
Properties of Gaseous Deprotonated L-Cysteine S-Sulfate Anion [cysS-SO3]−: Intramolecular H-Bond Network, Electron Affinity, Chemically Active Site, and Vibrational Fingerprints

 ,
,  , ,
, ,
Abstract
1. Introduction
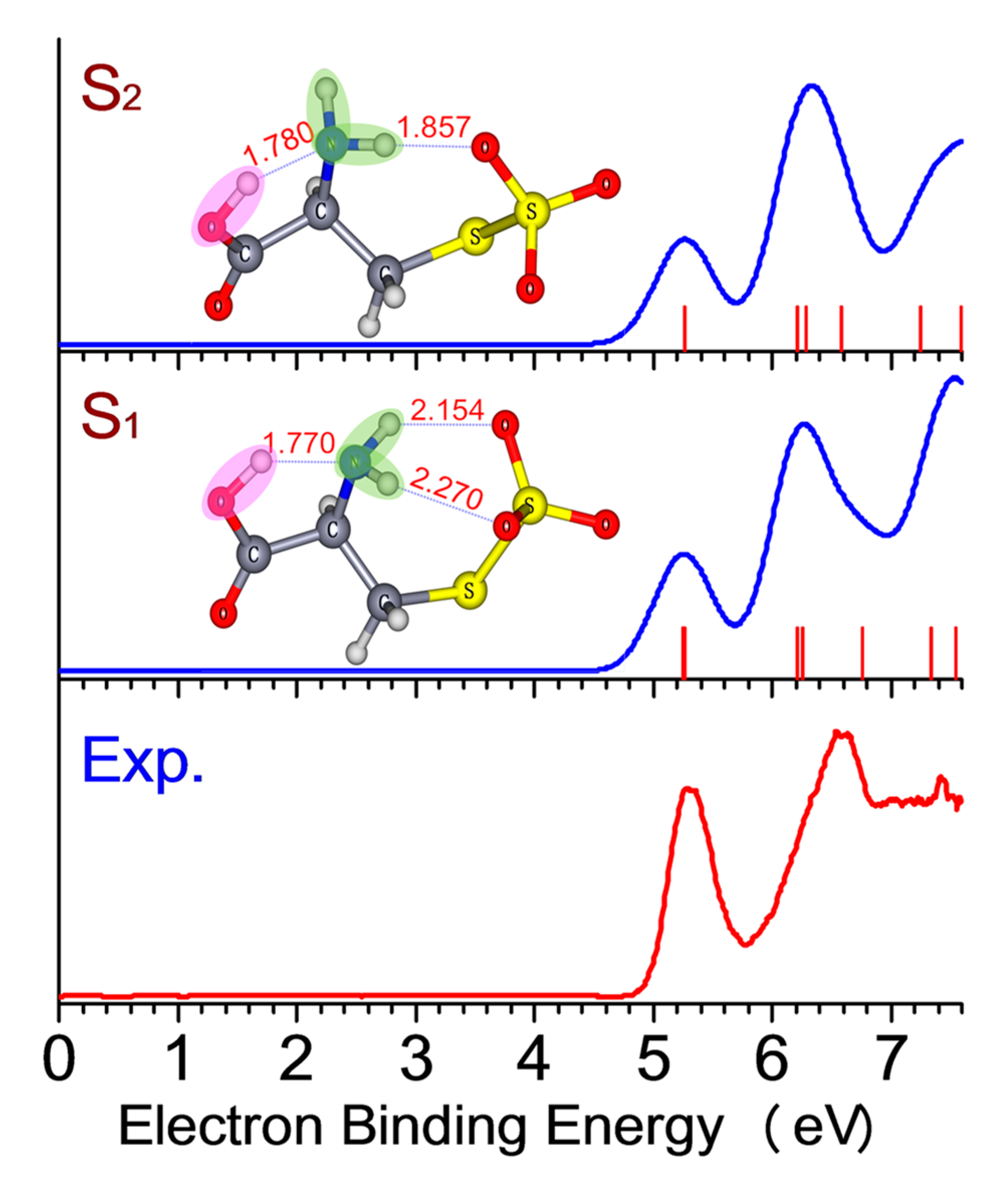
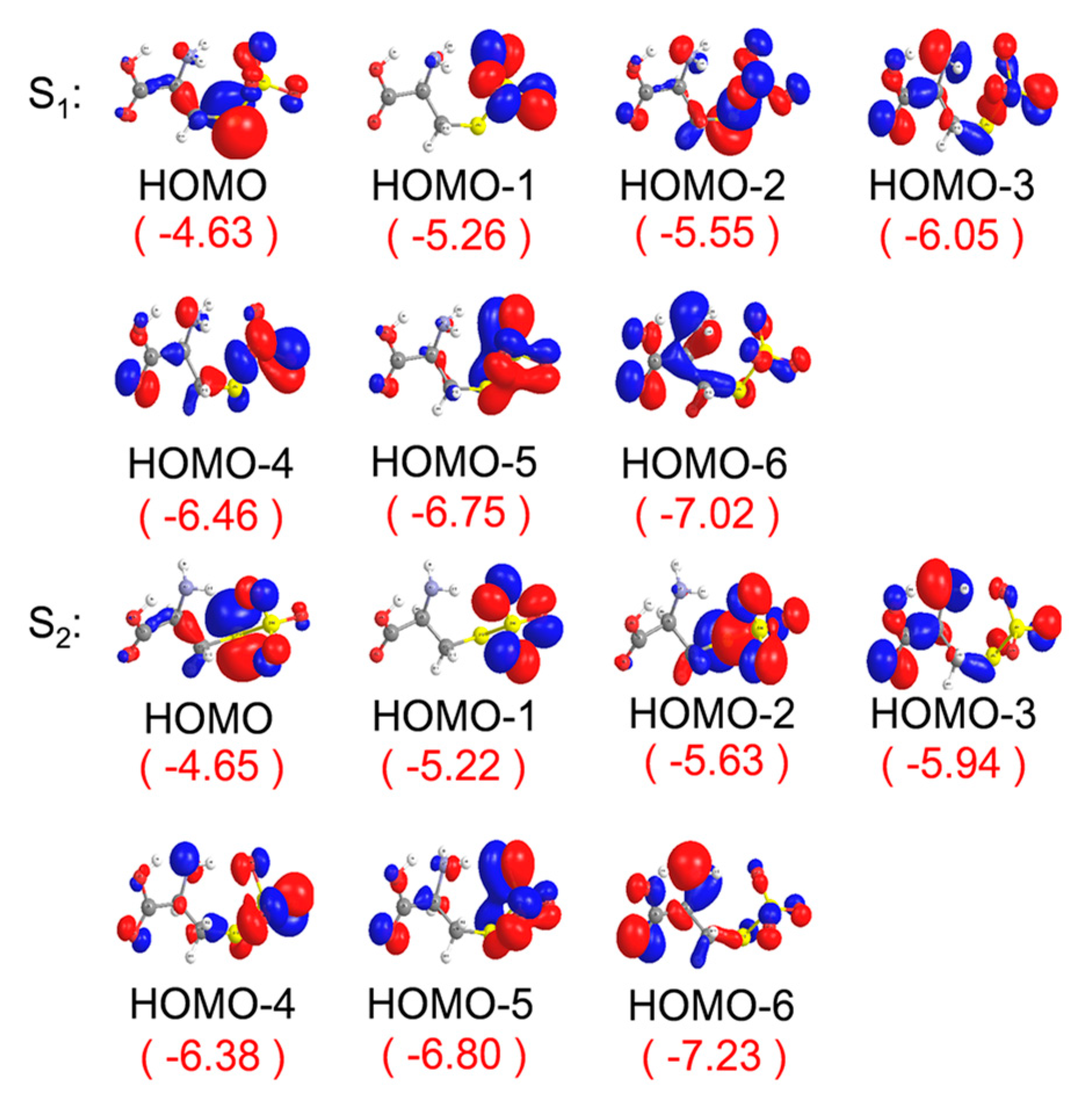
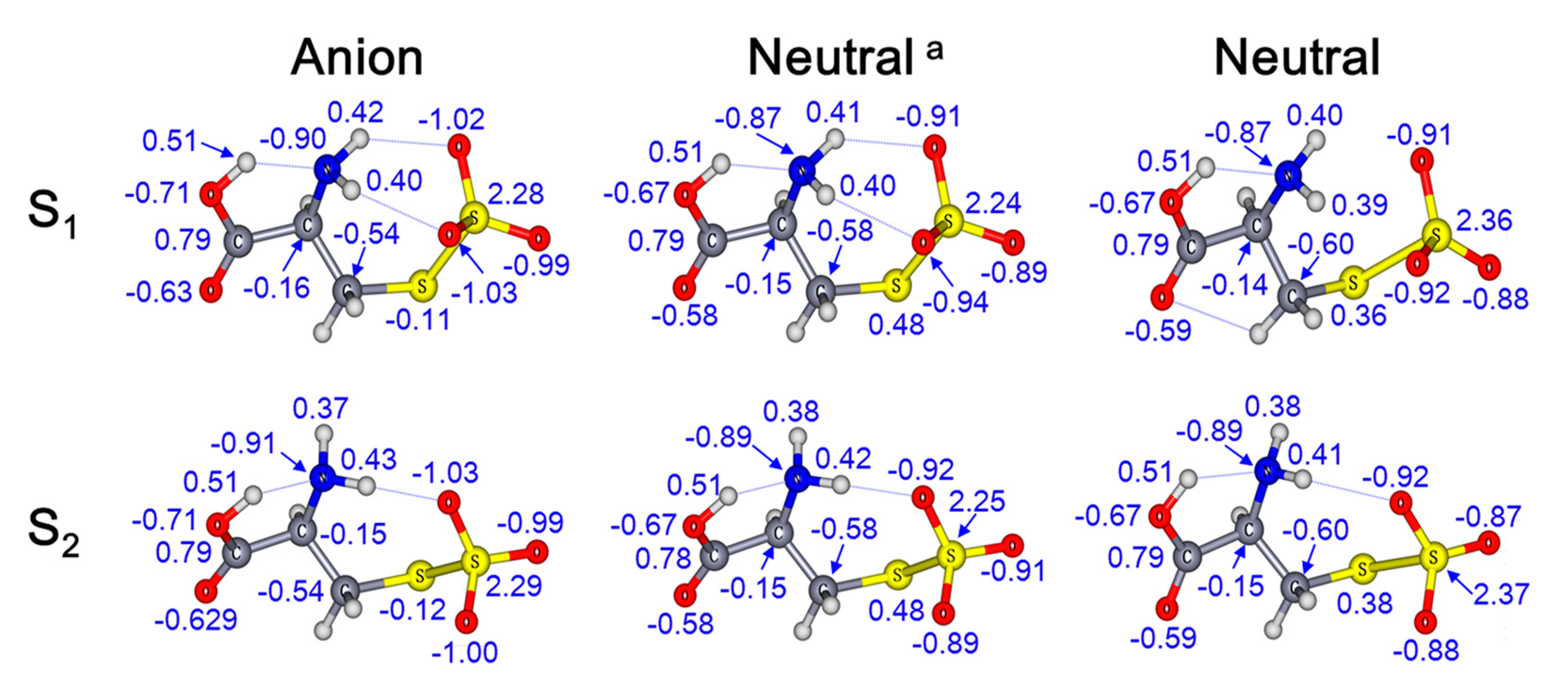
2. Results and Discussion

3. Materials and Methods
3.1. Experimental and Computational Methodologies
3.1.1. Negative Ion Photoelectron Spectroscopy
3.1.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, Y.; Mistretta, B.M.; Elsea, S.H.; Sun, Q. Development of a rapid UPLC–MS/MS determination of urine sulfocysteine for diagnosis of sulfocysteinuria and molybdenum co-factor deficiencies. Bioanalysis 2018, 10, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Strott, C.A. Sulfonation and molecular action. Endocr. Rev. 2002, 23, 703–732. [Google Scholar] [CrossRef] [PubMed]
