

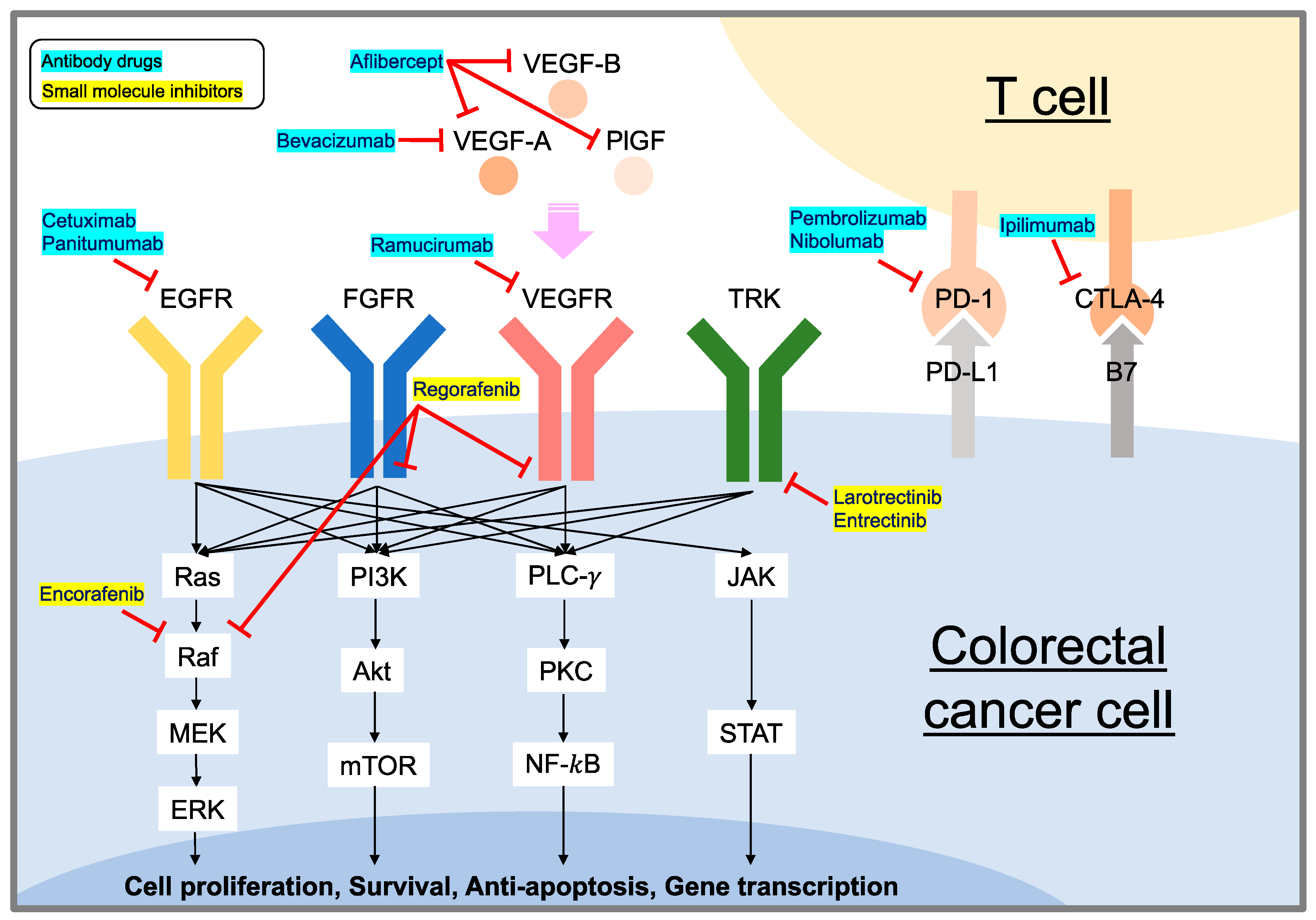
Current Targeted Therapy for Metastatic Colorectal Cancer
 ,
,  and
and
Abstract
1. Introduction
2. mCRC Treatment Strategies
3. EGFR-Targeting Strategy
3.1. Molecular Mechanism of EGFR Signaling
3.2. Cetuximab and Panitumumab
4. VEGF/VEGFR Targeting Strategy
4.1. Molecular Mechanism of VEGF/VEGFR Signaling
4.2. Bevacizumab
4.3. Aflibercept
4.4. Ramucirumab
4.5. Regorafenib
4.6. Fruquintinib
5. Immune Checkpoint Targeting Immunotherapies
5.1. Molecular Mechanism of Immune Checkpoints
5.2. Pembrolizumab, Nivolumab, and Ipilimumab
6. NTRK Signaling Pathway and Targeting Strategy
Larotrectinib and Entrectinib
7. BRAF Signaling Pathway and Targeting Strategy
Encorafenib
8. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Prim. 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed]
- van der Stok, E.P.; Spaander, M.C.W.; Grunhagen, D.J.; Verhoef, C.; Kuipers, E.J. Surveillance after curative treatment for colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.M.; Coyle, V.M.; Kennedy, R.D.; Wilson, R.H. Molecular Subtypes and Personalized Therapy in Metastatic Colorectal Cancer. Curr. Color. Cancer Rep. 2016, 12, 141–150. [Google Scholar] [CrossRef]
- Motta, R.; Cabezas-Camarero, S.; Torres-Mattos, C.; Riquelme, A.; Calle, A.; Montenegro, P.; Sotelo, M.J. Personalizing first-line treatment in advanced colorectal cancer: Present status and future perspectives. J. Clin. Transl. Res. 2021, 7, 771–785. [Google Scholar]
- Marques, R.P.; Duarte, G.S.; Sterrantino, C.; Pais, H.L.; Quintela, A.; Martins, A.P.; Costa, J. Triplet (FOLFOXIRI) versus doublet (FOLFOX or FOLFIRI) backbone chemotherapy as first-line treatment of metastatic colorectal cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2017, 118, 54–62. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Bekaii-Saab, T.; Stintzing, S. Reconsidering the benefit of intermittent versus continuous treatment in the maintenance treatment setting of metastatic colorectal cancer. Cancer Treat. Rev. 2016, 45, 97–104. [Google Scholar] [CrossRef]
- Miyo, M.; Kato, T.; Yoshino, T.; Yamanaka, T.; Bando, H.; Satake, H.; Yamazaki, K.; Taniguchi, H.; Oki, E.; Kotaka, M.; et al. Protocol of the QUATTRO-II study: A multicenter randomized phase II study comparing CAPOXIRI plus bevacizumab with FOLFOXIRI plus bevacizumab as a first-line treatment in patients with metastatic colorectal cancer. BMC Cancer 2020, 20, 687. [Google Scholar] [CrossRef]
- DeStefanis, R.A.; Kratz, J.D.; Emmerich, P.B.; Deming, D.A. Targeted Therapy in Metastatic Colorectal Cancer: Current Standards and Novel Agents in Review. Curr. Color. Cancer Rep. 2019, 15, 61–69. [Google Scholar] [CrossRef]
- Nappi, A.; Berretta, M.; Romano, C.; Tafuto, S.; Cassata, A.; Casaretti, R.; Silvestro, L.; Divitiis, C.; Alessandrini, L.; Fiorica, F.; et al. Metastatic Colorectal Cancer: Role of Target Therapies and Future Perspectives. Curr. Cancer Drug Targets 2018, 18, 421–429. [Google Scholar] [CrossRef]
- Baraibar, I.; Ros, J.; Mulet, N.; Salva, F.; Argiles, G.; Martini, G.; Cuadra, J.L.; Sardo, E.; Ciardiello, D.; Tabernero, J.; et al. Incorporating traditional and emerging biomarkers in the clinical management of metastatic colorectal cancer: An update. Expert Rev. Mol. Diagn. 2020, 20, 653–664. [Google Scholar] [CrossRef]
- Singh, M.P.; Rai, S.; Pandey, A.; Singh, N.K.; Srivastava, S. Molecular subtypes of colorectal cancer: An emerging therapeutic opportunity for personalized medicine. Genes Dis. 2021, 8, 133–145. [Google Scholar] [CrossRef]
- Wu, C.W.K.; Reid, M.; Leedham, S.; Lui, R.N. The emerging era of personalized medicine in advanced colorectal cancer. J. Gastroenterol. Hepatol. 2022, 37, 1411–1425. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Cunningham, D.; Roth, A.D.; Navarro, M.; James, R.D.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: A multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.M.; Sargent, D.J.; Morton, R.F.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Pitot, H.C.; Alberts, S.R. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol. 2004, 22, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Caballero Servin, I.A. Evaluation and application of microbiological methods to the sanitary control of restaurants. Salud Publica Mex. 1967, 9, 221–235. [Google Scholar] [PubMed]
- US Food and Drug Administration. New treatments for colorectal cancer. FDA Consum 2004, 38, 17. [Google Scholar]
- US Food and Drug Administration. The FDA approves drugs for colorectal cancer, lung cancer. FDA Consum 2007, 41, 5. [Google Scholar]
- FDA approves aflibercept (Zaltrap) for metastatic colorectal cancer. Oncology 2012, 26, 842–873.
- FDA approves regorafenib (Stivarga) for metastatic colorectal cancer. Oncology 2012, 26, 896.
- Venook, A.P. The value and effectiveness of angiogenesis inhibitors for colorectal cancer. Clin. Adv. Hematol. Oncol. 2015, 13, 561–563. [Google Scholar] [PubMed]
- Geanta, M.; Cioroboiu, C. The FDA Changed Everything. Biomed. Hub 2017, 2, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Goldberg, R.M. Hypermutated Tumors and Immune Checkpoint Inhibition. Drugs 2018, 78, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.R.; Jayswal, R.; Adams, V.; Anthony, L.B.; Villano, J.L. Multiple sclerosis outcomes after cancer immunotherapy. Clin. Transl. Oncol. 2019, 21, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Valeri, N. Streamlining Detection of Fusion Genes in Colorectal Cancer: Having “Faith” in Precision Oncology in the (Tissue) “Agnostic” Era. Cancer Res. 2019, 79, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA notches up third tissue-agnostic cancer approval. Nat. Rev. Drug Discov. 2019, 18, 737. [Google Scholar] [CrossRef]
- Koumaki, K.; Kontogianni, G.; Kosmidou, V.; Pahitsa, F.; Kritsi, E.; Zervou, M.; Chatziioannou, A.; Souliotis, V.L.; Papadodima, O.; Pintzas, A. BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600Epsilon CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166061. [Google Scholar] [CrossRef]
- Kheder, E.S.; Hong, D.S. Emerging Targeted Therapy for Tumors with NTRK Fusion Proteins. Clin. Cancer Res. 2018, 24, 5807–5814. [Google Scholar] [CrossRef]
- Guler, I.; Askan, G.; Klostergaard, J.; Sahin, I.H. Precision medicine for metastatic colorectal cancer: An evolving era. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 919–931. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef]
- Wang, S.C.; Hung, M.C. Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors. Clin. Cancer Res. 2009, 15, 6484–6489. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Stintzing, S.; Kirchner, T.; Boeck, S.; Jung, A. Clinical relevance of EGFR- and KRAS-status in colorectal cancer patients treated with monoclonal antibodies directed against the EGFR. Cancer Treat. Rev. 2009, 35, 262–271. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.J.; Cha, P.H.; Choi, K.Y. Strategies to overcome resistance to epidermal growth factor receptor monoclonal antibody therapy in metastatic colorectal cancer. World J. Gastroenterol. 2014, 20, 9862–9871. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Allegra, C.J.; Rumble, R.B.; Hamilton, S.R.; Mangu, P.B.; Roach, N.; Hantel, A.; Schilsky, R.L. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J. Clin. Oncol. 2016, 34, 179–185. [Google Scholar] [CrossRef]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar]
- Trotta, A.M.; Ottaiano, A.; Romano, C.; Nasti, G.; Nappi, A.; De Divitiis, C.; Napolitano, M.; Zanotta, S.; Casaretti, R.; D’Alterio, C.; et al. Prospective Evaluation of Cetuximab-Mediated Antibody-Dependent Cell Cytotoxicity in Metastatic Colorectal Cancer Patients Predicts Treatment Efficacy. Cancer Immunol. Res. 2016, 4, 366–374. [Google Scholar] [CrossRef]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med 2019, 7, 105. [Google Scholar] [CrossRef]
- Saltz, L.B.; Meropol, N.J.; Loehrer, P.J., Sr.; Needle, M.N.; Kopit, J.; Mayer, R.J. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J. Clin. Oncol. 2004, 22, 1201–1208. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Final results from PRIME: Randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann. Oncol. 2014, 25, 1346–1355. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Keating, G.M. Panitumumab: A review of its use in metastatic colorectal cancer. Drugs 2010, 70, 1059–1078. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.; Vincent, M. Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr. Oncol. 2010, 17 (Suppl. 1), S18–S30. [Google Scholar] [CrossRef]
- Yarom, N.; Jonker, D.J. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov. Med. 2011, 11, 95–105. [Google Scholar] [PubMed]
- Koefoed, K.; Steinaa, L.; Soderberg, J.N.; Kjaer, I.; Jacobsen, H.J.; Meijer, P.J.; Haurum, J.S.; Jensen, A.; Kragh, M.; Andersen, P.S.; et al. Rational identification of an optimal antibody mixture for targeting the epidermal growth factor receptor. MAbs 2011, 3, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Peeters, M.; Kim, T.W.; Li, J.; Cascinu, S.; Ruff, P.; Suresh, A.S.; Thomas, A.; Tjulandin, S.; Zhang, K.; et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): A randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014, 15, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Kaley, K.; Chu, E.; Copur, M.S. Safety and efficacy of panitumumab therapy after progression with cetuximab: Experience at two institutions. Clin. Color. Cancer 2010, 9, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Katakami, N.; Kitajima, N. Successful cetuximab therapy after failure of panitumumab rechallenge in a patient with metastatic colorectal cancer: Restoration of drug sensitivity after anti-EGFR monoclonal antibody-free interval. J. Gastrointest. Cancer 2014, 45, 506–507. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, F.; Blanchard, F.; Charbonnier, F.; Le Pessot, F.; Lamy, A.; Galais, M.P.; Bastit, L.; Killian, A.; Sesboue, R.; Tuech, J.J.; et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br. J. Cancer 2007, 96, 1166–1169. [Google Scholar] [CrossRef]
- Benvenuti, S.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007, 67, 2643–2648. [Google Scholar] [CrossRef]
- De Roock, W.; Piessevaux, H.; De Schutter, J.; Janssens, M.; De Hertogh, G.; Personeni, N.; Biesmans, B.; Van Laethem, J.L.; Peeters, M.; Humblet, Y.; et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann. Oncol. 2008, 19, 508–515. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Lievre, A.; Bachet, J.B.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouche, O.; Landi, B.; Louvet, C.; et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Hartmann, J.T.; de Braud, F.; Schuch, G.; Zubel, A.; Celik, I.; Schlichting, M.; Koralewski, P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: The OPUS study. Ann. Oncol. 2011, 22, 1535–1546. [Google Scholar] [CrossRef]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Bokemeyer, C.; Kohne, C.H.; Ciardiello, F.; Lenz, H.J.; Heinemann, V.; Klinkhardt, U.; Beier, F.; Duecker, K.; van Krieken, J.H.; Tejpar, S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 2015, 51, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Oliner, K.S.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; Andre, T.; Chan, E.; Lordick, F.; et al. Analysis of KRAS/NRAS Mutations in a Phase III Study of Panitumumab with FOLFIRI Compared with FOLFIRI Alone as Second-line Treatment for Metastatic Colorectal Cancer. Clin. Cancer Res. 2015, 21, 5469–5479. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Wilson, C.Y.; Tolias, P. Recent advances in cancer drug discovery targeting RAS. Drug Discov. Today 2016, 21, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef]
- Tejpar, S.; Celik, I.; Schlichting, M.; Sartorius, U.; Bokemeyer, C.; Van Cutsem, E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J. Clin. Oncol. 2012, 30, 3570–3577. [Google Scholar] [CrossRef]
- Mao, C.; Huang, Y.F.; Yang, Z.Y.; Zheng, D.Y.; Chen, J.Z.; Tang, J.L. KRAS p.G13D mutation and codon 12 mutations are not created equal in predicting clinical outcomes of cetuximab in metastatic colorectal cancer: A systematic review and meta-analysis. Cancer 2013, 119, 714–721. [Google Scholar] [CrossRef]
- Kumar, S.S.; Price, T.J.; Mohyieldin, O.; Borg, M.; Townsend, A.; Hardingham, J.E. KRAS G13D Mutation and Sensitivity to Cetuximab or Panitumumab in a Colorectal Cancer Cell Line Model. Gastrointest. Cancer Res. 2014, 7, 23–26. [Google Scholar]
- De Roock, W.; Jonker, D.J.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Aoyama, T.; Ishibashi, K.; Tsuji, A.; Takinishi, Y.; Shindo, Y.; Sakamoto, J.; Oba, K.; Mishima, H. Randomized phase II study of cetuximab versus irinotecan and cetuximab in patients with chemo-refractory KRAS codon G13D metastatic colorectal cancer (G13D-study). Cancer Chemother. Pharmacol. 2017, 79, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef]
- Loupakis, F.; Yang, D.; Yau, L.; Feng, S.; Cremolini, C.; Zhang, W.; Maus, M.K.; Antoniotti, C.; Langer, C.; Scherer, S.J.; et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J. Natl. Cancer Inst. 2015, 107, dju427. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Yu, Q.; Ning, R.; Zhao, W.; Wei, C. Efficacy of bevacizumab versus epidermal growth factor receptor inhibitors for wild-type RAS metastatic colorectal cancer: A meta-analysis. OncoTargets Ther. 2018, 11, 4271–4281. [Google Scholar] [CrossRef]
- Boeckx, N.; Koukakis, R.; Op de Beeck, K.; Rolfo, C.; Van Camp, G.; Siena, S.; Tabernero, J.; Douillard, J.Y.; Andre, T.; Peeters, M. Effect of Primary Tumor Location on Second- or Later-line Treatment Outcomes in Patients With RAS Wild-type Metastatic Colorectal Cancer and All Treatment Lines in Patients With RAS Mutations in Four Randomized Panitumumab Studies. Clin. Color. Cancer 2018, 17, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmuller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Schwartzberg, L.S.; Rivera, F.; Karthaus, M.; Fasola, G.; Canon, J.L.; Hecht, J.R.; Yu, H.; Oliner, K.S.; Go, W.Y. PEAK: A randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J. Clin. Oncol. 2014, 32, 2240–2247. [Google Scholar] [CrossRef]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for Patients With RAS and BRAF Wild-Type Metastatic Colorectal Cancer With Acquired Resistance to First-line Cetuximab and Irinotecan: A Phase 2 Single-Arm Clinical Trial. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Crisafulli, G.; Bartolini, A.; Fenocchio, E.; Amatu, A.; Manca, P.; et al. Circulating tumor DNA to guide rechallenge with panitumumab in metastatic colorectal cancer: The phase 2 CHRONOS trial. Nat. Med. 2022, 28, 1612–1618. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Mariotti, V.; Fiorotto, R.; Cadamuro, M.; Fabris, L.; Strazzabosco, M. New insights on the role of vascular endothelial growth factor in biliary pathophysiology. JHEP Rep. 2021, 3, 100251. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef]
- Byrne, A.M.; Bouchier-Hayes, D.J.; Harmey, J.H. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF). J. Cell Mol. Med. 2005, 9, 777–794. [Google Scholar] [CrossRef]
- Wada, S.; Tsunoda, T.; Baba, T.; Primus, F.J.; Kuwano, H.; Shibuya, M.; Tahara, H. Rationale for antiangiogenic cancer therapy with vaccination using epitope peptides derived from human vascular endothelial growth factor receptor 2. Cancer Res. 2005, 65, 4939–4946. [Google Scholar] [CrossRef]
- Ishizaki, H.; Tsunoda, T.; Wada, S.; Yamauchi, M.; Shibuya, M.; Tahara, H. Inhibition of tumor growth with antiangiogenic cancer vaccine using epitope peptides derived from human vascular endothelial growth factor receptor 1. Clin. Cancer Res. 2006, 12, 5841–5849. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fei, D.; Vanderlaan, M.; Song, A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis 2004, 7, 335–345. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Cunningham, D.; Lang, I.; Marcuello, E.; Lorusso, V.; Ocvirk, J.; Shin, D.B.; Jonker, D.; Osborne, S.; Andre, N.; Waterkamp, D.; et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): An open-label, randomised phase 3 trial. Lancet Oncol. 2013, 14, 1077–1085. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausova, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, R.; Yau, T.C.; Ma, B.; Pan, H.; Xu, J.; Bai, Y.; Chi, Y.; Wang, L.; et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015, 16, 619–629. [Google Scholar] [CrossRef]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.C.; Shi, Y.; Wang, Y.R.; Lv, Y.; Yan, H.; Mao, H.; Wang, Z.K.; Wu, Z.Y.; Shi, W.W.; Dai, G.H. KRAS mutation and primary tumor location do not affect efficacy of bevacizumab-containing chemotherapy in stagae IV colorectal cancer patients. Sci. Rep. 2017, 7, 14368. [Google Scholar] [CrossRef]
- Yoshimatsu, K.; Satake, M.; Sano, M.; Asaka, S.; Yamada, Y.; Okayama, S.; Yano, Y.; Yokomizo, H.; Usui, T.; Yamaguchi, K.; et al. Standard Chemotherapy with Bevacizumab as First-Line Therapy for Metastatic Colorectal Cancer with RAS Mutation. Gan Kagaku Ryoho 2017, 44, 918–920. [Google Scholar]
- Grothey, A.; Sugrue, M.M.; Purdie, D.M.; Dong, W.; Sargent, D.; Hedrick, E.; Kozloff, M. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: Results from a large observational cohort study (BRiTE). J. Clin. Oncol. 2008, 26, 5326–5334. [Google Scholar] [CrossRef]
- Grothey, A.; Flick, E.D.; Cohn, A.L.; Bekaii-Saab, T.S.; Bendell, J.C.; Kozloff, M.; Roach, N.; Mun, Y.; Fish, S.; Hurwitz, H.I. Bevacizumab exposure beyond first disease progression in patients with metastatic colorectal cancer: Analyses of the ARIES observational cohort study. Pharmacoepidemiol. Drug Saf. 2014, 23, 726–734. [Google Scholar] [CrossRef]
- Bennouna, J.; Sastre, J.; Arnold, D.; Osterlund, P.; Greil, R.; Van Cutsem, E.; von Moos, R.; Vieitez, J.M.; Bouche, O.; Borg, C.; et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): A randomised phase 3 trial. Lancet Oncol. 2013, 14, 29–37. [Google Scholar] [CrossRef]
- Bendell, J.C.; Bekaii-Saab, T.S.; Cohn, A.L.; Hurwitz, H.I.; Kozloff, M.; Tezcan, H.; Roach, N.; Mun, Y.; Fish, S.; Flick, E.D.; et al. Treatment patterns and clinical outcomes in patients with metastatic colorectal cancer initially treated with FOLFOX-bevacizumab or FOLFIRI-bevacizumab: Results from ARIES, a bevacizumab observational cohort study. Oncologist 2012, 17, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Joulain, F.; Hoff, P.M.; Mitchell, E.; Ruff, P.; Lakomy, R.; Prausova, J.; Moiseyenko, V.M.; van Hazel, G.; Cunningham, D.; et al. Aflibercept Plus FOLFIRI vs. Placebo Plus FOLFIRI in Second-Line Metastatic Colorectal Cancer: A Post Hoc Analysis of Survival from the Phase III VELOUR Study Subsequent to Exclusion of Patients who had Recurrence During or Within 6 Months of Completing Adjuvant Oxaliplatin-Based Therapy. Target. Oncol. 2016, 11, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.S. Aflibercept (AVE0005): An alternative strategy for inhibiting tumour angiogenesis by vascular endothelial growth factors. Expert Opin. Biol. Ther. 2009, 9, 263–271. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Satake, H.; Ando, K.; Oki, E.; Shimokawa, M.; Makiyama, A.; Saeki, H.; Tsuji, A.; Mori, M. Protocol of the EFFORT study: A prospective study of FOLFIRI plus aflibercept as second-line treatment after progression on FOLFOXIRI plus bevacizumab or during maintenance treatment in patients with unresectable/metastatic colorectal cancer. BMC Cancer 2020, 20, 1116. [Google Scholar] [CrossRef]
- Debeuckelaere, C.; Murgioni, S.; Lonardi, S.; Girardi, N.; Alberti, G.; Fano, C.; Gallimberti, S.; Magro, C.; Ahcene-Djaballah, S.; Daniel, F.; et al. Ramucirumab: The long and winding road toward being an option for mCRC treatment. Expert Opin. Biol. Ther. 2019, 19, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Yoshihiro, T.; Kusaba, H.; Makiyama, A.; Kobayashi, K.; Uenomachi, M.; Ito, M.; Doi, Y.; Mitsugi, K.; Aikawa, T.; Takayoshi, K.; et al. Efficacy and safety of ramucirumab plus modified FOLFIRI for metastatic colorectal cancer. Int. J. Clin. Oncol. 2019, 24, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Shinozaki, E.; Osumi, H.; Nakayama, I.; Ota, Y.; Ichimura, T.; Ogura, M.; Wakatsuki, T.; Ooki, A.; Takahari, D.; et al. Second-line FOLFIRI plus ramucirumab with or without prior bevacizumab for patients with metastatic colorectal cancer. Cancer Chemother. Pharmacol. 2019, 84, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J. Regorafenib: A review of its use in previously treated patients with progressive metastatic colorectal cancer. Drugs Aging 2014, 31, 67–78. [Google Scholar] [CrossRef]
- Dong, J.; Li, B.; Lin, D.; Zhou, Q.; Huang, D. Advances in Targeted Therapy and Immunotherapy for Non-small Cell Lung Cancer Based on Accurate Molecular Typing. Front. Pharmacol. 2019, 10, 230. [Google Scholar] [CrossRef]
- Sun, Q.; Zhou, J.; Zhang, Z.; Guo, M.; Liang, J.; Zhou, F.; Long, J.; Zhang, W.; Yin, F.; Cai, H.; et al. Discovery of fruquintinib, a potent and highly selective small molecule inhibitor of VEGFR 1, 2, 3 tyrosine kinases for cancer therapy. Cancer Biol. Ther. 2014, 15, 1635–1645. [Google Scholar] [CrossRef]
- Song, Y.; Qu, T.; Zhang, H.; Sun, Y.; Cui, C.; Chi, Y.; Zhang, W.; Wang, X.; Yang, L. The Real-World Practice of Fruquintinib for Chinese Patients with Metastatic Colorectal Cancer. Cancer Manag. Res. 2021, 13, 6199–6205. [Google Scholar] [CrossRef]
- Dasari, A.; Sobrero, A.; Yao, J.; Yoshino, T.; Schelman, W.; Yang, Z.; Chien, C.; Kania, M.; Tabernero, J.; Eng, C. FRESCO-2: A global Phase III study investigating the efficacy and safety of fruquintinib in metastatic colorectal cancer. Future Oncol. 2021, 17, 3151–3162. [Google Scholar] [CrossRef]
- Hu, L.F.; Lan, H.R.; Huang, D.; Li, X.M.; Jin, K.T. Personalized Immunotherapy in Colorectal Cancers: Where Do We Stand? Front. Oncol. 2021, 11, 769305. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Colle, R.; Cohen, R.; Cochereau, D.; Duval, A.; Lascols, O.; Lopez-Trabada, D.; Afchain, P.; Trouilloud, I.; Parc, Y.; Lefevre, J.H.; et al. Immunotherapy and patients treated for cancer with microsatellite instability. Bull Cancer 2017, 104, 42–51. [Google Scholar] [CrossRef]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef]
- Myint, Z.W.; Goel, G. Role of modern immunotherapy in gastrointestinal malignancies: A review of current clinical progress. J. Hematol. Oncol. 2017, 10, 86. [Google Scholar] [CrossRef]
- Wang, J.; Yuan, R.; Song, W.; Sun, J.; Liu, D.; Li, Z. PD-1, PD-L1 (B7-H1) and Tumor-Site Immune Modulation Therapy: The Historical Perspective. J. Hematol. Oncol. 2017, 10, 34. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Allison, J.P. Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol. Rev. 2008, 224, 141–165. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, Y.; Mamun, M.; Li, X.; Chen, X.; Gao, Y. Research progress of PD-1/PD-L1 immunotherapy in gastrointestinal tumors. Biomed. Pharmacother. 2020, 129, 110504. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Bencardino, K.; Pizzutilo, E.G.; Tosi, F.; Siena, S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann. Oncol. 2019, 30 (Suppl. 8), viii5–viii15. [Google Scholar] [CrossRef]
- Cho, H.; Kim, N.; Murakami, T.; Sim, T. Anti-Tumor Activity of AZD4547 Against NTRK1 Fusion Positive Cancer Cells Through Inhibition of NTRKs. Front. Oncol. 2021, 11, 757598. [Google Scholar] [CrossRef] [PubMed]
- Davare, M.A.; Tognon, C.E. Detecting and targetting oncogenic fusion proteins in the genomic era. Biol. Cell 2015, 107, 111–129. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27 (Suppl. 3), iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Akhoundova, D.; Hussung, S.; Sivakumar, S.; Topfer, A.; Rechsteiner, M.; Kahraman, A.; Arnold, F.; Angst, F.; Britschgi, C.; Zoche, M.; et al. ROS1 genomic rearrangements are rare actionable drivers in microsatellite stable colorectal cancer. Int. J. Cancer 2022, 151, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Tejpar, S.; Sartore-Bianchi, A.; Hechtman, J.F.; Christiansen, J.; Novara, L.; Tebbutt, N.; et al. ALK, ROS1, and NTRK Rearrangements in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djx089. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Ratti, M.; Grizzi, G.; Passalacqua, R.; Lampis, A.; Cereatti, F.; Grassia, R.; Hahne, J.C. NTRK fusions in colorectal cancer: Clinical meaning and future perspective. Expert Opin. Ther. Targets 2021, 25, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- He, Z.; Thorrez, L.; Siegfried, G.; Meulemans, S.; Evrard, S.; Tejpar, S.; Khatib, A.M.; Creemers, J.W.M. The proprotein convertase furin is a pro-oncogenic driver in KRAS and BRAF driven colorectal cancer. Oncogene 2020, 39, 3571–3587. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. (Non-V600) BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef]
- Chu, J.E.; Johnson, B.; Kugathasan, L.; Morris, V.K.; Raghav, K.; Swanson, L.; Lim, H.J.; Renouf, D.J.; Gill, S.; Wolber, R.; et al. Population-based Screening for BRAF (V600E) in Metastatic Colorectal Cancer Reveals Increased Prevalence and Poor Prognosis. Clin. Cancer Res. 2020, 26, 4599–4605. [Google Scholar] [CrossRef]
- Takeda, H.; Sunakawa, Y. Management of BRAF Gene Alterations in Metastatic Colorectal Cancer: From Current Therapeutic Strategies to Future Perspectives. Front. Oncol. 2021, 11, 602194. [Google Scholar] [CrossRef] [PubMed]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef] [PubMed]
- Das, V.; Kalita, J.; Pal, M. Predictive and prognostic biomarkers in colorectal cancer: A systematic review of recent advances and challenges. Biomed. Pharmacother. 2017, 87, 8–19. [Google Scholar] [CrossRef]
- Castro, F.; Leite Pereira, C.; Helena Macedo, M.; Almeida, A.; Jose Silveira, M.; Dias, S.; Patricia Cardoso, A.; Jose Oliveira, M.; Sarmento, B. Advances on colorectal cancer 3D models: The needed translational technology for nanomedicine screening. Adv. Drug Deliv. Rev. 2021, 175, 113824. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, Y.; Lu, J.; He, K.; Chen, Y.; Ding, Y.; Jin, K.; Wang, H.; Zhang, H.; Wang, H.; et al. Patient-derived xenograft models for gastrointestinal tumors: A single-center retrospective study. Front. Oncol. 2022, 12, 985154. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wei, Y.; Fu, Y.; Li, J.; Han, L. A novel enterocyte-related 4-gene signature for predicting prognosis in colon adenocarcinoma. Front. Immunol. 2022, 13, 1052182. [Google Scholar] [CrossRef]
- Petty, A.J.; Heyman, B.; Yang, Y. Chimeric Antigen Receptor Cell Therapy: Overcoming Obstacles to Battle Cancer. Cancers 2020, 12, 842. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Spreafico, A.; Vashisht, K.; Hinrichs, M.J. Patient Selection Strategies to Maximize Therapeutic Index of Antibody-Drug Conjugates: Prior Approaches and Future Directions. Mol. Cancer Ther. 2020, 19, 1770–1783. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H. Radioimmunotherapy: A specific treatment protocol for cancer by cytotoxic radioisotopes conjugated to antibodies. Sci. World J. 2014, 2014, 492061. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Mitsunaga, M.; Sawada, R.; Saruta, M.; Kobayashi, H.; Matsumoto, N.; Kanke, T.; Yanai, H.; Nakamura, K. Photoimmunotherapy targeting biliary-pancreatic cancer with humanized anti-TROP2 antibody. Cancer Med. 2019, 8, 7781–7792. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kaneko, M.K. A cancer-specific monoclonal antibody recognizes the aberrantly glycosylated podoplanin. Sci. Rep. 2014, 4, 5924. [Google Scholar] [CrossRef]
- Itai, S.; Yamada, S.; Kaneko, M.K.; Chang, Y.W.; Harada, H.; Kato, Y. Establishment of EMab-134, a Sensitive and Specific Anti-Epidermal Growth Factor Receptor Monoclonal Antibody for Detecting Squamous Cell Carcinoma Cells of the Oral Cavity. Monoclon. Antibodies Immunodiagn. Immunother. 2017, 36, 272–281. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Yamada, S.; Itai, S.; Chang, Y.W.; Nakamura, T.; Yanaka, M.; Kato, Y. Elucidation of the critical epitope of an anti-EGFR monoclonal antibody EMab-134. Biochem. Biophys. Rep. 2018, 14, 54–57. [Google Scholar] [CrossRef]
- Sano, M.; Kaneko, M.K.; Aasano, T.; Kato, Y. Epitope Mapping of an Antihuman EGFR Monoclonal Antibody (EMab-134) Using the REMAP Method. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 191–195. [Google Scholar] [CrossRef]
- Hosono, H.; Takei, J.; Ohishi, T.; Sano, M.; Asano, T.; Sayama, Y.; Nakamura, T.; Yanaka, M.; Kawada, M.; Harada, H.; et al. AntiEGFR monoclonal antibody 134mG2a exerts antitumor effects in mouse xenograft models of oral squamous cell carcinoma. Int. J. Mol. Med. 2020, 46, 1443–1452. [Google Scholar] [CrossRef]
- Tateyama, N.; Nanamiya, R.; Ohishi, T.; Takei, J.; Nakamura, T.; Yanaka, M.; Hosono, H.; Saito, M.; Asano, T.; Tanaka, T.; et al. Defucosylated Anti-Epidermal Growth Factor Receptor Monoclonal Antibody 134-mG2a-f Exerts Antitumor Activities in Mouse Xenograft Models of Dog Epidermal Growth Factor Receptor-Overexpressed Cells. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 177–183. [Google Scholar] [CrossRef]
- Li, G.; Ohishi, T.; Kaneko, M.K.; Takei, J.; Mizuno, T.; Kawada, M.; Saito, M.; Suzuki, H.; Kato, Y. Defucosylated Mouse-Dog Chimeric Anti-EGFR Antibody Exerts Antitumor Activities in Mouse Xenograft Models of Canine Tumors. Cells 2021, 10, 3599. [Google Scholar] [CrossRef] [PubMed]
- Itai, S.; Kaneko, M.K.; Fujii, Y.; Yamada, S.; Nakamura, T.; Yanaka, M.; Saidoh, N.; Handa, S.; Chang, Y.W.; Suzuki, H.; et al. Development of EMab-51, a Sensitive and Specific Anti-Epidermal Growth Factor Receptor Monoclonal Antibody in Flow Cytometry, Western Blot, and Immunohistochemistry. Monoclon. Antibodies Immunodiagn. Immunother. 2017, 36, 214–219. [Google Scholar] [CrossRef]
- Nanamiya, R.; Sano, M.; Asano, T.; Yanaka, M.; Nakamura, T.; Saito, M.; Tanaka, T.; Hosono, H.; Tateyama, N.; Kaneko, M.K.; et al. Epitope Mapping of an Anti-Human Epidermal Growth Factor Receptor Monoclonal Antibody (EMab-51) Using the RIEDL Insertion for Epitope Mapping Method. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Takei, J.; Kaneko, M.K.; Ohishi, T.; Kawada, M.; Harada, H.; Kato, Y. A novel anti-EGFR monoclonal antibody (EMab-17) exerts antitumor activity against oral squamous cell carcinomas via antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity. Oncol. Lett 2020, 19, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, T.; Kato, Y.; Kaneko, M.K.; Ohba, S.I.; Inoue, H.; Harakawa, A.; Kawada, M. Anti-Metastatic Activity of an Anti-EGFR Monoclonal Antibody against Metastatic Colorectal Cancer with KRAS p.G13D Mutation. Int. J. Mol. Sci. 2020, 21, 6037. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wang, X.; Xin, Y.; Wang, Z.; Gong, J.; Zhang, X.; Li, Y.; Ji, C.; Sun, Y.; Zhao, F.; et al. Trastuzumab combined with irinotecan in patients with HER2-positive metastatic colorectal cancer: A phase II single-arm study and exploratory biomarker analysis. Cancer Res. Treat. 2022. [Google Scholar] [CrossRef]
- Patelli, G.; Tosi, F.; Amatu, A.; Mauri, G.; Curaba, A.; Patane, D.A.; Pani, A.; Scaglione, F.; Siena, S.; Sartore-Bianchi, A. Strategies to tackle RAS-mutated metastatic colorectal cancer. ESMO Open 2021, 6, 100156. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Tabata, J.; Nakaoku, T. REToma: A cancer subtype with a shared driver oncogene. Carcinogenesis 2020, 41, 123–129. [Google Scholar] [CrossRef]
- Crutcher, M.; Waldman, S. Biomarkers in the development of individualized treatment regimens for colorectal cancer. Front. Med. 2022, 9, 1062423. [Google Scholar] [CrossRef]


{kind=link}
| Year Approved by the FDA | Drugs | Targets | Drug Details | Ref |
|---|---|---|---|---|
| 2004 | Cetuximab | EGFR | Chimeric mouse/human mAb (IgG1) | [18] |
| Bevacizumab | VEGF-A | Humanized mAb (IgG1) | [18] | |
| 2006 | Panitumumab | EGFR | Fully human mAb (IgG1) | [19] |
| 2012 | Aflibercept | VEGF-A, VEGF-B, PlGF | Fusion protein which consists of the binding portions of VEGF from VEGF-1 and 2 fused to the Fc portion ofimmunoglobulin G1 (IgG1) | [20] |
| Regorafenib | VEGFR, FGFR, KIT, PDGFR, BRAF | Small molecule inhibitor of membrane-bound and intracellular receptor tyrosine kinases | [21] | |
| 2015 | Ramucirumab | VEGFR-2 | Fully human mAb (IgG1) | [22] |
| 2017 | Pembrolizumab | PD-1 | Humanized mAb (IgG4) | [23] |
| Nivolumab | PD-1 | Fully human mAb (IgG4) | [24] | |
| 2018 | Ipilimumab | CTLA-4 | Fully human mAb (IgG1) | [25] |
| Larotrectinib | TRK | Small molecule of tyrosine kinase inhibitor | [26] | |
| 2019 | Entrectinib | TRK, ALK, ROS1 | Small molecule of tyrosine kinase inhibitor | [27] |
| 2020 | Encorafenib | BRAF (V600E-mutant) | Small molecule kinase inhibitor | [28] |
| Targeted Drugs | Study | Phase | Study Regimen | Number | Results | Ref |
|---|---|---|---|---|---|---|
| Cetuximab | BOND | II | Cetuximab + Irinotecan /Irinotecan (Refractory to Irinotecan) (Third-line) | 218/111 | OS: 22.9/10.8 months (p = 0.007) Disease control: 55.5%/32.4% (p < 0.001) | Cunningham et al. [43] |
| Panitumumab | PRIME | III | Panitumumab + FOLFOX4 /FOLFOX4 (First-line) | 325/331 | OS: 23.8/19.4 months (p = 0.03) PFS: 10.0/8.6 months (p = 0.01) | Douillard et al. [44] |
| Cetuximab + Encorafenib | BEACON | III | Cetuximab + Encorafenib /Cetuximab + Chemotherapy 1 (Second- or later-line) | 220/221 | Median OS: 8.4/5.4 months (p < 0.001) Confirmed RR: 20%/2% (p < 0.001) | Kopetz et al. [45] |
| Cetuximab + Encorafenib + Binimetinib | BEACON | III | Cetuximab + Encorafenib + Binimetinib /Cetuximab + Chemotherapy 1 (Second- or later-line) | 224/221 | Median OS: 9.0/5.4 months (p < 0.001) Confirmed RR: 26%/2% (p < 0.001) | Kopetz et al. [45] |
| Targeted Drugs | Study | Phase | Study Regimen | Number | Results | Ref |
|---|---|---|---|---|---|---|
| Bevacizumab | Clinical study | III | Bevacizumab + IFL/Placebo + IFL (First-line) | 402/411 | OS: 20.3/15.6 months (p < 0.001) PFS: 10.6/6.2 months (p < 0.001) RR: 44.8%/34.8% (p = 0.004) 1-year survival rate: 74.3%/63.4% (p < 0.001) | Hurwitz et al. [88] |
| Bevacizumab | AVEX | III | Bevacizumab + Capecitabine/Capecitabine (First-line) | 140/140 | OS: 20.7/16.8 months (p = 0.18) PFS: 9.1/5.1 months (p < 0.001) | Cunningham et al. [89] |
| Aflibercept | VELOUR (NCT00561470) | III | Aflibercept + FOLFILI/Placebo + FOLFILI (Second-line) | 612/614 | OS: 13.5/12.06 months (p = 0.0032) PFS: 6.9/4.67 months (p < 0.0001) RR: 19.8%/11.1% (p = 0.0001) | Van Cutsem et al. [91] |
| Regorafenib | CORRECT (NCT01103323) | III | Regorafenib/Placebo (Third- or later-line) | 505/255 | OS: 6.4/5.0 months (p = 0.0052) PFS: 1.9/1.7 months (p < 0.0001) | Grothey et al. [92] |
| Regorafenib | CONCUR (NCT01103323) | III | Regorafenib/Placebo (Third- or later-line) | 138/68 | OS: 8.8/6.3 months (p = 0.00016) PFS: 3.2/1.7 months (p < 0.0001) | Li et al. [93] |
| Ramucirumab | RAISE (NCT01183780) | III | Ramucirumab + FOLFIRI/Placebo + FOLFIRI (Second-line) | 536/536 | OS: 13.3/11.7 months (p = 0.0219) PFS: 5.7/4.5 months (p < 0.0005) | Tabernero et al. [94] |
| Targeted Drugs | Study | Phase | Study Regimen | Number | Results | Ref |
|---|---|---|---|---|---|---|
| Pembrolizumab | Clinical study (NCT01876511) | II | Pembrolizumab (Second-line) (Patients with MSI-high/dMMR mCRC) | 40 | ORR: 52% Disease control rate: 82% 2-year OS: 72% 2-year PFS: 59% | Le et al. [125] |
| Pembrolizumab | KEYNOTE-177 (NCT02563002) | II | Pembrolizumab/Chemotherapy (First-line) (Patients with MSI-high/dMMR mCRC) | 153/154 | PFS: 16.5/8.2 months ORR: 43.8%/33.1% | Andre et al. [126] |
| Nivolumab | CheckMate-142 (NCT02060188) | II | Nivolumab (Second-line) (Patients with MSI-high/dMMR mCRC) | 74 | ORR: 31% Disease control for 12 weeks or longer: 69% | Overman et al. [127] |
| Nivolumab +Ipilimumab | CheckMate-142 (NCT02060188) | II | Nivolumab + Ipilimumab/Nivolumab (Second-line) (Patients with MSI-high/dMMR mCRC) | 119/74 | 1-year OS: 85%/73% ORR: 55%/31% Disease control for 12 weeks or longer: 80%/69% | Overman et al. [128] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohishi, T.; Kaneko, M.K.; Yoshida, Y.; Takashima, A.; Kato, Y.; Kawada, M. Current Targeted Therapy for Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 1702. https://doi.org/10.3390/ijms24021702
Ohishi T, Kaneko MK, Yoshida Y, Takashima A, Kato Y, Kawada M. Current Targeted Therapy for Metastatic Colorectal Cancer. International Journal of Molecular Sciences. 2023; 24(2):1702. https://doi.org/10.3390/ijms24021702
Chicago/Turabian StyleOhishi, Tomokazu, Mika K. Kaneko, Yukihiro Yoshida, Atsuo Takashima, Yukinari Kato, and Manabu Kawada. 2023. "Current Targeted Therapy for Metastatic Colorectal Cancer" International Journal of Molecular Sciences 24, no. 2: 1702. https://doi.org/10.3390/ijms24021702
APA StyleOhishi, T., Kaneko, M. K., Yoshida, Y., Takashima, A., Kato, Y., & Kawada, M. (2023). Current Targeted Therapy for Metastatic Colorectal Cancer. International Journal of Molecular Sciences, 24(2), 1702. https://doi.org/10.3390/ijms24021702