
Molecular Mechanisms of High-Altitude Acclimatization

 , and
, and 
Abstract
1. Introduction
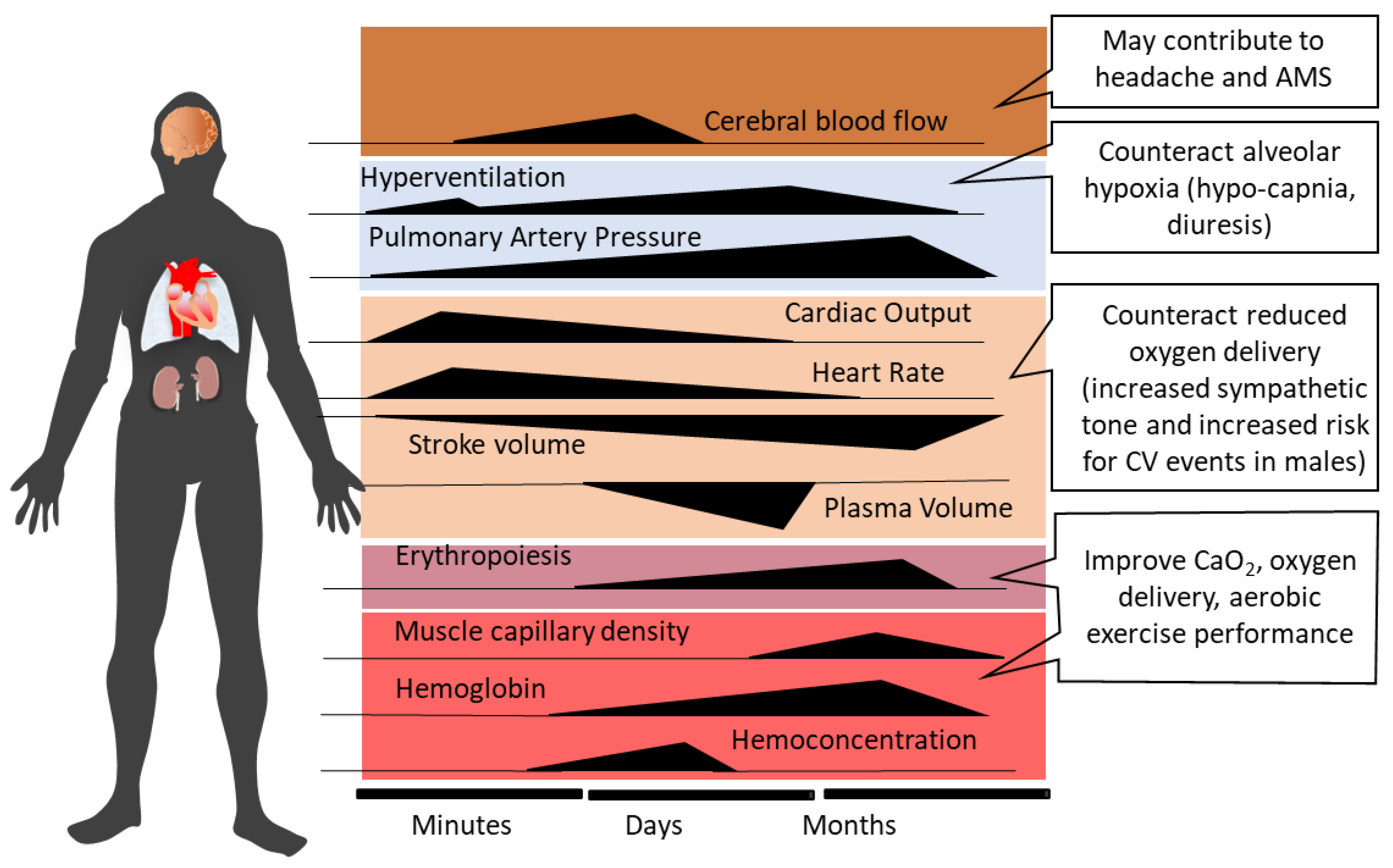
2. Systemic Physiological Responses to Acute High-Altitude Exposure and Acclimatization
3. High Altitude Illnesses: Epidemiology and Pathophysiology
3.1. Acute Mountain Sickness and High-Altitude Cerebral Edema
3.2. High-Altitude Pulmonary Edema
4. Contributions of ROS to the Development of HAIs
4.1. Oxidative Stress in Hypoxia
4.2. Sources of ROS in Hypoxia
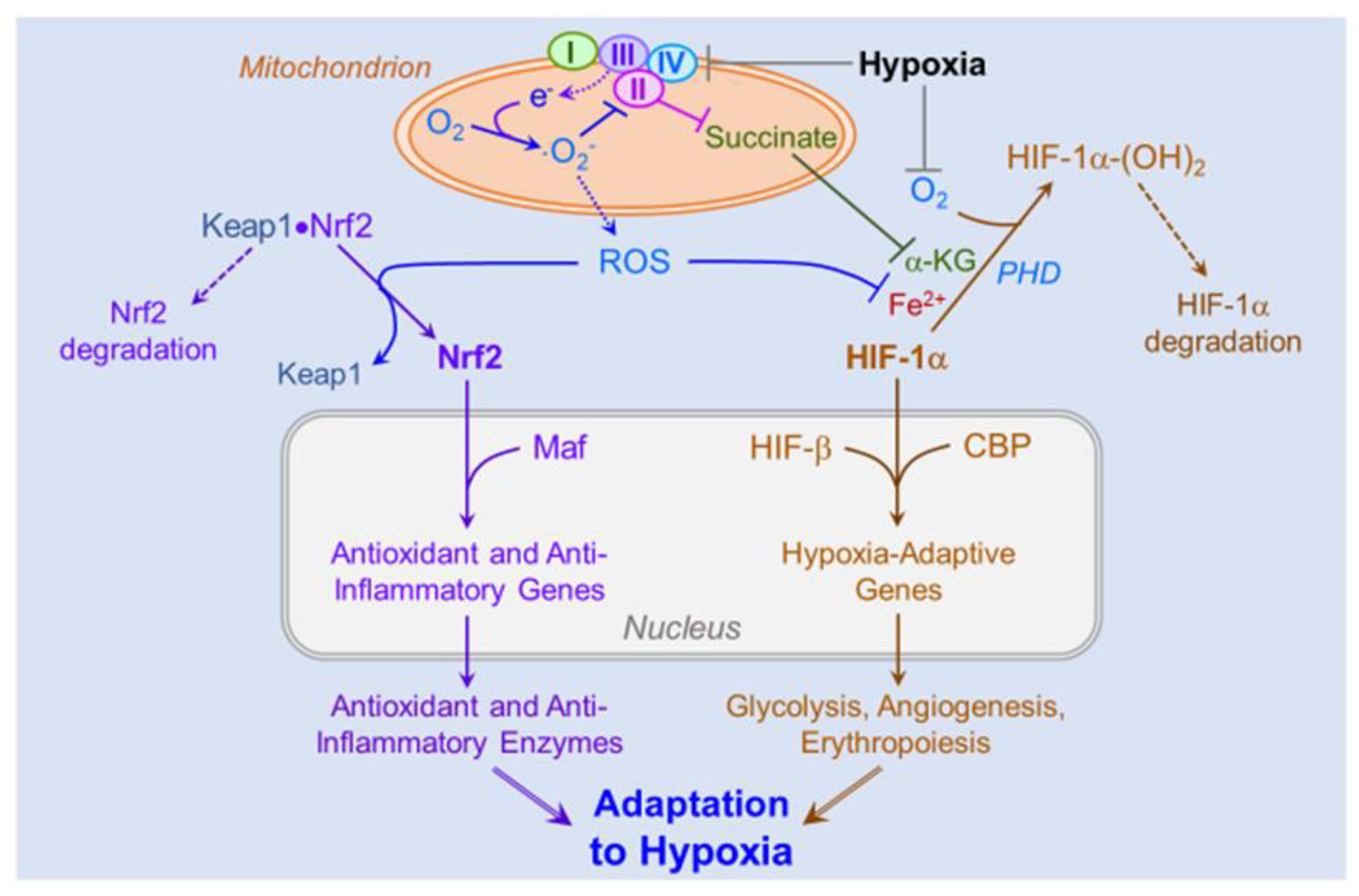
4.3. Role of ROS in Hypoxic Signaling and Acclimatization
4.4. Role of ROS on the Pathogenesis of High-Altitude Illnesses
4.5. Role of ROS-Induced Activation of the HIFs and Nrf2 on Cellular and Systemic Acclimatization
5. How Do Mitochondria Contribute to Successful Acclimatization?
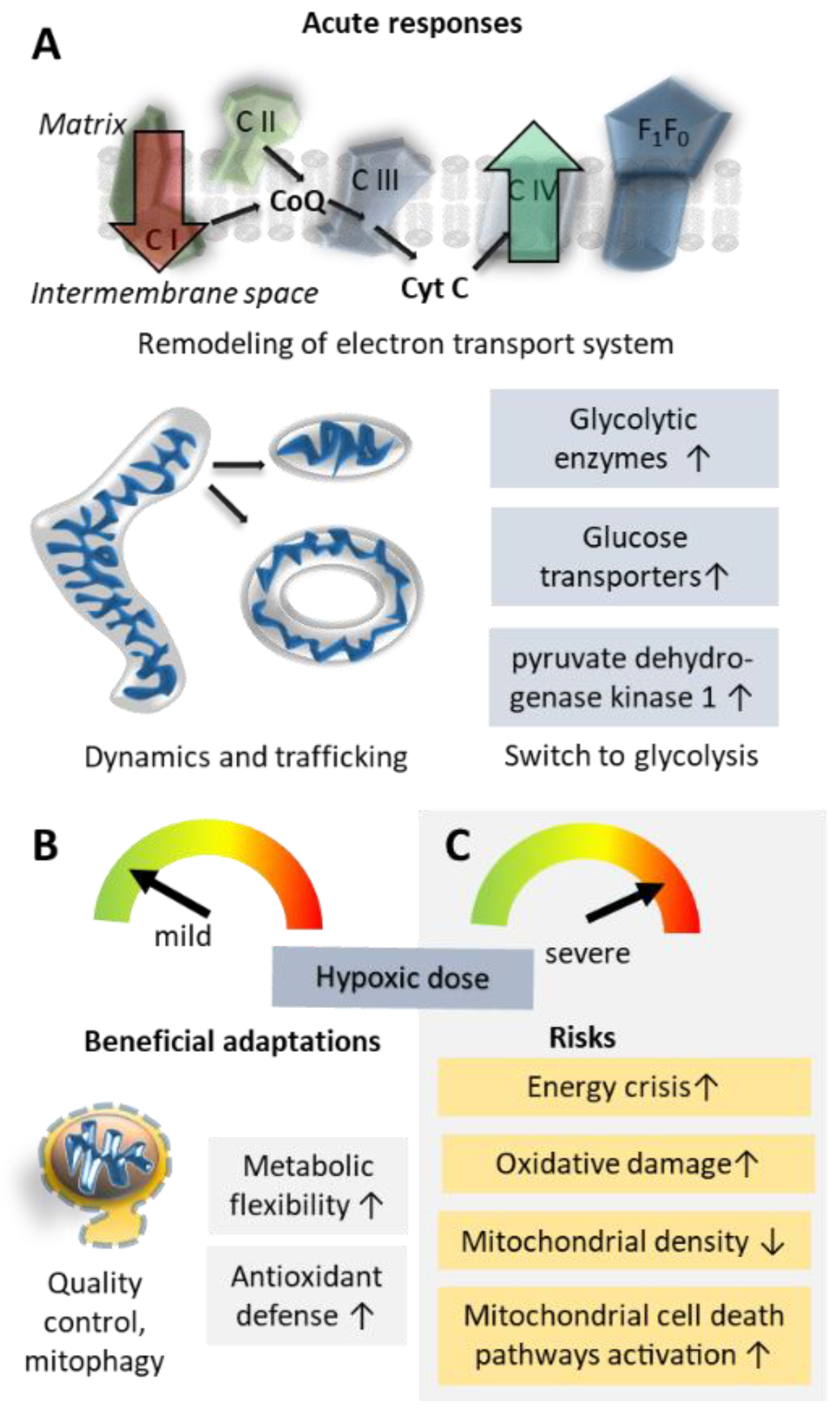
5.1. Oxidative Phosphorylation Efficiency and Metabolic Re-Modelling
5.2. Mitochondrial Damage; Reactive Oxygen Species and Inflammation
5.3. Mitochondrial Shape Changes and Localization
5.4. Mitochondrial Biogenesis Versus Clearance of Mitochondria
6. A Potential Role of Systemic Energy and REDOX Homeostatic Processes Modulated by the STAT3-RXR-Nrf2 Pathway
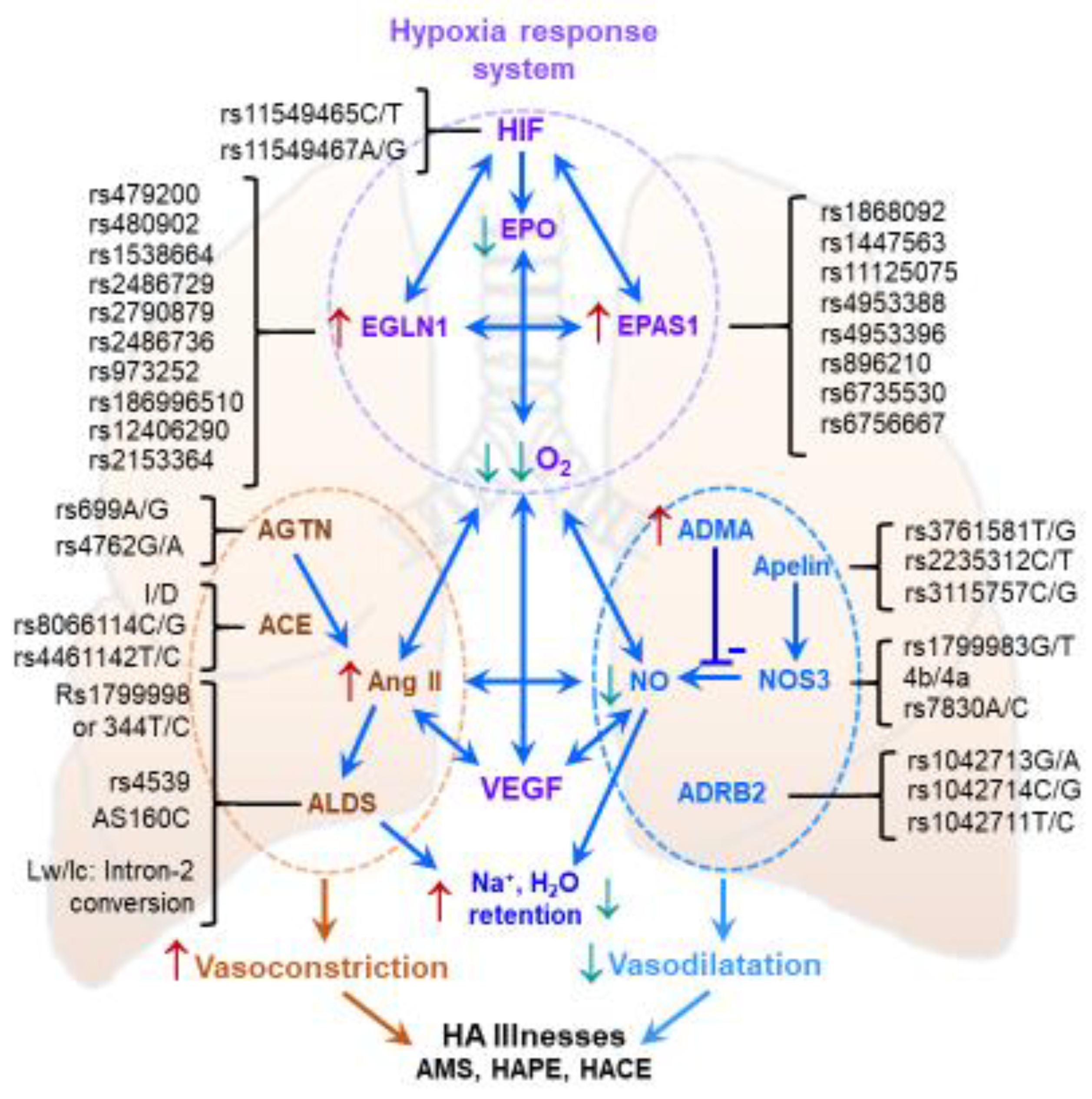
7. Genetic Aspects of High-Altitude Acclimatization
8. Time Course of Acclimatization and Memory Effects after De-Acclimatization
9. Practical Applications of Acclimatization and Altitude Training Strategies
9.1. Consequences of Ventilatory Acclimatization
9.2. Consequences of Increased Diuresis
9.3. Consequences of Shifts in Substrate Oxidation
9.4. Consequences of Sympatho-Adrenal Activation
9.5. Consequences of the Shift from Oxidative Energy Metabolism to Glycolysis
9.6. Consequences of Hypoxia-Activated Erythropoiesis
9.7. Consequences of Energy Metabolism Optimization
9.8. Consequences of Oxidative Stress
10. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hackett, P.H.; Roach, R.C. High-altitude illness. N. Engl. J. Med. 2001, 345, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, M.; Hefti, U.; Hefti, J.P. High-altitude illnesses: Old stories and new insights into the pathophysiology, treatment and prevention. Sport. Med. Health Sci. 2021, 3, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Mallet, R.T.; Pialoux, V.; Millet, G.P.; Burtscher, M. Adaptive responses to hypoxia and/or hyperoxia in humans. Antioxid. Redox Signal. 2022, 37, 887–912. [Google Scholar] [CrossRef]
- Millet, G.P.; Debevec, T. CrossTalk proposal: Barometric pressure, independent of PO2, is the forgotten parameter in altitude physiology and mountain medicine. J. Physiol. 2020, 598, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Loeppky, J.A.; Roach, R.C.; Maes, D.; Hinghofer-Szalkay, H.; Roessler, A.; Gates, L.; Fletcher, E.R.; Icenogle, M.V. Role of hypobaria in fluid balance response to hypoxia. High Alt. Med. Biol. 2005, 6, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Bärtsch, P.; Gibbs, J.S. Effect of altitude on the heart and the lungs. Circulation 2007, 116, 2191–2202. [Google Scholar] [CrossRef]
- West, J.B.; Richalet, J.P. Denis Jourdanet (1815–1892) and the early recognition of the role of hypoxia at high altitude. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 305, L333–L340. [Google Scholar] [CrossRef]
- Luks, A.M.; Swenson, E.R.; Bartsch, P. Acute high-altitude sickness. Eur. Respir. Rev. 2017, 26, 160096. [Google Scholar] [CrossRef]
- Tannheimer, M.; Lechner, R. Rapid ascents of Mt Everest: Normobaric hypoxic preacclimatization. J. Travel Med. 2020, 27, taaa099. [Google Scholar] [CrossRef]
- Burtscher, M.; Millet, G.P.; Burtscher, J. Hypoxia conditioning for high-altitude pre-acclimatization. J. Sci. Sport Exerc. 2022, 4, 331–345. [Google Scholar] [CrossRef]
- Houston, C.S. Acute pulmonary edema of high altitude. N. Engl. J. Med. 1960, 263, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Lenfant, C.; Sullivan, K. Adaptation to high altitude. N. Engl. J. Med. 1971, 284, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- West, J.B.; Hackett, P.H.; Maret, K.H.; Milledge, J.S.; Peters, R.M.; Pizzo, C.J.; Winslow, R.M. Pulmonary gas exchange on the summit of Mount Everest. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 55, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.R.; Reeves, J.T.; Wagner, P.D.; Groves, B.M.; Cymerman, A.; Malconian, M.K.; Rock, P.B.; Young, P.M.; Walter, S.D.; Houston, C.S. Operation Everest II: Oxygen transport during exercise at extreme simulated altitude. J. Appl. Physiol. 1988, 64, 1309–1321. [Google Scholar] [CrossRef]
- Hackett, P.H.; Roach, R.C.; Schoene, R.B.; Harrison, G.L.; Mills, W.J. Abnormal control of ventilation in high-altitude pulmonary edema. J. Appl. Physiol. 1988, 64, 1268–1272. [Google Scholar] [CrossRef]
- Oelz, O.; Maggiorini, M.; Ritter, M.; Waber, U.; Jenni, R.; Vock, P.; Bärtsch, P. Nifedipine for high altitude pulmonary oedema. Lancet 1989, 2, 1241–1244. [Google Scholar] [CrossRef]
- Bärtsch, P.; Maggiorini, M.; Ritter, M.; Noti, C.; Vock, P.; Oelz, O. Prevention of high-altitude pulmonary edema by nifedipine. N. Engl. J. Med. 1991, 325, 1284–1289. [Google Scholar] [CrossRef]
- Roach, R.C.; Greene, E.R.; Schoene, R.B.; Hackett, P.H. Arterial oxygen saturation for prediction of acute mountain sickness. Aviat. Space Environ. Med. 1998, 69, 1182–1185. [Google Scholar]
- Burtscher, M.; Likar, R.; Nachbauer, W.; Philadelphy, M. Aspirin for prophylaxis against headache at high altitudes: Randomised, double blind, placebo controlled trial. BMJ 1998, 316, 1057–1058. [Google Scholar] [CrossRef]
- Hackett, P.H. High altitude cerebral edema and acute mountain sickness. A pathophysiology update. Adv. Exp. Med. Biol. 1999, 474, 23–45. [Google Scholar] [CrossRef]
- Sartori, C.; Allemann, Y.; Trueb, L.; Delabays, A.; Nicod, P.; Scherrer, U. Augmented vasoreactivity in adult life associated with perinatal vascular insult. Lancet 1999, 353, 2205–2207. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, U.; Sartori, C.; Lepori, M.; Allemann, Y.; Duplain, H.; Trueb, L.; Nicod, P. High-altitude pulmonary edema: From exaggerated pulmonary hypertension to a defect in transepithelial sodium transport. Adv. Exp. Med. Biol. 1999, 474, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Maggiorini, M.; Melot, C.; Pierre, S.; Pfeiffer, F.; Greve, I.; Sartori, C.; Lepori, M.; Hauser, M.; Scherrer, U.; Naeije, R. High-altitude pulmonary edema is initially caused by an increase in capillary pressure. Circulation 2001, 103, 2078–2083. [Google Scholar] [CrossRef]
- Calbet, J.A.; Rådegran, G.; Boushel, R.; Søndergaard, H.; Saltin, B.; Wagner, P.D. Effect of blood haemoglobin concentration on V(O2,max) and cardiovascular function in lowlanders acclimatised to 5260 m. J. Physiol. 2002, 545, 715–728. [Google Scholar] [CrossRef]
- Swenson, E.R.; Maggiorini, M.; Mongovin, S.; Gibbs, J.S.; Greve, I.; Mairbäurl, H.; Bärtsch, P. Pathogenesis of high-altitude pulmonary edema: Inflammation is not an etiologic factor. JAMA 2002, 287, 2228–2235. [Google Scholar] [CrossRef]
- Bernardi, L.; Schneider, A.; Pomidori, L.; Paolucci, E.; Cogo, A. Hypoxic ventilatory response in successful extreme altitude climbers. Eur. Respir. J. 2006, 27, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Bloch, J.; Duplain, H.; Rimoldi, S.F.; Stuber, T.; Kriemler, S.; Allemann, Y.; Sartori, C.; Scherrer, U. Prevalence and time course of acute mountain sickness in older children and adolescents after rapid ascent to 3450 meters. Pediatrics 2009, 123, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Naeije, R.; Huez, S.; Lamotte, M.; Retailleau, K.; Neupane, S.; Abramowicz, D.; Faoro, V. Pulmonary artery pressure limits exercise capacity at high altitude. Eur. Respir. J. 2010, 36, 1049–1055. [Google Scholar] [CrossRef]
- Berger, M.M.; Sareban, M.; Bärtsch, P. Acute mountain sickness: Do different time courses point to different pathophysiological mechanisms? J. Appl. Physiol. 2020, 128, 952–959. [Google Scholar] [CrossRef]
- Julian, C.G. Epigenomics and human adaptation to high altitude. J. Appl. Physiol. 2017, 123, 1362–1370. [Google Scholar] [CrossRef]
- Gassmann, M.; Muckenthaler, M.U. Adaptation of iron requirement to hypoxic conditions at high altitude. J. Appl. Physiol. 2015, 119, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef] [PubMed]
- Pham, K.; Frost, S.; Parikh, K.; Puvvula, N.; Oeung, B.; Heinrich, E.C. Inflammatory gene expression during acute high-altitude exposure. J. Physiol. 2022, 600, 4169–4186. [Google Scholar] [CrossRef] [PubMed]
- Samaja, M.; Milano, G. Adaptation to Hypoxia: A Chimera? Int. J. Mol. Sci. 2020, 21, 1527. [Google Scholar] [CrossRef]
- Samaja, M.; Milano, G. Hypoxia and reoxygenation: From basic science to bedside. Front. Pediatr. 2015, 3, 86. [Google Scholar] [CrossRef]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Semenza, G.L. Life with oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef]
- Lee, C.C.; Wu, C.Y.; Yang, H.Y. Discoveries of how cells sense oxygen win the 2019 Nobel Prize in physiology or medicine. Biomed. J. 2020, 43, 434–437. [Google Scholar] [CrossRef]
- Weil, J.V.; Byrne-Quinn, E.; Sodal, I.E.; Friesen, W.O.; Underhill, B.; Filley, G.F.; Grover, R.F. Hypoxic ventilatory drive in normal man. J. Clin. Investig. 1970, 49, 1061–1072. [Google Scholar] [CrossRef]
- Reeves, J.T. Sympathetics and Hypoxia: A Brief Review; Burlington VT: Queen City, TX, USA, 1993; pp. 1–6. [Google Scholar]
- Zouboules, S.M.; Lafave, H.C.; O’Halloran, K.D.; Brutsaert, T.D.; Nysten, H.E.; Nysten, C.E.; Steinback, C.D.; Sherpa, M.T.; Day, T.A. Renal reactivity: Acid-base compensation during incremental ascent to high altitude. J. Physiol. 2018, 596, 6191–6203. [Google Scholar] [CrossRef]
- Burtscher, M.; Faulhaber, M.; Flatz, M.; Likar, R.; Nachbauer, W. Effects of short-term acclimatization to altitude (3200 m) on aerobic and anaerobic exercise performance. Int. J. Sports Med. 2006, 27, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Savourey, G.; Garcia, N.; Besnard, Y.; Hanniquet, A.M.; Fine, M.O.; Bittel, J. Physiological changes induced by pre-adaptation to high altitude. Eur. J. Appl. Physiol. Occup. Physiol. 1994, 69, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Wolff, C.B.; Barry, P.; Collier, D.J. Cardiovascular and respiratory adjustments at altitude sustain cerebral oxygen delivery—Severinghaus revisited. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2002, 132, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Samaja, M.; di Prampero, P.E.; Cerretelli, P. The role of 2,3-DPG in the oxygen transport at altitude. Respir. Physiol. 1986, 64, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Severinghaus, J.W.; Bickler, P. Time course of augmentation and depression of hypoxic ventilatory responses at altitude. J. Appl. Physiol. 1994, 77, 313–316. [Google Scholar] [CrossRef]
- Bilo, G.; Caravita, S.; Torlasco, C.; Parati, G. Blood pressure at high altitude: Physiology and clinical implications. Kardiol. Pol. 2019, 77, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Dünnwald, T.; Kienast, R.; Niederseer, D.; Burtscher, M. The use of pulse oximetry in the assessment of acclimatization to high altitude. Sensors 2021, 21, 1263. [Google Scholar] [CrossRef]
- Rimoldi, S.F.; Sartori, C.; Seiler, C.; Delacrétaz, E.; Mattle, H.P.; Scherrer, U.; Allemann, Y. High-altitude exposure in patients with cardiovascular disease: Risk assessment and practical recommendations. Prog. Cardiovasc. Dis. 2010, 52, 512–524. [Google Scholar] [CrossRef]
- Swenson, E.R. Hypoxic pulmonary vasoconstriction. High Alt. Med. Biol. 2013, 14, 101–110. [Google Scholar] [CrossRef]
- Naeije, R. Physiological adaptation of the cardiovascular system to high altitude. Prog. Cardiovasc. Dis. 2010, 52, 456–466. [Google Scholar] [CrossRef]
- Garvican, L.; Martin, D.; Quod, M.; Stephens, B.; Sassi, A.; Gore, C. Time course of the hemoglobin mass response to natural altitude training in elite endurance cyclists. Scand. J. Med. Sci. Sports 2012, 22, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Breen, E.; Tang, K.; Olfert, M.; Knapp, A.; Wagner, P. Skeletal muscle capillarity during hypoxia: VEGF and its activation. High Alt. Med. Biol. 2008, 9, 158–166. [Google Scholar] [CrossRef]
- Fulco, C.S.; Beidleman, B.A.; Muza, S.R. Effectiveness of preacclimatization strategies for high-altitude exposure. Exerc. Sport Sci. Rev. 2013, 41, 55–63. [Google Scholar] [CrossRef]
- Maggiorini, M.; Bühler, B.; Walter, M.; Oelz, O. Prevalence of acute mountain sickness in the Swiss Alps. BMJ 1990, 301, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Mairer, K.; Wille, M.; Bucher, T.; Burtscher, M. Prevalence of acute mountain sickness in the Eastern Alps. High Alt. Med. Biol. 2009, 10, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Richalet, J.P.; Larmignat, P.; Poitrine, E.; Letournel, M.; Canouï-Poitrine, F. Physiological risk factors for severe high-altitude illness: A prospective cohort study. Am. J. Respir. Crit. Care Med. 2012, 185, 192–198. [Google Scholar] [CrossRef]
- Burtscher, M.; Wille, M.; Menz, V.; Faulhaber, M.; Gatterer, H. Symptom progression in acute mountain sickness during a 12-hour exposure to normobaric hypoxia equivalent to 4500 m. High Alt. Med. Biol. 2014, 15, 446–451. [Google Scholar] [CrossRef]
- Wu, T.; Ding, S.; Liu, J.; Jia, J.; Dai, R.; Liang, B.; Zhao, J.; Qi, D. Ataxia: An early indicator in high altitude cerebral edema. High Alt. Med. Biol. 2006, 7, 275–280. [Google Scholar] [CrossRef]
- Davis, C.; Hackett, P. Advances in the prevention and treatment of high altitude illness. Emerg. Med. Clin. N. Am. 2017, 35, 241–260. [Google Scholar] [CrossRef]
- Roach, R.C.; Bärtsch, P.; Hackett, P.H.; Oelz, O. The Lake Louise acute mountain sickness scoring system. In Hypoxia and Molecular Medicine, Sutton, J.R., Coates, G., Eds.; Queen City Press: Burlington, NJ, USA, 1993; pp. 272–274. [Google Scholar]
- Roach, R.C.; Hackett, P.H.; Oelz, O.; Bärtsch, P.; Luks, A.M.; MacInnis, M.J.; Baillie, J.K.; Committee, L.L.A.S.C. The 2018 Lake Louise acute mountain sickness score. High Alt. Med. Biol. 2018, 19, 4–6. [Google Scholar] [CrossRef]
- Sagoo, R.S.; Hutchinson, C.E.; Wright, A.; Handford, C.; Parsons, H.; Sherwood, V.; Wayte, S.; Nagaraja, S.; Ng’Andwe, E.; Wilson, M.H.; et al. Magnetic Resonance investigation into the mechanisms involved in the development of high-altitude cerebral edema. J. Cereb. Blood Flow Metab. 2017, 37, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Kallenberg, K.; Bailey, D.M.; Christ, S.; Mohr, A.; Roukens, R.; Menold, E.; Steiner, T.; Bärtsch, P.; Knauth, M. Magnetic resonance imaging evidence of cytotoxic cerebral edema in acute mountain sickness. J. Cereb. Blood Flow Metab. 2007, 27, 1064–1071. [Google Scholar] [CrossRef]
- Schoonman, G.G.; Sándor, P.S.; Nirkko, A.C.; Lange, T.; Jaermann, T.; Dydak, U.; Kremer, C.; Ferrari, M.D.; Boesiger, P.; Baumgartner, R.W. Hypoxia-induced acute mountain sickness is associated with intracellular cerebral edema: A 3 T magnetic resonance imaging study. J. Cereb. Blood Flow Metab. 2008, 28, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Sanchez del Rio, M.; Moskowitz, M.A. High altitude headache. Lessons from headaches at sea level. Adv. Exp. Med. Biol. 1999, 474, 145–153. [Google Scholar] [PubMed]
- Serrano-Dueñas, M. High-altitude headache. Expert Rev. Neurother. 2007, 7, 245–248. [Google Scholar] [CrossRef]
- Burtscher, M.; Mairer, K.; Wille, M.; Broessner, G. Risk factors for high-altitude headache in mountaineers. Cephalalgia 2011, 31, 706–711. [Google Scholar] [CrossRef]
- Gallagher, S.A.; Hackett, P.H. High-altitude illness. Emerg. Med. Clin. N. Am. 2004, 22, 329–355. [Google Scholar] [CrossRef]
- Lafuente, J.V.; Bermudez, G.; Camargo-Arce, L.; Bulnes, S. Blood-brain barrier changes in high altitude. CNS Neurol. Disord. Drug Targets 2016, 15, 1188–1197. [Google Scholar] [CrossRef]
- Bärtsch, P.; Swenson, E.R. Clinical practice: Acute high-altitude illnesses. N. Engl. J. Med. 2013, 368, 2294–2302. [Google Scholar] [CrossRef]
- Swenson, E.R.; Bärtsch, P. High-altitude pulmonary edema. Compr. Physiol. 2012, 2, 2753–2773. [Google Scholar] [CrossRef]
- Eichstaedt, C.A.; Mairbäurl, H.; Song, J.; Benjamin, N.; Fischer, C.; Dehnert, C.; Schommer, K.; Berger, M.M.; Bärtsch, P.; Grünig, E.; et al. Genetic predisposition to high-altitude pulmonary edema. High Alt. Med. Biol. 2020, 21, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, U.; Turini, P.; Thalmann, S.; Hutter, D.; Salmon, C.S.; Stuber, T.; Shaw, S.; Jayet, P.Y.; Sartori-Cucchial, C.; Villena, M.; et al. Pulmonary hypertension in high-altitude dwellers: Novel mechanisms, unsuspected predisposing factors. Adv. Exp. Med. Biol. 2006, 588, 277–291. [Google Scholar] [CrossRef]
- El Alam, S.; Pena, E.; Aguilera, D.; Siques, P.; Brito, J. Inflammation in pulmonary hypertension and edema induced by hypobaric hypoxia exposure. Int. J. Mol. Sci. 2022, 23, 2656. [Google Scholar] [CrossRef]
- Oelz, O.; Maggiorini, M.; Ritter, M.; Noti, C.; Waber, U.; Vock, P.; Bärtsch, P. Prevention and treatment of high altitude pulmonary edema by a calcium channel blocker. Int. J. Sports Med. 1992, 13 (Suppl. S1), S65–S68. [Google Scholar] [CrossRef]
- Deshwal, R.; Iqbal, M.; Basnet, S. Nifedipine for the treatment of high altitude pulmonary edema. Wilderness Environ. Med. 2012, 23, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Boggild, A.K.; Costiniuk, C.; Kain, K.C.; Pandey, P. Environmental hazards in Nepal: Altitude illness, environmental exposures, injuries, and bites in travelers and expatriates. J. Travel Med. 2007, 14, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Nerín, M.A.; Palop, J.; Montaño, J.A.; Morandeira, J.R.; Vázquez, M. Acute mountain sickness: Influence of fluid intake. Wilderness Environ. Med. 2006, 17, 215–220. [Google Scholar] [CrossRef]
- Whayne, T.F., Jr. Altitude and cold weather: Are they vascular risks? Curr. Opin. Cardiol. 2014, 29, 396–402. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Chen, C.C.; Lin, S.H. Case report: Severe rhabdomyolysis and acute liver injury in a high-altitude mountain climber. Front. Med. 2022, 9, 917355. [Google Scholar] [CrossRef]
- Roach, R.C.; Maes, D.; Sandoval, D.; Robergs, R.A.; Icenogle, M.; Hinghofer-Szalkay, H.; Lium, D.; Loeppky, J.A. Exercise exacerbates acute mountain sickness at simulated high altitude. J. Appl. Physiol. 2000, 88, 581–585. [Google Scholar] [CrossRef]
- Schoene, R.B.; Roach, R.C.; Hackett, P.H.; Harrison, G.; Mills, W.J. High altitude pulmonary edema and exercise at 4400 meters on mount McKinley. Effect of expiratory positive airway pressure. Chest 1985, 87, 330–333. [Google Scholar] [CrossRef]
- Swenson, E.R. Early hours in the development of high-altitude pulmonary edema: Time course and mechanisms. J. Appl. Physiol. 2020, 128, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Faiss, R.; Pialoux, V.; Sartori, C.; Faes, C.; Dériaz, O.; Millet, G.P. Ventilation, oxidative stress, and nitric oxide in hypobaric versus normobaric hypoxia. Med. Sci. Sports Exerc. 2013, 45, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Pialoux, V.; Mounier, R.; Brown, A.D.; Steinback, C.D.; Rawling, J.M.; Poulin, M.J. Relationship between oxidative stress and HIF-1 alpha mRNA during sustained hypoxia in humans. Free Radic. Biol. Med. 2009, 46, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Quindry, J.; Dumke, C.; Slivka, D.; Ruby, B. Impact of extreme exercise at high altitude on oxidative stress in humans. J. Physiol. 2016, 594, 5093–5104. [Google Scholar] [CrossRef]
- Vij, A.G.; Dutta, R.; Satija, N.K. Acclimatization to oxidative stress at high altitude. High Alt. Med. Biol. 2005, 6, 301–310. [Google Scholar] [CrossRef]
- Horscroft, J.A.; Kotwica, A.O.; Laner, V.; West, J.A.; Hennis, P.J.; Levett, D.Z.H.; Howard, D.J.; Fernandez, B.O.; Burgess, S.L.; Ament, Z.; et al. Metabolic basis to Sherpa altitude adaptation. Proc. Natl. Acad. Sci. USA 2017, 114, 6382–6387. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef]
- Paddenberg, R.; Ishaq, B.; Goldenberg, A.; Faulhammer, P.; Rose, F.; Weissmann, N.; Braun-Dullaeus, R.C.; Kummer, W. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2003, 284, L710–L719. [Google Scholar] [CrossRef]
- Rathore, R.; Zheng, Y.-M.; Niu, C.-F.; Liu, Q.-H.; Korde, A.; Ho, Y.-S.; Wang, Y.-X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.; Mamary, A.J.; Verhoeven, A.J.; Marshall, B.E. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am. J. Respir. Cell Mol. Biol. 1996, 15, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Dawson, V.L.; Dawson, T.M.; Snyder, S.H.; Zweier, J.L. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc. Natl. Acad. Sci. USA 1996, 93, 6770–6774. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Vivar, J.; Martásek, P.; Whitsett, J.; Joseph, J.; Kalyanaraman, B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: An EPR spin trapping study. Biochem. J. 2002, 362, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, L.; Stankevicius, E.; Andersen, M.R.; Eskildsen-Helmond, Y.; Ledet, T.; Mulvany, M.J.; Simonsen, U. Diminished NO release in chronic hypoxic human endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2894–H2903. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef]
- Gao, L.; Ortega-Sáenz, P.; Moreno-Domínguez, A.; López-Barneo, J. Mitochondrial Redox Signaling in O(2)-sensing chemoreceptor cells. Antioxid. Redox Signal. 2022, 37, 274–289. [Google Scholar] [CrossRef]
- Fernández-Agüera, M.C.; Gao, L.; González-Rodríguez, P.; Pintado, C.O.; Arias-Mayenco, I.; García-Flores, P.; García-Pergañeda, A.; Pascual, A.; Ortega-Sáenz, P.; López-Barneo, J. Oxygen sensing by arterial chemoreceptors depends on mitochondrial complex I signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef]
- Ortega-Sáenz, P.; Pardal, R.; García-Fernandez, M.; López-Barneo, J. Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. J. Physiol. 2003, 548, 789–800. [Google Scholar] [CrossRef]
- Swiderska, A.; Coney, A.M.; Alzahrani, A.A.; Aldossary, H.S.; Batis, N.; Ray, C.J.; Kumar, P.; Holmes, A.P. Mitochondrial succinate metabolism and reactive oxygen species are important but not essential for eliciting carotid body and ventilatory responses to hypoxia in the rat. Antioxidants 2021, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.M.; Taudorf, S.; Berg, R.M.; Lundby, C.; McEneny, J.; Young, I.S.; Evans, K.A.; James, P.E.; Shore, A.; Hullin, D.A.; et al. Increased cerebral output of free radicals during hypoxia: Implications for acute mountain sickness? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1283–R1292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, Z.; Yang, F.; Ji, L.; Yang, Y.; Liu, C.; Liu, H.; Ma, J.; Liu, J.; Dang, Z.; et al. Novel insights into plasma biomarker candidates in patients with chronic mountain sickness based on proteomics. Biosci. Rep. 2021, 41, BSR20202219. [Google Scholar] [CrossRef] [PubMed]
- Sarada, S.; Himadri, P.; Mishra, C.; Geetali, P.; Ram, M.S.; Ilavazhagan, G. Role of oxidative stress and NFkB in hypoxia-induced pulmonary edema. Exp. Biol. Med. 2008, 233, 1088–1098. [Google Scholar] [CrossRef]
- Lüneburg, N.; Siques, P.; Brito, J.; Arriaza, K.; Pena, E.; Klose, H.; Leon-Velarde, F.; Böger, R.H. Long-term chronic intermittent hypobaric hypoxia in rats causes an imbalance in the asymmetric dimethylarginine/nitric oxide pathway and ROS activity: A possible synergistic mechanism for altitude pulmonary hypertension? Pulm. Med. 2016, 2016, 6578578. [Google Scholar] [CrossRef] [PubMed]
- Irarrázaval, S.; Allard, C.; Campodónico, J.; Pérez, D.; Strobel, P.; Vásquez, L.; Urquiaga, I.; Echeverría, G.; Leighton, F. Oxidative stress in acute hypobaric hypoxia. High Alt. Med. Biol. 2017, 18, 128–134. [Google Scholar] [CrossRef]
- Bailey, D.M.; Rimoldi, S.F.; Rexhaj, E.; Pratali, L.; Salinas Salmòn, C.; Villena, M.; McEneny, J.; Young, I.S.; Nicod, P.; Allemann, Y.; et al. Oxidative-nitrosative stress and systemic vascular function in highlanders with and without exaggerated hypoxemia. Chest 2013, 143, 444–451. [Google Scholar] [CrossRef]
- Pena, E.; El Alam, S.; Siques, P.; Brito, J. Oxidative stress and diseases associated with high-altitude exposure. Antioxidants 2022, 11, 267. [Google Scholar] [CrossRef]
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Siques, P.; López de Pablo, A.L.; Brito, J.; Arribas, S.M.; Flores, K.; Arriaza, K.; Naveas, N.; González, M.C.; Hoorntje, A.; León-Velarde, F.; et al. Nitric oxide and superoxide anion balance in rats exposed to chronic and long term intermittent hypoxia. Biomed. Res. Int. 2014, 2014, 610474. [Google Scholar] [CrossRef]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol. 2011, 31, 3531–3545. [Google Scholar] [CrossRef] [PubMed]
- Siques, P.; Brito, J.; Pena, E. Reactive oxygen species and pulmonary vasculature during hypobaric hypoxia. Front. Physiol. 2018, 9, 865. [Google Scholar] [CrossRef] [PubMed]
- Himadri, P.; Kumari, S.S.; Chitharanjan, M.; Dhananjay, S. Role of oxidative stress and inflammation in hypoxia-induced cerebral edema: A molecular approach. High Alt. Med. Biol. 2010, 11, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Yin, L.; Yuan, L.; Sui, D.; Sun, Y.; Fu, H.; Chen, L.; Wang, X. Ganglioside GM1 protects against high altitude cerebral edema in rats by suppressing the oxidative stress and inflammatory response via the PI3K/AKT-Nrf2 pathway. Mol. Immunol. 2018, 95, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free. Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Taylor, C.T. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 2008, 409, 19–26. [Google Scholar] [CrossRef]
- Martínez-Ruiz, A. Post-translational nitric oxide-dependent modifications in immune system. Redox Biol. 2015, 5, 418–419. [Google Scholar] [CrossRef]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Shvetsova, A.N.; Mennerich, D.; Kerätär, J.M.; Hiltunen, J.K.; Kietzmann, T. Non-electron transfer chain mitochondrial defects differently regulate HIF-1α degradation and transcription. Redox Biol. 2017, 12, 1052–1061. [Google Scholar] [CrossRef]
- Pan, Y.; Mansfield, K.D.; Bertozzi, C.C.; Rudenko, V.; Chan, D.A.; Giaccia, A.J.; Simon, M.C. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell. Biol. 2007, 27, 912–925. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Wittig, I.; Brüne, B. TMEM126B deficiency reduces mitochondrial SDH oxidation by LPS, attenuating HIF-1α stabilization and IL-1β expression. Redox Biol. 2019, 20, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Lukyanova, L.D.; Dudchenko, A.M.; Tsybina, T.A.; Germanova, E.L.; Tkachuk, E.N.; Erenburg, I.V. Effect of intermittent normobaric hypoxia on kinetic properties of mitochondrial enzymes. Bull. Exp. Biol. Med. 2007, 144, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Goyal, P.; Weissmann, N.; Grimminger, F.; Hegel, C.; Bader, L.; Rose, F.; Fink, L.; Ghofrani, H.A.; Schermuly, R.T.; Schmidt, H.H.; et al. Upregulation of NAD(P)H oxidase 1 in hypoxia activates hypoxia-inducible factor 1 via increase in reactive oxygen species. Free. Radic. Biol. Med. 2004, 36, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Emerling, B.M.; Chandel, N.S. Mitochondrial regulation of oxygen sensing. Mitochondrion 2005, 5, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free. Radic. Biol. Med. 2019, 133, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wu, W.S.; Su, L.; Zheng, X.; Wu, W.Y.; Santambrogio, P.; Gou, Y.J.; Hao, Q.; Wang, P.N.; Li, Y.R.; et al. Mitochondrial ferritin is a hypoxia-inducible factor 1α-inducible gene that protects from hypoxia-induced cell death in brain. Antioxid. Redox Signal. 2019, 30, 198–212. [Google Scholar] [CrossRef]
- Reddy, N.M.; Potteti, H.R.; Vegiraju, S.; Chen, H.J.; Tamatam, C.M.; Reddy, S.P. PI3K-AKT Signaling via Nrf2 Protects against Hyperoxia-Induced Acute Lung Injury, but Promotes Inflammation Post-Injury Independent of Nrf2 in Mice. PLoS ONE 2015, 10, e0129676. [Google Scholar] [CrossRef]
- Wang, L.; Yang, S.; Yan, L.; Wei, H.; Wang, J.; Yu, S.; Kong, A.T.; Zhang, Y. Hypoxia preconditioning promotes endurance exercise capacity of mice by activating skeletal muscle Nrf2. J. Appl. Physiol. 2019, 127, 1267–1277. [Google Scholar] [CrossRef]
- Syapin, P. Regulation of haeme oxygenase-1 for treatment of neuroinflammation and brain disorders. Br. J. Pharmacol. 2008, 155, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Inose, Y.; Izumi, Y.; Takada-Takatori, Y.; Akaike, A.; Koyama, Y.; Kaneko, S.; Kume, T. Protective effects of Nrf2–ARE activator on dopaminergic neuronal loss in Parkinson disease model mice: Possible involvement of heme oxygenase-1. Neurosci. Lett. 2020, 736, 135268. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial quality control: From molecule to organelle. Cell. Mol. Life Sci. 2021, 78, 3853–3866. [Google Scholar] [CrossRef]
- Green, H.J.; Sutton, J.R.; Wolfel, E.E.; Reeves, J.T.; Butterfield, G.E.; Brooks, G.A. Altitude acclimatization and energy metabolic adaptations in skeletal muscle during exercise. J. Appl. Physiol. 1992, 73, 2701–2708. [Google Scholar] [CrossRef]
- Gnaiger, E.; Méndez, G.; Hand, S.C. High phosphorylation efficiency and depression of uncoupled respiration in mitochondria under hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 11080–11085. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef]
- Thomas, L.W.; Ashcroft, M. Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell. Mol. Life Sci. 2019, 76, 1759–1777. [Google Scholar] [CrossRef]
- Hayashi, T.; Asano, Y.; Shintani, Y.; Aoyama, H.; Kioka, H.; Tsukamoto, O.; Hikita, M.; Shinzawa-Itoh, K.; Takafuji, K.; Higo, S. Higd1a is a positive regulator of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 1553–1558. [Google Scholar] [CrossRef]
- Lendahl, U.; Lee, K.L.; Yang, H.; Poellinger, L. Generating specificity and diversity in the transcriptional response to hypoxia. Nat. Rev. Genet. 2009, 10, 821–832. [Google Scholar] [CrossRef]
- González, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The yin and yang of cellular nutrient sensing and growth control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Zhang, Y.-Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.C.; Kuppusamy, P.; Parinandi, N. Oxygen, the lead actor in the pathophysiologic drama: Enactment of the trinity of normoxia, hypoxia, and hyperoxia in disease and therapy. Antioxid. Redox Signal. 2007, 9, 1717–1730. [Google Scholar] [CrossRef]
- Yakes, F.M.; van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Samanta, D.; Semenza, G.L. Maintenance of redox homeostasis by hypoxia-inducible factors. Redox Biol. 2017, 13, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Ding, K.; Oktay, Y.; Gaur, A.; Thurmond, F.; Yan, L.J.; Marck, B.T.; Matsumoto, A.M.; Shelton, J.M.; Richardson, J.A.; et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat. Genet. 2003, 35, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M. Heme oxygenase-1: Molecular mechanisms of gene expression in oxygen-related stress. Antioxid. Redox Signal. 2002, 4, 625–632. [Google Scholar] [CrossRef]
- Liu, X.; Hajnoczky, G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia–reoxygenation stress. Cell Death Differ. 2011, 18, 1561–1572. [Google Scholar] [CrossRef]
- Al-Mehdi, A.B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012, 5, ra47. [Google Scholar] [CrossRef]
- Levett, D.Z.; Radford, E.J.; Menassa, D.A.; Graber, E.F.; Morash, A.J.; Hoppeler, H.; Clarke, K.; Martin, D.S.; Ferguson-Smith, A.C.; Montgomery, H.E.; et al. Acclimatization of skeletal muscle mitochondria to high-altitude hypoxia during an ascent of Everest. FASEB J. 2012, 26, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Paul, S.; Gangwar, A.; Bhargava, K.; Ahmad, Y. STAT3-RXR-Nrf2 activates systemic redox and energy homeostasis upon steep decline in pO. Redox Biol. 2018, 14, 423–438. [Google Scholar] [CrossRef]
- Yang, J.; Jia, Z.; Song, X.; Shi, J.; Wang, X.; Zhao, X.; He, K. Proteomic and clinical biomarkers for acute mountain sickness in a longitudinal cohort. Commun. Biol. 2022, 5, 548. [Google Scholar] [CrossRef] [PubMed]
- Chanana, N.; Palmo, T.; Newman, J.H.; Pasha, M.A.Q. Vascular homeostasis at high-altitude: Role of genetic variants and transcription factors. Pulm. Circ. 2020, 10, 2045894020913475. [Google Scholar] [CrossRef] [PubMed]
- Pasha, M.A.; Newman, J.H. High-altitude disorders: Pulmonary hypertension: Pulmonary vascular disease: The global perspective. Chest 2010, 137, 13s–19s. [Google Scholar] [CrossRef]
- Lorenzo, V.F.; Yang, Y.; Simonson, T.S.; Nussenzveig, R.; Jorde, L.B.; Prchal, J.T.; Ge, R.L. Genetic adaptation to extreme hypoxia: Study of high-altitude pulmonary edema in a three-generation Han Chinese family. Blood Cells Mol. Dis. 2009, 43, 221–225. [Google Scholar] [CrossRef] [PubMed]
- MacInnis, M.J.; Koehle, M.S. Evidence for and against genetic predispositions to acute and chronic altitude illnesses. High Alt. Med. Biol. 2016, 17, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, A.; Charu, R.; Pasha, M.A.; Norboo, T.; Afrin, F.; Baig, M.A. eNOS allelic variants at the same locus associate with HAPE and adaptation. Thorax 2004, 59, 1000–1002. [Google Scholar] [CrossRef]
- Ahsan, A.; Norboo, T.; Baig, M.A.; Qadar Pasha, M.A. Simultaneous selection of the wild-type genotypes of the G894T and 4B/4A polymorphisms of NOS3 associate with high-altitude adaptation. Ann. Hum. Genet. 2005, 69, 260–267. [Google Scholar] [CrossRef]
- Droma, Y.; Hanaoka, M.; Basnyat, B.; Arjyal, A.; Neupane, P.; Pandit, A.; Sharma, D.; Miwa, N.; Ito, M.; Katsuyama, Y.; et al. Genetic contribution of the endothelial nitric oxide synthase gene to high altitude adaptation in sherpas. High Alt. Med. Biol. 2006, 7, 209–220. [Google Scholar] [CrossRef]
- Erzurum, S.C.; Ghosh, S.; Janocha, A.J.; Xu, W.; Bauer, S.; Bryan, N.S.; Tejero, J.; Hemann, C.; Hille, R.; Stuehr, D.J.; et al. Higher blood flow and circulating NO products offset high-altitude hypoxia among Tibetans. Proc. Natl. Acad. Sci. USA 2007, 104, 17593–17598. [Google Scholar] [CrossRef]
- Snyder, E.M.; Beck, K.C.; Turner, S.T.; Hoffman, E.A.; Joyner, M.J.; Johnson, B.D. Genetic variation of the beta2-adrenergic receptor is associated with differences in lung fluid accumulation in humans. J. Appl. Physiol. 2007, 102, 2172–2178. [Google Scholar] [CrossRef]
- Stobdan, T.; Kumar, R.; Mohammad, G.; Thinlas, T.; Norboo, T.; Iqbal, M.; Pasha, M.A. Probable role of beta2-adrenergic receptor gene haplotype in high-altitude pulmonary oedema. Respirology 2010, 15, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Negi, S.; Jha, P.; Singh, P.K.; Stobdan, T.; Pasha, M.A.; Ghosh, S.; Agrawal, A.; Prasher, B.; Mukerji, M. EGLN1 involvement in high-altitude adaptation revealed through genetic analysis of extreme constitution types defined in Ayurveda. Proc. Natl. Acad. Sci. USA 2010, 107, 18961–18966. [Google Scholar] [CrossRef] [PubMed]
- Beall, C.M.; Cavalleri, G.L.; Deng, L.; Elston, R.C.; Gao, Y.; Knight, J.; Li, C.; Li, J.C.; Liang, Y.; McCormack, M.; et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc. Natl. Acad. Sci. USA 2010, 107, 11459–11464. [Google Scholar] [CrossRef]
- Simonson, T.S.; Yang, Y.; Huff, C.D.; Yun, H.; Qin, G.; Witherspoon, D.J.; Bai, Z.; Lorenzo, F.R.; Xing, J.; Jorde, L.B.; et al. Genetic evidence for high-altitude adaptation in Tibet. Science 2010, 329, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Bigham, A.; Bauchet, M.; Pinto, D.; Mao, X.; Akey, J.M.; Mei, R.; Scherer, S.W.; Julian, C.G.; Wilson, M.J.; López Herráez, D.; et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010, 6, e1001116. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Mohammad, G.; Thinlas, T.; Pasha, M.A. EGLN1 variants influence expression and SaO2 levels to associate with high-altitude pulmonary oedema and adaptation. Clin. Sci. 2013, 124, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, F.R.; Huff, C.; Myllymäki, M.; Olenchock, B.; Swierczek, S.; Tashi, T.; Gordeuk, V.; Wuren, T.; Ri-Li, G.; McClain, D.A.; et al. A genetic mechanism for Tibetan high-altitude adaptation. Nat. Genet. 2014, 46, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Li, L.S.; Arsenault, P.R.; Tan, Q.; Bigham, A.W.; Heaton-Johnson, K.J.; Master, S.R.; Lee, F.S. Defective Tibetan PHD2 binding to p23 links high altitude adaption to altered oxygen sensing. J. Biol. Chem. 2014, 289, 14656–14665. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.D.; Qiu, Y.Q.; Xu, J.; Irwin, D.M.; Tam, S.C.; Tang, N.L.; Zhang, Y.P. Genetic adaptation of the hypoxia-inducible factor pathway to oxygen pressure among eurasian human populations. Mol. Biol. Evol. 2012, 29, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Shen, Y.; Liu, C.; Yang, J.; Yang, Y.Q.; Zhang, C.; Bian, S.Z.; Yu, J.; Gao, X.B.; Zhang, L.P.; et al. EPAS1 and VEGFA gene variants are related to the symptoms of acute mountain sickness in Chinese Han population: A cross-sectional study. Mil. Med. Res. 2020, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zeng, Y.; Chen, G.; Bian, S.; Qiu, Y.; Liu, X.; Xu, B.; Song, P.; Zhang, J.; Qin, J.; et al. Analysis of high-altitude syndrome and the underlying gene polymorphisms associated with acute mountain sickness after a rapid ascent to high-altitude. Sci. Rep. 2016, 6, 38323. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.I.; Zhang, J.; Jin, J.; Gao, X.; Yu, J.; Geng, Q.; Li, H.; Huang, L. Genetic variants of endothelial PAS domain protein 1 are associated with susceptibility to acute mountain sickness in individuals unaccustomed to high altitude: A nested case-control study. Exp. Ther. Med. 2015, 10, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Zhang, J.; Jin, J.; Qin, J.; Li, H.; Huang, L. Variants of the low oxygen sensors EGLN1 and HIF-1AN associated with acute mountain sickness. Int. J. Mol. Sci. 2014, 15, 21777–21787. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Ali, Z.; Vibhuti, A.; Kumar, R.; Alam, P.; Ram, R.; Thinlas, T.; Mohammad, G.; Pasha, M.A. CYBA and GSTP1 variants associate with oxidative stress under hypobaric hypoxia as observed in high-altitude pulmonary oedema. Clin. Sci. 2012, 122, 299–309. [Google Scholar] [CrossRef]
- Beall, C.M. Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc. Natl. Acad. Sci. USA 2007, 104 (Suppl. S1), 8655–8660. [Google Scholar] [CrossRef] [PubMed]
- Simonson, T.S. Altitude adaptation: A glimpse through various lenses. High Alt. Med. Biol. 2015, 16, 125–137. [Google Scholar] [CrossRef]
- Bigham, A.W. Genetics of human origin and evolution: High-altitude adaptations. Curr. Opin. Genet. Dev. 2016, 41, 8–13. [Google Scholar] [CrossRef] [PubMed]
- West, J.B. Rate of ventilatory acclimatization to extreme altitude. Respir. Physiol. 1988, 74, 323–333. [Google Scholar] [CrossRef]
- Schmidt, W.; Heinicke, K.; Rojas, J.; Manuel Gomez, J.; Serrato, M.; Mora, M.; Wolfarth, B.; Schmid, A.; Keul, J. Blood volume and hemoglobin mass in endurance athletes from moderate altitude. Med. Sci. Sports Exerc. 2002, 34, 1934–1940. [Google Scholar] [CrossRef]
- Hauser, A.; Schmitt, L.; Troesch, S.; Saugy, J.J.; Cejuela-Anta, R.; Faiss, R.; Robinson, N.; Wehrlin, J.P.; Millet, G.P. Similar Hemoglobin Mass Response in Hypobaric and Normobaric Hypoxia in Athletes. Med. Sci. Sports Exerc. 2016, 48, 734–741. [Google Scholar] [CrossRef]
- Lyons, T.P.; Muza, S.R.; Rock, P.B.; Cymerman, A. The effect of altitude pre-acclimatization on acute mountain sickness during reexposure. Aviat. Space Environ. Med. 1995, 66, 957–962. [Google Scholar] [PubMed]
- Beidleman, B.A.; Fulco, C.S.; Cadarette, B.S.; Cymerman, A.; Buller, M.J.; Salgado, R.M.; Posch, A.M.; Staab, J.E.; Sils, I.V.; Yurkevicius, B.R.; et al. Is normobaric hypoxia an effective treatment for sustaining previously acquired altitude acclimatization? J. Appl. Physiol. 2017, 123, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Pamenter, M.E.; Powell, F.L. Time domains of the hypoxic ventilatory response and their molecular basis. Compr. Physiol. 2016, 6, 1345–1385. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Zhang, Y.; Han, L.; Yegutkin, G.G.; Liu, H.; Sun, K.; D’Alessandro, A.; Li, J.; Karmouty-Quintana, H.; Iriyama, T.; et al. Erythrocytes retain hypoxic adenosine response for faster acclimatization upon re-ascent. Nat. Commun. 2017, 8, 14108. [Google Scholar] [CrossRef] [PubMed]
- Forster, H.V. Plasticity in the control of breathing following sensory denervation. J. Appl. Physiol. 2003, 94, 784–794. [Google Scholar] [CrossRef]
- McGregor, K.H.; Gil, J.; Lahiri, S. A morphometric study of the carotid body in chronically hypoxic rats. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 57, 1430–1438. [Google Scholar] [CrossRef]
- Powell, F.L.; Fu, Z. HIF-1 and ventilatory acclimatization to chronic hypoxia. Respir. Physiol. Neurobiol. 2008, 164, 282–287. [Google Scholar] [CrossRef]
- Vizcardo-Galindo, G.; León-Velarde, F.; Villafuerte, F.C. High-Altitude Hypoxia Decreases Plasma Erythropoietin Soluble Receptor Concentration in Lowlanders. High Alt. Med. Biol. 2020, 21, 92–98. [Google Scholar] [CrossRef]
- Zon, L.I.; Youssoufian, H.; Mather, C.; Lodish, H.F.; Orkin, S.H. Activation of the erythropoietin receptor promoter by transcription factor GATA-1. Proc. Natl. Acad. Sci. USA 1991, 88, 10638–10641. [Google Scholar] [CrossRef]
- Reynafarje, C.; Lozano, R.; Valdivieso, J. The polycythemia of high altitudes: Iron metabolism and related aspects. Blood 1959, 14, 433–455. [Google Scholar] [CrossRef]
- Klein, M.; Kaestner, L.; Bogdanova, A.Y.; Minetti, G.; Rudloff, S.; Lundby, C.; Makhro, A.; Seiler, E.; van Cromvoirt, A.; Fenk, S.; et al. Absence of neocytolysis in humans returning from a 3-week high-altitude sojourn. Acta Physiol. 2021, 232, e13647. [Google Scholar] [CrossRef] [PubMed]
- Beidleman, B.A.; Muza, S.R.; Fulco, C.S.; Cymerman, A.; Ditzler, D.T.; Stulz, D.; Staab, J.E.; Robinson, S.R.; Skrinar, G.S.; Lewis, S.F.; et al. Intermittent altitude exposures improve muscular performance at 4300 m. J. Appl. Physiol. 2003, 95, 1824–1832. [Google Scholar] [CrossRef]
- Beidleman, B.A.; Muza, S.R.; Fulco, C.S.; Cymerman, A.; Sawka, M.N.; Lewis, S.F.; Skrinar, G.S. Seven intermittent exposures to altitude improves exercise performance at 4300 m. Med. Sci. Sports Exerc. 2008, 40, 141–148. [Google Scholar] [CrossRef]
- Fulco, C.S.; Muza, S.R.; Beidleman, B.; Jones, J.; Staab, J.; Rock, P.B.; Cymerman, A. Exercise performance of sea-level residents at 4300 m after 6 days at 2200 m. Aviat. Space Environ. Med. 2009, 80, 955–961. [Google Scholar] [CrossRef]
- Beidleman, B.A.; Muza, S.R.; Fulco, C.S.; Cymerman, A.; Ditzler, D.; Stulz, D.; Staab, J.E.; Skrinar, G.S.; Lewis, S.F.; Sawka, M.N. Intermittent altitude exposures reduce acute mountain sickness at 4300 m. Clin. Sci. 2004, 106, 321–328. [Google Scholar] [CrossRef]
- Furian, M.; Bitos, K.; Hartmann, S.E.; Muralt, L.; Lichtblau, M.; Bader, P.R.; Rawling, J.M.; Ulrich, S.; Poulin, M.J.; Bloch, K.E. Acute high altitude exposure, acclimatization and re-exposure on nocturnal breathing. Front. Physiol. 2022, 13, 965021. [Google Scholar] [CrossRef] [PubMed]
- Fulco, C.S.; Muza, S.R.; Beidleman, B.A.; Demes, R.; Staab, J.E.; Jones, J.E.; Cymerman, A. Effect of repeated normobaric hypoxia exposures during sleep on acute mountain sickness, exercise performance, and sleep during exposure to terrestrial altitude. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R428–R436. [Google Scholar] [CrossRef]
- Beidleman, B.A.; Muza, S.R.; Fulco, C.S.; Jones, J.E.; Lammi, E.; Staab, J.E.; Cymerman, A. Intermittent hypoxic exposure does not improve endurance performance at altitude. Med. Sci. Sports Exerc. 2009, 41, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Millet, G.P.; Faiss, R.; Pialoux, V. Evidence for differences between hypobaric and normobaric hypoxia is conclusive. Exerc. Sport Sci. Rev. 2013, 41, 133. [Google Scholar] [CrossRef] [PubMed]
- Millet, G.P.; Jornet, K. On Top to the top-acclimatization strategy for the “fastest known time” to Mount Everest. Int. J. Sports Physiol. Perform. 2019, 14, 1438–1441. [Google Scholar] [CrossRef]
- Katayama, K.; Fujita, H.; Sato, K.; Ishida, K.; Iwasaki, K.; Miyamura, M. Effect of a repeated series of intermittent hypoxic exposures on ventilatory response in humans. High Alt. Med. Biol. 2005, 6, 50–59. [Google Scholar] [CrossRef]
- Kayser, B. Nutrition and high altitude exposure. Int. J. Sport. Med. 1992, 13 (Suppl. S1), S129–S132. [Google Scholar] [CrossRef] [PubMed]
- Stellingwerff, T.; Peeling, P.; Garvican-Lewis, L.A.; Hall, R.; Koivisto, A.E.; Heikura, I.A.; Burke, L.M. Nutrition and altitude: Strategies to enhance adaptation, improve performance and maintain health: A narrative review. Sports Med. 2019, 49, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, G.E.; Gates, J.; Fleming, S.; Brooks, G.A.; Sutton, J.R.; Reeves, J.T. Increased energy intake minimizes weight loss in men at high altitude. J. Appl. Physiol. 1992, 72, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Meeuwsen, T.; Hendriksen, I.J.; Holewijn, M. Training-induced increases in sea-level performance are enhanced by acute intermittent hypobaric hypoxia. Eur. J. Appl. Physiol. Occup. Physiol. 2001, 84, 283–290. [Google Scholar] [CrossRef]
- Butterfield, G.E. Nutrient requirements at high altitude. Clin. Sports Med. 1999, 18, 607–621. [Google Scholar] [CrossRef]
- Roberts, A.C.; Butterfield, G.E.; Cymerman, A.; Reeves, J.T.; Wolfel, E.E.; Brooks, G.A. Acclimatization to 4300-m altitude decreases reliance on fat as a substrate. J. Appl. Physiol. 1996, 81, 1762–1771. [Google Scholar] [CrossRef]
- Roels, B.; Thomas, C.; Bentley, D.J.; Mercier, J.; Hayot, M.; Millet, G. Effects of intermittent hypoxic training on amino and fatty acid oxidative combustion in human permeabilized muscle fibers. J. Appl. Physiol. 2007, 102, 79–86. [Google Scholar] [CrossRef]
- Brooks, G.A.; Butterfield, G.E.; Wolfe, R.R.; Groves, B.M.; Mazzeo, R.S.; Sutton, J.R.; Wolfel, E.E.; Reeves, J.T. Increased dependence on blood glucose after acclimatization to 4300 m. J. Appl. Physiol. 1991, 70, 919–927. [Google Scholar] [CrossRef]
- Koivisto-Mork, A.E.; Paur, I.; Paulsen, G.; Garthe, I.; Raastad, T.; Bastani, N.E.; Blomhoff, R.; Bohn, S.K. Dietary adjustments to altitude training in elite endurance athletes; impact of a randomized clinical trial with antioxidant-rich foods. Front. Sports Act. Living 2020, 2, 106. [Google Scholar] [CrossRef]
- Tschop, M.; Strasburger, C.J.; Hartmann, G.; Biollaz, J.; Bartsch, P. Raised leptin concentrations at high altitude associated with loss of appetite. Lancet 1998, 352, 1119–1120. [Google Scholar] [CrossRef] [PubMed]
- Ainslie, P.N.; Lucas, S.J.; Fan, J.L.; Thomas, K.N.; Cotter, J.D.; Tzeng, Y.C.; Burgess, K.R. Influence of sympathoexcitation at high altitude on cerebrovascular function and ventilatory control in humans. J. Appl. Physiol. 2012, 113, 1058–1067. [Google Scholar] [CrossRef]
- Farinelli, C.C.; Kayser, B.; Binzoni, T.; Cerretelli, P.; Girardier, L. Autonomic nervous control of heart rate at altitude (5050 m). Eur. J. Appl. Physiol. Occup. Physiol. 1994, 69, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L.; Bouthiaux, S.; Millet, G.P. Eleven years’ monitoring of the world’s most successful male biathlete of the last decade. Int. J. Sports Physiol. Perform. 2021, 16, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L.; Regnard, J.; Millet, G.P. Monitoring fatigue status with HRV measures in elite athletes: An avenue beyond RMSSD? Front. Physiol. 2015, 6, 343. [Google Scholar] [CrossRef]
- Schmitt, L.; Fouillot, J.P.; Millet, G.P.; Robach, P.; Nicolet, G.; Brugniaux, J.; Richalet, J.P. Altitude, heart rate variability and aerobic capacities. Int. J. Sports Med. 2008, 29, 300–306. [Google Scholar] [CrossRef]
- Perini, R.; Milesi, S.; Biancardi, L.; Veicsteinas, A. Effects of high altitude acclimatization on heart rate variability in resting humans. Eur. J. Appl. Physiol. Occup. Physiol. 1996, 73, 521–528. [Google Scholar] [CrossRef]
- Schmitt, L.; Willis, S.J.; Fardel, A.; Coulmy, N.; Millet, G.P. Live high-train low guided by daily heart rate variability in elite Nordic-skiers. Eur. J. Appl. Physiol. 2018, 118, 419–428. [Google Scholar] [CrossRef]
- West, J.B. Point: The lactate paradox does/does not occur during exercise at high altitude. J. Appl. Physiol. 2007, 102, 2398–2399. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Beatty, C.L.; Burelle, Y.; Trump, M.E.; McKenzie, D.C.; Matheson, G.O. The lactate paradox in human high-altitude physiological performance. News Physiol. Sci. 2002, 17, 122–126. [Google Scholar]
- Kayser, B. Lactate during exercise at high altitude. Eur. J. Appl. Physiol. Occup. Physiol. 1996, 74, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Millet, G.P.; Roels, B.; Schmitt, L.; Woorons, X.; Richalet, J.P. Combining hypoxic methods for peak performance. Sports Med. 2010, 40, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.; Prommer, N. The optimised CO-rebreathing method: A new tool to determine total haemoglobin mass routinely. Eur. J. Appl. Physiol. 2005, 95, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.; Troesch, S.; Steiner, T.; Brocherie, F.; Girard, O.; Saugy, J.J.; Schmitt, L.; Millet, G.P.; Wehrlin, J.P. Do male athletes with already high initial haemoglobin mass benefit from ‘live high-train low’ altitude training? Exp. Physiol. 2018, 103, 68–76. [Google Scholar] [CrossRef]
- Millet, G.P.; Chapman, R.F.; Girard, O.; Brocherie, F. Is live high-train low altitude training relevant for elite athletes? Flawed analysis from inaccurate data. Br. J. Sports Med. 2019, 53, 923–925. [Google Scholar] [CrossRef]
- Eastwood, A.; Sharpe, K.; Bourdon, P.C.; Woolford, S.M.; Saunders, P.U.; Robertson, E.Y.; Clark, S.A.; Gore, C.J. Within-subject variation in hemoglobin mass in elite athletes. Med. Sci. Sports Exerc. 2012, 44, 725–732. [Google Scholar] [CrossRef]
- Koivisto, A.E.; Olsen, T.; Paur, I.; Paulsen, G.; Bastani, N.E.; Garthe, I.; Raastad, T.; Matthews, J.; Blomhoff, R.; Bøhn, S.K. Effects of antioxidant-rich foods on altitude-induced oxidative stress and inflammation in elite endurance athletes: A randomized controlled trial. PLoS ONE 2019, 14, e0217895. [Google Scholar] [CrossRef]
- Govus, A.D.; Garvican-Lewis, L.A.; Abbiss, C.R.; Peeling, P.; Gore, C.J. Pre-altitude serum ferritin levels and daily oral iron supplement dose mediate iron parameter and hemoglobin mass responses to altitude exposure. PLoS ONE 2015, 10, e0135120. [Google Scholar] [CrossRef]
- Koivisto-Mork, A.E.; Svendsen, I.S.; Skattebo, O.; Hallen, J.; Paulsen, G. Impact of baseline serum ferritin and supplemental iron on altitude-induced hemoglobin mass response in elite athletes. Scand. J. Med. Sci. Sports 2021, 31, 1764–1773. [Google Scholar] [CrossRef]
- Foster, C.; Lucia, A. Running economy: The forgotten factor in elite performance. Sport. Med. 2007, 37, 316–319. [Google Scholar] [CrossRef]
- Saunders, P.U.; Telford, R.D.; Pyne, D.B.; Cunningham, R.B.; Gore, C.J.; Hahn, A.G.; Hawley, J.A. Improved running economy in elite runners after 20 days of simulated moderate-altitude exposure. J. Appl. Physiol. 2004, 96, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L.; Millet, G.; Robach, P.; Nicolet, G.; Brugniaux, J.V.; Fouillot, J.P.; Richalet, J.P. Influence of “living high-training low” on aerobic performance and economy of work in elite athletes. Eur. J. Appl. Physiol. 2006, 97, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Mallet, R.T.; Manukhina, E.B.; Ruelas, S.S.; Caffrey, J.L.; Downey, H.F. Cardioprotection by intermittent hypoxia conditioning: Evidence, mechanisms and therapeutic potential. Am. J. Physiol. Heart Circ. Physiol. 2018, 318, H332–H344. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Mallet, R.T.; Burtscher, M.; Millet, G.P. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing Res. Rev. 2021, 68, 101343. [Google Scholar] [CrossRef]
- Burtscher, J.; Niedermeier, M.; Hüfner, K.; van den Burg, E.; Kopp, M.; Stoop, R.; Burtscher, M.; Gatterer, H.; Millet, G.P. The interplay of hypoxic and mental stress: Implications for anxiety and depressive disorders. Neurosci. Biobehav. Rev. 2022, 138, 104718. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxygen-Sensing System | ||
|---|---|---|
| Gene | Single-Nucleotide Polymorphisms | Physiological Function |
| HIF-1α | rs11549465C/T, rs11549467A/G | Regulates oxygen transport and delivery, glycolysis and many more |
| EPAS1 | rs1868092A/G, 1447563C/A, rs11125075G/A, rs4953388A/G, rs4953396A/C, rs896210G/A, rs6735530C/T, rs6756667A/G | Expresses HIF-2α |
| EGLN1 | rs1538664G/A, rs479200C/T, rs2486729G/A, rs2790879T/G, rs480902T/C, rs2486736G/A, rs973252A/G, rs186996510C/G, rs12406290A/G, rs2153364A/G | Regulates HIF-1α by promoting its hydroxylation and degradation |
| EPO | rs1617640A/C | Activates erythrocyte production |
| Endothelial system | ||
| AGT | Rs699A/G, rs4762G/A | Angiotensinogen yields the precursor pentapeptide, a substrate for angiotensin converting enzyme (ACE) |
| ACE | I/D, 8066114C/G rs4461142T/C | ACE produces angiotensin II, a potent vasoconstrictor and activator of several signaling molecules, e.g., VEGF |
| CYP11B2 | rs1799998 or –344T/C rs4539 5160C Iw/Ic: Intron-2 conversion | Mediates production of aldosterone, which activates sodium retention in the alimentary tract and kidneys to expand the extracellular fluid volume |
| AGTR1 | Rs275651T/A, 275652T/G | Expresses a receptor that plays a major role in blood pressure homeostasis by governing angiotensin II signaling |
| EDN1 | rs10478694 or -3A/-4A rs2070699G/T, rs5370 | This gene produces a preproprotein that is cleaved to endothelin-1, a powerful vasoconstrictor |
| APLN | rs3761581T/G, rs2235312C/T, rs3115757C/G | Apelin stimulates NOS3 to produce NO |
| NOS3 | rs1799983G/T, 4b/4a, rs7830A/C | The enzyme produces NO, a powerful vasodilator |
| ADRB2 | rs1042713G/A rs1042714C/G rs1042711T/C | The protein stimulates vasodilatation. Several exonic SNPs have been reported |
| CYBA | rs9932581A/G or −930A/G rs4673C/T or H72Y | Mitochondrial respiratory complex component involved in ROS generation |
| GSTP1 | rs1695A/G or I105V rs1138272C/T or A114V | The enzyme scavenges ROS to maintain homeostasis |
| Vascular smooth muscle system | ||
| VEGF | rs3025039C/T | A factor promoting angiogenesis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallet, R.T.; Burtscher, J.; Pialoux, V.; Pasha, Q.; Ahmad, Y.; Millet, G.P.; Burtscher, M. Molecular Mechanisms of High-Altitude Acclimatization. Int. J. Mol. Sci. 2023, 24, 1698. https://doi.org/10.3390/ijms24021698
Mallet RT, Burtscher J, Pialoux V, Pasha Q, Ahmad Y, Millet GP, Burtscher M. Molecular Mechanisms of High-Altitude Acclimatization. International Journal of Molecular Sciences. 2023; 24(2):1698. https://doi.org/10.3390/ijms24021698
Chicago/Turabian StyleMallet, Robert T., Johannes Burtscher, Vincent Pialoux, Qadar Pasha, Yasmin Ahmad, Grégoire P. Millet, and Martin Burtscher. 2023. "Molecular Mechanisms of High-Altitude Acclimatization" International Journal of Molecular Sciences 24, no. 2: 1698. https://doi.org/10.3390/ijms24021698
APA StyleMallet, R. T., Burtscher, J., Pialoux, V., Pasha, Q., Ahmad, Y., Millet, G. P., & Burtscher, M. (2023). Molecular Mechanisms of High-Altitude Acclimatization. International Journal of Molecular Sciences, 24(2), 1698. https://doi.org/10.3390/ijms24021698