- Macaluso, V.; Scuderi, D.; Crestoni, M.E.; Fornarini, S.; Corinti, D.; Dalloz, E.; Martínez-Núñez, E.; Hase, W.L.; Spezia, R. L-Cysteine modified by S-sulfation: Consequence on fragmentation processes elucidated by tandem mass spectrometry and chemical dynamics simulations. J. Phys. Chem. A 2019, 123, 3685–3696. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.B.; Hou, G.L.; Yang, Z.; Valiev, M.; Wang, X.B. Distonic radical anion species in cysteine oxidation processes. Phys. Chem. Chem. Phys. 2020, 22, 17554–17558. [Google Scholar] [CrossRef] [PubMed]
- Wolk, A.B.; Leavitt, C.M.; Garand, E.; Johnson, M.A. Cryogenic ion chemistry and spectroscopy. Acc. Chem. Res. 2014, 47, 202–210. [Google Scholar] [CrossRef]
- Wang, X.-B. Cluster model studies of anion and molecular specificities via electrospray ionization photoelectron spectroscopy. J. Phys. Chem. A 2017, 121, 1389–1401. [Google Scholar] [CrossRef]
- Asmis, K.R.; Neumark, D.M. Vibrational spectroscopy of microhydrated conjugate base anions. Acc. Chem. Res. 2012, 45, 43–52. [Google Scholar] [CrossRef]
- Meot-Ner, M. Update 1 of: Strong ionic hydrogen bonds. Chem. Rev. 2012, 112, PR22–PR103. [Google Scholar] [CrossRef]
- Wang, X.-B.; Woo, H.-K.; Kiran, B.; Wang, L.-S. Observation of weak C–H⋯O hydrogen bonding to unactivated alkanes. Angew. Chem. Int. Ed. 2005, 44, 4968–4972. [Google Scholar] [CrossRef]
- Herschlag, D.; Pinney, M.M. Hydrogen bonds: Simple after all? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef]
- Tozer, D.J.; Handy, N.C. