macrophage polarization in castration-resistant prostate cancer. Cell Death Dis. 2022, 13, 813. [Google Scholar] [CrossRef]
- Paquissi, F.; Abensur, H. The Th17/IL-17 Axis and Kidney Diseases, With Focus on Lupus Nephritis. Front Med. 2021, 8, 654912. [Google Scholar] [CrossRef]
- Zhang, H.; Phan, S. Inhibition of myofibroblast apoptosis by transforming growth factor beta. Am. J. Respir. Cell Mol. Biol. 1999, 21, 658–665. [Google Scholar] [CrossRef]
- Tang, W.W.; Ulich, T.R.; Lacey, D.L.; Hill, D.C.; Qi, M.; Kaufman, S.A.; Van, G.Y.; Tarpley, J.E.; Yee, J.S. Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am. J. Pathol. 1996, 148, 1169–1180. [Google Scholar]
- Iekushi, K.; Taniyama, Y.; Azuma, J.; Sanada, F.; Kusunoki, H.; Yokoi, T.; Koibuchi, N.; Okayama, K.; Rakugi, H.; Morishita, R. Hepatocyte growth factor attenuates renal fibrosis through TGF-β1 suppression by apoptosis of myofibroblasts. J. Hypertens. 2010, 28, 2454–2461. [Google Scholar] [CrossRef]
- Yoshimine, H.; Tanoue, S.; Ibi, Y.; Minami, M.; Nakahara, M.; Tokunaga, K.; Kanmura, S.; Ido, A. Hepatocyte growth factor ameliorates methyl-glyoxal-induced peritoneal inflammation and fibrosis in mouse model. Clin. Exp. Nephrol. 2021, 25, 935–943. [Google Scholar] [CrossRef]
- Jeffers, A.; Qin, W.; Owens, S.; Koenig, K.B.; Komatsu, S.; Giles, F.J.; Schmitt, D.M.; Idell, S.; Tucker, T.A. Glycogen Synthase Kinase-3β Inhibition with 9-ING-41 At-tenuates the Progression of Pulmonary Fibrosis. Sci. Rep. 2019, 9, 18925. [Google Scholar] [CrossRef]
- Kochtebane, N.; Choqueux, C.; Passefort, S.; Nataf, P.; Messika-Zeitoun, D.; Bartagi, A.; Michel, J.B.; Anglés-Cano, E.; Jacob, M.P. Plasmin induces apoptosis of aortic valvular myofibroblasts. J. Pathol. 2009, 221, 37–48. [Google Scholar] [CrossRef]
- He, Y.; Tsou, P.; Khanna, D.; Sawalha, A. Methyl-CpG-binding protein 2 mediates antifibrotic effects in scleroderma fibroblasts. Ann. Rheum. Dis. 2018, 77, 1208–1218. [Google Scholar] [CrossRef]
- Sugioka, K.; Nishida, T.; Kodama-Takahashi, A.; Murakami, J.; Mano, F.; Okada, K.; Fukuda, M.; Kusaka, S. Urokinase-type plasminogen activator negatively regulates α-smooth muscle actin expression via Endo180 and the uPA receptor in corneal fibroblasts. Am. J. Physiol. Physiol. 2022, 323, C104–C115. [Google Scholar] [CrossRef]
- Wang, L.; Ly, C.M.; Ko, C.-Y.; Meyers, E.E.; Lawrence, D.A.; Bernstein, A.M. uPA Binding to PAI-1 Induces Corneal Myofibroblast Differentiation on Vitronectin. Investig. Opthalmology Vis. Sci. 2012, 53, 4765–4775. [Google Scholar] [CrossRef]
- Vorstandlechner, V.; Laggner, M.; Copic, D.; Klas, K.; Direder, M.; Chen, Y.; Golabi, B.; Haslik, W.; Radtke, C.; Tschachler, E. The serine proteases dipeptidyl-peptidase 4 and urokinase are key molecules in human and mouse scar formation. Nat. Commun. 2021, 12, 6242. [Google Scholar] [CrossRef]
- Bernstein, A.M.; Twining, S.S.; Warejcka, D.J.; Tall, E.; Masur, S.K. Urokinase Receptor Cleavage: A Crucial Step in Fibroblast-to-Myofibroblast Differentiation. Mol. Biol. Cell 2007, 18, 2716–2727. [Google Scholar] [CrossRef]
- Manetti, M.; Romano, E.; Rosa, I.; Guiducci, S.; Bellando-Randone, S.; De Paulis, A.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Semina, E.V.; Rubina, K.A.; Shmakova, A.A.; Rysenkova, K.D.; Klimovich, P.S.; Aleksanrushkina, N.A.; Sysoeva, V.Y.; Karagyaur, M.N.; Tkachuk, V.A. Downregulation of uPAR promotes urokinase translocation into the nucleus and epithelial to mesenchymal transition in neuroblastoma. J. Cell. Physiol. 2020, 235, 6268–6286. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Ma, M.; Zhang, S. EGF-induced urokinase plasminogen activator receptor promotes epithelial to mesenchymal tran-sition in human gastric cancer cells. Oncol. Rep. 2017, 38, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, A.; Biagioni, A.; Bianchini, F.; Peppicelli, S.; Chillà, A.; Margheri, F.; Luciani, C.; Pimpinelli, N.; Del Rosso, M.; Calorini, L. Inhibition of uPAR-TGFβ crosstalk blocks MSC-dependent EMT in melanoma cells. J. Mol. Med. 2015, 93, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Y.; Zhang, Y.; Zhang, Y.; Xiao, W. The role of uPAR in epithelial-mesenchymal transition in small airway epi-thelium of patients with chronic obstructive pulmonary disease. Respir Res. 2013, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.D.; Jo, M.; Montel, V.; Takimoto, S.; Gonias, S.L. uPAR induces epithelial–mesenchymal transition in hypoxic breast cancer cells. J. Cell Biol. 2007, 178, 425–436. [Google Scholar] [CrossRef]
- Hannan, R.T.; Miller, A.E.; Hung, R.-C.; Sano, C.; Peirce, S.M.; Barker, T.H. Extracellular matrix remodeling associated with bleomycin-induced lung injury supports pericyte-to-myofibroblast transition. Matrix Biol. Plus 2020, 10, 100056. [Google Scholar] [CrossRef]
- Katoh, D.; Kozuka, Y.; Noro, A.; Ogawa, T.; Imanaka-Yoshida, K.; Yoshida, T. Tenascin-C Induces Phenotypic Changes in Fibroblasts to Myofibroblasts with High Contractility through the Integrin αvβ1/Transforming Growth Factor β/SMAD Signaling Axis in Human Breast Cancer. Am. J. Pathol. 2020, 190, 2123–2135. [Google Scholar] [CrossRef]
- Bianchini, F.; Peppicelli, S.; Fabbrizzi, P.; Biagioni, A.; Mazzanti, B.; Menchi, G.; Calorini, L.; Pupi, A.; Trabocchi, A. Triazole RGD antagonist reverts TGFβ1-induced endothelial-to-mesenchymal transition in endothelial precursor cells. Mol. Cell. Biochem. 2016, 424, 99–110. [Google Scholar] [CrossRef]
- Borok, Z. Role for α3 integrin in EMT and pulmonary fibrosis. J. Clin. Investig. 2008, 119, 7–10. [Google Scholar] [CrossRef]
- Shochet, G.E.; Brook, E.; Bardenstein-Wald, B.; Grobe, H.; Edelstein, E.; Israeli-Shani, L.; Shitrit, D. Integrin alpha-5 silencing leads to my-ofibroblastic differentiation in IPF-derived human lung fibroblasts. Ther. Adv. Chronic Dis. 2020, 24, 2040622320936023. [Google Scholar]
- Overstreet, J.M.; Wang, Y.; Wang, X.; Niu, A.; Gewin, L.S.; Yao, B.; Harris, R.C.; Zhang, M.Z. Selective activation of epidermal growth factor receptor in renal proximal tubule induces tubulointerstitial fibrosis. FASEB J. 2017, 31, 4407–4421. [Google Scholar] [CrossRef]
- Xu, H.; Liu, L.; Cong, M.; Liu, T.; Sun, S.; Ma, H.; You, H.; Jia, J.; Wang, P. EGF neutralization antibodies attenuate liver fibrosis by inhibiting myofi-broblast proliferation in bile duct ligation mice. Histochem. Cell Biol. 2020, 154, 107–116. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Enhanced EGF receptor-signaling potentiates TGFβ-induced lens epithelial-mesenchymal transition. Exp. Eye Res. 2019, 185, 107693. [Google Scholar] [CrossRef]
- Wu, S.; Yang, S.; Qu, H. circ_CHFR regulates ox-LDL-mediated cell proliferation, apoptosis, and EndoMT by miR-15a-5p/EGFR axis in human brain microvessel endothelial cells. Open Life Sci. 2021, 16, 1053–1063. [Google Scholar] [CrossRef]
- Thooyamani, A.S.; Mukhopadhyay, A. PDGFRα mediated survival of myofibroblasts inhibit satellite cell proliferation during aberrant regeneration of lacerated skeletal muscle. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grünert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Investig. 2006, 116, 1561–1570. [Google Scholar] [CrossRef]
- Yan, D.; Liu, X.; Xu, H.; Guo, S.-W. Platelets induce endothelial–mesenchymal transition and subsequent fibrogenesis in endometriosis. Reprod. Biomed. Online 2020, 41, 500–517. [Google Scholar] [CrossRef]
- Chen, L.; Lin, G.; Chen, K.; Liang, R.; Wan, F.; Zhang, C.; Tian, G.; Zhu, X. VEGF promotes migration and invasion by regulating EMT and MMPs in nasopharyngeal carcinoma. J. Cancer 2020, 11, 7291–7301. [Google Scholar] [CrossRef]
- Modi, S.; Tiwari, A.; Kulkarni, V. Reversal of TGF-β-induced epithelial-mesenchymal transition in hepatocellular carcinoma by sorafenib, a VEGFR-2 and Raf kinase inhibitor. Curr. Res. Pharmacol. Drug. Discov. 2021, 2, 100014. [Google Scholar] [CrossRef]
- Rossato, F.A.; Su, Y.; Mackey, A.; Ng, Y.S.E. Fibrotic Changes and Endothelial-to-Mesenchymal Transition Promoted by VEGFR2 Antagonism Alter the Therapeutic Effects of VEGFA Pathway Blockage in a Mouse Model of Choroidal Neovascularization. Cells 2020, 9, 2057. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Cui, Z.; Le Saux, C.J.; Horowitz, J.C.; Rangarajan, S.; Kurundkar, A.; Antony, V.B.; Thannickal, V.J. SMAD-Independent Down-Regulation of Caveolin-1 by TGF-β: Effects on Proliferation and Survival of Myofibroblasts. PLoS ONE 2015, 10, e0116995. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhao, J.; Huang, S.; Shu, B.; Yang, R.; Chen, L.; Xu, Y.; Xie, J.; Liu, X.; Jia, J. Reduced hydration-induced decreased caveolin-1 expression causes epithelial-to-mesenchymal transition. Am. J. Transl. Res. 2020, 12, 8067–8083. [Google Scholar] [PubMed]
- Jung, A.C.; Ray, A.M.; Ramolu, L.; Macabre, C.; Simon, F.; Noulet, F.; Blandin, A.F.; Renner, G.; Lehmann, M.; Choulier, L. Caveolin-1-negative head and neck squamous cell carcinoma primary tumors display increased epithelial to mesenchymal transition and prometastatic properties. Oncotarget 2015, 6, 41884–41901. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wermuth, P.J.; Benn, B.S.; Lisanti, M.P.; Jimenez, S.A. Caveolin-1 Deficiency Induces Spontaneous Endothelial-to-Mesenchymal Transition in Murine Pulmonary Endothelial Cells in Vitro. Am. J. Pathol. 2012, 182, 325–331. [Google Scholar] [CrossRef]
- Schnieder, J.; Mamazhakypov, A.; Birnhuber, A.; Wilhelm, J.; Kwapiszewska, G.; Ruppert, C.; Markart, P.; Wujak, L.; Rubio, K.; Barreto, G. Loss of LRP1 promotes acquisition of contractile-myofibroblast phenotype and release of active TGF-β1 from ECM stores. Matrix Biol. 2019, 88, 69–88. [Google Scholar] [CrossRef]
- Hu, K.; Lin, L.; Tan, X.; Yang, J.; Bu, G.; Mars, W.M.; Liu, Y. tPA Protects Renal Interstitial Fibroblasts and Myofibroblasts from Apoptosis. J. Am. Soc. Nephrol. 2008, 19, 503–514. [Google Scholar] [CrossRef]
- Hu, K.; Wu, C.; Mars, W.M.; Liu, Y. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor–related protein 1–mediated integrin signaling. J. Clin. Investig. 2007, 117, 3821–3832. [Google Scholar] [CrossRef]
- Omori, K.; Hattori, N.; Senoo, T.; Takayama, Y.; Masuda, T.; Nakashima, T.; Iwamoto, H.; Fujitaka, K.; Hamada, H.; Kohno, N. Inhibition of Plasminogen Activator Inhibitor-1 Attenuates Transforming Growth Factor-β-Dependent Epithelial Mesenchymal Transition and Differentiation of Fibroblasts to Myofibroblasts. PLoS ONE 2016, 11, e0148969. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Wang, W.L.; Liu, J.; Li, W.B.; Bai, L.L.; Yuan, Y.D.; Song, S.X. Plasminogen activator inhibitor-1 promotes the proliferation and inhibits the apoptosis of pulmonary fibroblasts by Ca2+ signaling. Thromb. Res. 2012, 131, 64–71. [Google Scholar] [CrossRef]
- Masuda, T.; Nakashima, T.; Namba, M.; Yamaguchi, K.; Sakamoto, S.; Horimasu, Y.; Miyamoto, S.; Iwamoto, H.; Fujitaka, K.; Miyata, Y. Inhibition of PAI-1 limits chemotherapy resistance in lung cancer through suppressing myofibroblast characteristics of cancer-associated fibroblasts. J. Cell. Mol. Med. 2019, 23, 2984–2994. [Google Scholar] [CrossRef]
- Pedroja, B.; Kang, L.; Imas, A.; Carmeliet, P.; Bernstein, A. Plasminogen activator inhibitor-1 regulates integrin alphavbeta3 ex-pression and autocrine transforming growth factor beta signaling. J. Biol. Chem. 2009, 284, 20708–20717. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bradham, W.S.; Gleaves, L.A.; De Taeye, B.; Murphy, S.B.; Covington, J.W.; Vaughan, D.E. Genetic deficiency of plasminogen activator inhibitor-1 promotes cardiac fibrosis in aged mice: Involvement of constitutive transforming growth factor-beta signaling and endothelial-to-mesenchymal transition. Circulation 2010, 122, 1200–1209. [Google Scholar] [CrossRef]
- Baumeier, C.; Escher, F.; Aleshcheva, G.; Pietsch, H.; Schultheiss, H.-P. Plasminogen activator inhibitor-1 reduces cardiac fibrosis and promotes M2 macrophage polarization in inflammatory cardiomyopathy. Basic Res. Cardiol. 2021, 116, 1–9. [Google Scholar] [CrossRef]
- Kanno, Y.; Kawashita, E.; Kokado, A.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. Alpha2-antiplasmin regulates the development of dermal fibrosis in mice by prostaglandin F2α synthesis through adipose triglyceride lipase/calcium-independent phospho-lipase. Arthritis Rheum. 2013, 65, 492–502. [Google Scholar] [CrossRef]
- Kanno, Y.; Shu, E.; Niwa, H.; Seishima, M.; Ozaki, K.-I. MicroRNA-30c attenuates fibrosis progression and vascular dysfunction in systemic sclerosis model mice. Mol. Biol. Rep. 2021, 48, 3431–3437. [Google Scholar] [CrossRef]
- Kanno, Y.; Hirota, M.; Matsuo, O.; Ozaki, K.-I. α2-antiplasmin positively regulates endothelial-to-mesenchymal transition and fibrosis progression in diabetic nephropathy. Mol. Biol. Rep. 2021, 49, 205–215. [Google Scholar] [CrossRef]
- Michalczyk, K.; Cymbaluk-Płoska, A. Metalloproteinases in Endometrial Cancer-Are They Worth Measuring? Int. J. Mol. Sci. 2021, 22, 12472. [Google Scholar] [CrossRef]
- Lijnen, H.R. Matrix Metalloproteinases and Cellular Fibrinolytic Activity. Biochemistry 2002, 67, 92–98. [Google Scholar] [CrossRef]
- Newby, A.C. Metalloproteinase production from macrophages—A perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 2016, 101, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, I.; Noro, T.; Tsutsui, N.; Yamanouchi, E.; Kuroda, H.; Nakano, M.; Yokomori, H.; Inagaki, Y. Fibrogenesis and Carcinogenesis in Nonalcoholic Steatohepatitis (NASH): Involvement of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinase (TIMPs). Cancers 2014, 6, 1220–1255. [Google Scholar] [CrossRef] [PubMed]
- Young-Min, A.S.; Beeton, C.; Laughton, R.; Plumpton, T.; Bartram, S.; Murphy, G.; Black, C.; Cawston, E.T. Serum TIMP-1, TIMP-2, and MMP-1 in patients with systemic sclerosis, primary Raynaud’s phenomenon, and in normal controls. Ann. Rheum. Dis. 2001, 60, 846–851. [Google Scholar] [PubMed]
- Yao, H.; Yang, X.; Yan, M.; Fang, X.; Wang, Y.; Qi, H.; Sun, L. Correlation of Serum M-CSF, CER, and TIMP-1 Levels with Liver Fibrosis in Viral Hepatitis. Comput. Math. Methods Med. 2022, 2022, 6736225. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Iwasa, M.; Sugimoto, R.; Tempaku, M.; Yoshikawa, K.; Yoshizawa, N.; Povero, D.; Sugimoto, K.; Hasegawa, H.; Takei, Y. Complement complex 1 subunit q-mediated hepatic stellate cell activation with connective tissue growth factor elevation is a prognostic factor for survival in rat and human chronic liver diseases. Hepatol. Commun. 2022, 6, 3515–3527. [Google Scholar] [CrossRef]
- Lefeuvre, C.; Roux, M.; Blanchard, S.; Le Guillou-Guillemette, H.; Boursier, J.; Lunel-Fabiani, F.; Jeannin, P.; Pivert, A.; Ducancelle, A. Analysis of hepatic fibrosis markers in the serum of chronic hepatitis B patients according to basal core promoter/precore mutants. Sci. Rep. 2022, 12, 10261. [Google Scholar] [CrossRef]
- Yang, K.; Palm, J.; König, J.; Seeland, U.; Rosenkranz, S.; Feiden, W.; Rübe, C.; Rübe, C.E. Matrix-Metallo-Proteinases and their tissue inhibitors in radiation-induced lung injury. Int. J. Radiat. Biol. 2007, 83, 665–676. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, G.; Mo, B.; Wang, C. Artesunate modulates expression of matrix metalloproteinases and their inhibitors as well as collagen-IV to attenuate pulmonary fibrosis in rats. Genet. Mol. Res. 2016, 15, 530. [Google Scholar] [CrossRef]
- Kökény, G.; Németh, Á.; Kopp, J.B.; Chen, W.; Oler, A.J.; Manzéger, A.; Rosivall, L.; Mózes, M.M. Susceptibility to kidney fibrosis in mice is associated with early growth response-2 protein and tissue inhibitor of metalloproteinase-1 expression. Kidney Int. 2022, 102, 337–354. [Google Scholar] [CrossRef]
- Takawale, A.; Zhang, P.; Patel, V.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myo-cardial Fibrosis by Mediating CD63-Integrin β1 Interaction. Hypertension 2017, 69, 1092–1103. [Google Scholar] [CrossRef]
- Kanno, Y.; Shu, E.; Niwa, H.; Kanoh, H.; Seishima, M. Alternatively activated macrophages are associated with the α2AP production that occurs with the development of dermal fibrosis: The role of alternatively activated macrophages on the development of fibrosis. Arthritis Res Ther. 2020, 22, 76. [Google Scholar] [CrossRef]
- Rabieian, R.; Boshtam, M.; Zareei, M.; Kouhpayeh, S.; Masoudifar, A.; Mirzaei, H. Plasminogen Activator Inhibitor Type-1 as a Regulator of Fibrosis. J. Cell Biochem. 2018, 119, 17–27. [Google Scholar] [CrossRef]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef]
- Ma, L.; Fogo, A. PAI-1 and kidney fibrosis. Front. Biosci. 2009, 14, 2028–2041. [Google Scholar] [CrossRef]
- Liakouli, V.; Cipriani, P.; Marrelli, A.; Alvaro, S.; Ruscitti, P.; Giacomelli, R. Angiogenic cytokines and growth factors in systemic sclerosis. Autoimmun. Rev. 2011, 10, 590–594. [Google Scholar] [CrossRef]
- Mostmans, Y.; Cutolo, M.; Giddelo, C.; Decuman, S.; Melsens, K.; Declercq, H.; Vandecasteele, E.; De Keyser, F.; Distler, O.; Gutermuth, J. The role of endothelial cells in the vasculopathy of systemic sclerosis: A systematic review. Autoimmun. Rev. 2017, 16, 774–786. [Google Scholar] [CrossRef]
- Zanin-Silva, D.C.; Santana-Gonçalves, M.; Kawashima-Vasconcelos, M.Y.; Oliveira, M.C. Management of Endothelial Dysfunction in Systemic Sclerosis: Current and Developing Strategies. Front. Med. 2021, 8, 250. [Google Scholar] [CrossRef]
- Plow, E.F.; Hoover-Plow, J. The Functions of Plasminogen in Cardiovascular Disease. Trends Cardiovasc. Med. 2004, 14, 180–186. [Google Scholar] [CrossRef]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef]
- Yan, Q.; Sage, E. Transforming growth factor-beta1 induces apoptotic cell death in cultured retinal endothelial cells but not pericytes: Association with decreased expression of p21waf1/cip. J. Cell Biochem. 1998, 70, 70–83. [Google Scholar] [CrossRef]
- Long, D.; Yang, J.; Wu, X.; Gui, Y.; Yu, L. Urokinase-type plasminogen activator protects human umbilical vein endothelial cells from apoptosis in sepsis. Int. J. Clin. Exp. Pathol. 2019, 12, 77–86. [Google Scholar] [PubMed]
- Prager, G.W.; Mihaly, J.; Brunner, P.M.; Koshelnick, Y.; Hoyer-Hansen, G.; Binder, B.R. Urokinase mediates endothelial cell survival via induction of the X-linked inhibitor of apoptosis protein. Blood 2009, 113, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Meng, S.; Xu, S.-T.; Jin, S.-J.; Zeng, Q.-Z.; Gu, G.-J. The overexpression of uPA promotes the proliferation and fibrinolytic activity of human umbilical vein endothelial cells. Int. J. Clin. Exp. Pathol. 2019, 12, 2959–2966. [Google Scholar] [PubMed]
- Beloglazova, I.B.; Zubkova, E.S.; Stambol’skii, D.V.; Plekhanova, O.S.; Men’shikov, M.Y.; Akopyan, Z.A.; Bibilashvili, R.S.; Parfenova, E.V.; Tkachuk, V.A. Proteolytically inactive recom-binant forms of urokinase suppress migration of endothelial cells. Bull. Exp. Biol. Med. 2014, 156, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Balsara, R.D.; Merryman, R.; Virjee, F.; Northway, C.; Castellino, F.J.; Ploplis, A.V. A deficiency of uPAR alters endothelial angiogenic function and cell morphology. Vasc. Cell 2011, 3, 10. [Google Scholar] [CrossRef]
- Lu, H.; Mabilat, C.; Yeh, P.; Guitton, J.D.; Li, H.; Pouchelet, M.; Shoevaert, D.; Legrand, Y.; Soria, J.; Soria, C. Blockage of urokinase receptor reduces in vitro the motility and the deformability of endothelial cells. FEBS Lett. 1996, 380, 21–24. [Google Scholar] [CrossRef]
- Beloglazova, I.; Stepanova, V.; Zubkova, E.; Dergilev, K.; Koptelova, N.; Tyurin-Kuzmin, P.A.; Dyikanov, D.; Plekhanova, O.; Cines, D.B.; Mazar, A.P. Mesenchymal stromal cells enhance self-assembly of a HUVEC tubular network through uPA-uPAR/VEGFR2/integrin/NOTCH crosstalk. Biochim. Biophys. Acta. Mol. Cell Res. 2022, 1869, 119157. [Google Scholar] [CrossRef]
- Alexander, R.A.; Prager, G.W.; Mihaly-Bison, J.; Uhrin, P.; Sunzenauer, S.; Binder, B.R.; Schütz, G.J.; Freissmuth, M.; Breuss, J.M. VEGF-induced endothelial cell migration requires urokinase receptor (uPAR)-dependent integrin redistribution. Cardiovasc. Res. 2012, 94, 125–135. [Google Scholar] [CrossRef]
- Reuning, U.; Sperl, S.; Kopitz, C.; Kessler, H.; Krüger, A.; Schmitt, M.; Magdolen, V. Urokinase-type Plasminogen Activator (uPA) and its Receptor (uPAR): Development of Antagonists of uPA / uPAR Interaction and their Effects In Vitro and In Vivo. Curr. Pharm. Des. 2003, 9, 1529–1543. [Google Scholar] [CrossRef]
- Poettler, M.; Unseld, M.; Mihaly-Bison, J.; Uhrin, P.; Koban, F.; Binder, B.R.; Zielinski, C.C.; Prager, G.W. The urokinase receptor (CD87) represents a central mediator of growth factor-induced endothelial cell migration. Thromb. Haemost. 2012, 108, 357–366. [Google Scholar] [CrossRef]
- Stepanova, V.; Jayaraman, P.S.; Zaitsev, S.V.; Lebedeva, T.; Bdeir, K.; Kershaw, R.; Holman, K.R.; Parfyonova, Y.V.; Semina, E.V.; Beloglazova, I.B. Urokinase-type Plasminogen Activator (uPA) Promotes Angiogenesis by Attenuating Proline-rich Homeodomain Protein (PRH) Transcription Factor Activity and De-repressing Vascular Endothelial Growth Factor (VEGF) Receptor Expression. J. Biol. Chem. 2016, 291, 15029–15045. [Google Scholar] [CrossRef]
- Raghu, H.; Nalla, A.K.; Gondi, C.S.; Gujrati, M.; Dinh, D.H.; Rao, J.S. uPA and uPAR shRNA inhibit angiogenesis via enhanced secretion of SVEGFR1 independent of GM-CSF but dependent on TIMP-1 in endothelial and glioblastoma cells. Mol. Oncol. 2011, 6, 33–47. [Google Scholar] [CrossRef]
- Park, J.; Keller, G.; Ferrara, N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subep-ithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell. 1993, 4, 1317–1326. [Google Scholar] [CrossRef]
- Kanno, Y.; Shu, E.; Kanoh, H.; Matsuda, A.; Seishima, M. α2AP regulates vascular alteration by inhibiting VEGF signaling in systemic sclerosis: The roles of α2AP in vascular dysfunction in systemic sclerosis. Arthritis Res. Ther. 2017, 19, 1–8. [Google Scholar] [CrossRef]
- Herkenne, S.; Paques, C.; Nivelles, O.; Lion, M.; Bajou, K.; Pollenus, T.; Fontaine, M.; Carmeliet, P.; Martial, J.A.; Nguyen, N.Q. The interaction of uPAR with VEGFR2 promotes VEGF-induced angiogenesis. Sci. Signal. 2015, 8, ra117. [Google Scholar] [CrossRef]
- Larusch, G.A.; Merkulova, A.; Mahdi, F.; Shariat-Madar, Z.; Sitrin, R.G.; Cines, D.B.; Schmaier, A.H. Domain 2 of uPAR regulates single-chain uro-kinase-mediated angiogenesis through β1-integrin and VEGFR. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H305–H320. [Google Scholar] [CrossRef]
- LaRusch, G.A.; Mahdi, F.; Shariat-Madar, Z.; Adams, G.; Sitrin, R.G.; Zhang, W.M.; McCrae, K.R.; Schmaier, A.H. Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood 2010, 115, 5111–5120. [Google Scholar] [CrossRef]
- D’Alessio, S.; Fibbi, G.; Cinelli, M.; Guiducci, S.; Del Rosso, A.; Margheri, F.; Serratì, S.; Pucci, M.; Kahaleh, B.; Fan, P. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004, 50, 3275–3285. [Google Scholar] [CrossRef]
- Bifulco, K.; Longanesi-Cattani, I.; Gala, M.; DICarluccio, G.; Masucci, M.T.; Pavone, V.; Lista, L.; Arra, C.; Stoppelli, M.P.; Carriero, M.V. The soluble form of urokinase receptor promotes angiogenesis through its Ser⁸⁸-Arg-Ser-Arg-Tyr⁹² chemotactic sequence. J. Thromb. Haemost. 2010, 8, 2789–2799. [Google Scholar] [CrossRef]
- Fuchs, P.Ö.; Calitz, C.; Pavlović, N.; Binet, F.; Solbak, S.M.Ø.; Danielson, U.H.; Kreuger, J.; Heindryckx, F.; Gerwins, P. Fibrin fragment E potentiates TGF-β-induced myofibroblast activation and recruitment. Cell Signal. 2020, 72, 109661. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, K.; Ichimura, K.; Reddy, S.; Haddad, F.; Spiekerkoetter, E. Cardiac Fibrosis in the Pressure Overloaded Left and Right Ventricle as a Therapeutic Target. Front. Cardiovasc. Med. 2022, 9, 6553. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-F.; Veetil, N.N.; Li, Q.; Kucherenko, M.M.; Knosalla, C.; Kuebler, W.M. Pulmonary hypertension: Linking inflammation and pulmonary arterial stiffening. Front. Immunol. 2022, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- Christou, H.; Khalil, R.A. Mechanisms of pulmonary vascular dysfunction in pulmonary hypertension and implications for novel therapies. Am. J. Physiol. Circ. Physiol. 2022, 322, H702–H724. [Google Scholar] [CrossRef] [PubMed]
- Ban, C.; Wang, T.; Zhang, S.; Xin, P.; Liang, L.; Wang, C.; Dai, H. Fibrinolytic system related to pulmonary arterial pressure and lung function of patients with idiopathic pulmonary fibrosis. Clin. Respir. J. 2015, 11, 640–647. [Google Scholar] [CrossRef]
- Levi, M.; Moons, L.; Bouché, A.; Shapiro, S.D.; Collen, D.; Carmeliet, P. Deficiency of Urokinase-Type Plasminogen Activator–Mediated Plasmin Generation Impairs Vascular Remodeling During Hypoxia-Induced Pulmonary Hypertension in Mice. Circulation 2001, 103, 2014–2020. [Google Scholar] [CrossRef]
- Manetti, M.; Allanore, Y.; Revillod, L.; Fatini, C.; Guiducci, S.; Cuomo, G.; Bonino, C.; Riccieri, V.; Bazzichi, L.; Liakouli, V. A genetic variation located in the promoter region of the UPAR (CD87) gene is associated with the vascular complications of systemic sclerosis. Arthritis Rheum. 2010, 63, 247–256. [Google Scholar] [CrossRef]
- Makarova, A.M.; Lebedeva, T.V.; Nassar, T.; Higazi, A.A.; Xue, J.; Carinato, M.E.; Bdeir, K.; Cines, D.B.; Stepanova, V. Urokinase-type Plasminogen Activator (uPA) Induces Pulmonary Microvascular Endothelial Permeability through Low Density Lipoprotein Receptor-related Protein (LRP)-dependent Activation of Endothelial Nitric-oxide Synthase. J. Biol. Chem. 2011, 286, 23044–23053. [Google Scholar] [CrossRef]
- Boccella, S.; Panza, E.; Lista, L.; Belardo, C.; Ianaro, A.; De Rosa, M.; de Novellis, V.; Pavone, V. Preclinical evaluation of the urokinase receptor-derived peptide UPARANT as an anti-inflammatory drug. Inflamm. Res. 2017, 66, 701–709. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Lee, Y.J.; Seo, J.H.; Sung, H.J.; Park, K.H.; Choi, I.K.; Kim, S.J.; Oh, S.C.; Choi, C.W.; Kim, B.S. uPAR expression under hypoxic conditions depends on iNOS modulated ERK phosphorylation in the MDA-MB-231 breast carcinoma cell line. Cell Res. 2006, 16, 75–81. [Google Scholar] [CrossRef]
- Huang, E.; Peng, N.; Xiao, F.; Hu, D.; Wang, X.; Lu, L. The Roles of Immune Cells in the Pathogenesis of Fibrosis. Int. J. Mol. Sci. 2020, 21, 5203. [Google Scholar] [CrossRef]
- Abdul, S.; Leebeek, F.; Rijken, D.; Uitte de Willige, S. Natural heterogeneity of α2-antiplasmin: Functional and clinical conse-quences. Blood 2016, 127, 538–545. [Google Scholar] [CrossRef]
- Van Geffen, C.; Deißler, A.; Quante, M.; Renz, H.; Hartl, D.; Kolahian, S. Regulatory Immune Cells in Idiopathic Pulmonary Fibrosis: Friends or Foes? Front. Immunol. 2021, 22, 663203. [Google Scholar] [CrossRef]
- Brown, M.; O’Reilly, S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin. Exp. Immunol. 2018, 195, 310–321. [Google Scholar] [CrossRef]
- Pattanaik, D.; Brown, M.; Postlethwaite, B.; Postlethwaite, A. Pathogenesis of Systemic Sclerosis. Front Immunol. 2015, 6, 272. [Google Scholar] [CrossRef]
- Numajiri, H.; Kuzumi, A.; Fukasawa, T.; Ebata, S.; Yoshizaki-Ogawa, A.; Asano, Y.; Kazoe, Y.; Mawatari, K.; Kitamori, T.; Yoshizaki, A. B Cell Depletion Inhibits Fibrosis via Suppression of Profibrotic Macrophage Differentiation in a Mouse Model of Systemic Sclerosis. Arthritis Rheumatol. 2021, 73, 2086–2095. [Google Scholar] [CrossRef]
- Nakayama, W.; Jinnin, M.; Makino, K.; Kajihara, I.; Makino, T.; Fukushima, S.; Inoue, Y.; Ihn, H. Serum levels of soluble CD163 in patients with systemic sclerosis. Rheumatol. Int. 2010, 32, 403–407. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Xu, J.; Xie, J.; Harris, D.C.H.; Zheng, G. The Role of Macrophages in Kidney Fibrosis. Front. Physiol. 2021, 12, 5838. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Mondino, A.; Blasi, F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004, 25, 450–455. [Google Scholar] [CrossRef]
- Syrovets, T.; Lunov, O.; Simmet, T. Plasmin as a proinflammatory cell activator. J. Leukoc. Biol. 2012, 92, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Medcalf, R.; Keragala, C. Fibrinolysis: A Primordial System Linked to the Immune Response. Int. J. Mol. Sci. 2021, 22, 3406. [Google Scholar] [CrossRef] [PubMed]
- Hastings, S.; Myles, P.; Medcalf, R. Plasmin, Immunity, and Surgical Site Infection. J. Clin. Med. 2021, 10, 2070. [Google Scholar] [CrossRef] [PubMed]
- Vago, J.P.; Sugimoto, M.A.; Lima, K.M.; Negreiros-Lima, G.L.; Baik, N.; Teixeira, M.M.; Perretti, M.; Parmer, R.J.; Miles, L.A.; Sousa, L.P. Plasminogen and the Plasminogen Receptor, Plg-RKT, Regulate Macrophage Phenotypic, and Functional Changes. Front. Immunol. 2019, 10, 1458. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, H.; Munakata, S.; Tashiro, Y.; Salama, Y.; Dhahri, D.; Eiamboonsert, S.; Ota, Y.; Onoda, H.; Tsuda, Y.; Okada, Y. Pharmacological targeting of plasmin prevents lethality in a murine model of macrophage activation syndrome. Blood 2017, 130, 59–72. [Google Scholar] [CrossRef]
- Li, X.; Syrovets, T.; Genze, F.; Pitterle, K.; Oberhuber, A.; Orend, K.H.; Simmet, T. Plasmin Triggers Chemotaxis of Monocyte-Derived Dendritic Cells Through an Akt2-Dependent Pathway and Promotes a T-Helper Type-1 Response. Arter. Thromb. Vasc. Biol. 2010, 30, 582–590. [Google Scholar] [CrossRef]
- Bryer, S.C.; Fantuzzi, G.; Van Rooijen, N.; Koh, T.J. Urokinase-Type Plasminogen Activator Plays Essential Roles in Macrophage Chemotaxis and Skeletal Muscle Regeneration. J. Immunol. 2008, 180, 1179–1188. [Google Scholar] [CrossRef]
- Meznarich, J.; Malchodi, L.; Helterline, D.; Ramsey, S.A.; Bertko, K.; Plummer, T.; Plawman, A.; Gold, E.; Stempien-Otero, A. Urokinase Plasminogen Activator Induces Pro-Fibrotic/M2 Phenotype in Murine Cardiac Macrophages. PLoS ONE 2013, 8, e57837. [Google Scholar] [CrossRef]
- Liu, G.; Yang, Y.; Yang, S.; Banerjee, S.; De Freitas, A.; Friggeri, A.; Davis, K.I.; Abraham, E. The receptor for urokinase regulates TLR2 mediated in-flammatory responses in neutrophils. PLoS ONE 2011, 6, e25843. [Google Scholar]
- Li, J.; Pan, Y.; Li, D.; Xia, X.; Jiang, Q.; Dou, H.; Hou, Y. Urokinase-type plasminogen activator receptor is required for impairing toll-like receptor 7 signaling on macrophage efferocytosis in lupus. Mol. Immunol. 2020, 127, 38–45. [Google Scholar] [CrossRef]
- Kiyan, Y.; Tkachuk, S.; Rong, S.; Gorrasi, A.; Ragno, P.; Dumler, I.; Haller, H.; Shushakova, N. TLR4 Response to LPS Is Reinforced by Urokinase Receptor. Front. Immunol. 2020, 11, 3550. [Google Scholar] [CrossRef]
- Rasmussen, L.J.H.; Petersen, J.E.V.; Eugen-Olsen, J. Soluble Urokinase Plasminogen Activator Receptor (suPAR) as a Biomarker of Systemic Chronic Inflammation. Front. Immunol. 2021, 12, 641. [Google Scholar] [CrossRef]
- Yousif, A.M.; Minopoli, M.; Bifulco, K.; Ingangi, V.; Di Carluccio, G.; Merlino, F.; Motti, M.L.; Grieco, P.; Carriero, M.V. Cyclization of the Urokinase Receptor-Derived Ser-Arg-Ser-Arg-Tyr Peptide Generates a Potent Inhibitor of Trans-Endothelial Migration of Monocytes. PLoS ONE 2015, 10, e0126172. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, Y. The uPA/uPAR System Orchestrates the Inflammatory Response, Vascular Homeostasis, and Immune System in Fibrosis Progression. Int. J. Mol. Sci. 2023, 24, 1796. https://doi.org/10.3390/ijms24021796
Kanno Y. The uPA/uPAR System Orchestrates the Inflammatory Response, Vascular Homeostasis, and Immune System in Fibrosis Progression. International Journal of Molecular Sciences. 2023; 24(2):1796. https://doi.org/10.3390/ijms24021796
Chicago/Turabian StyleKanno, Yosuke. 2023. "The uPA/uPAR System Orchestrates the Inflammatory Response, Vascular Homeostasis, and Immune System in Fibrosis Progression" International Journal of Molecular Sciences 24, no. 2: 1796. https://doi.org/10.3390/ijms24021796
APA StyleKanno, Y. (2023). The uPA/uPAR System Orchestrates the Inflammatory Response, Vascular Homeostasis, and Immune System in Fibrosis Progression. International Journal of Molecular Sciences, 24(2), 1796. https://doi.org/10.3390/ijms24021796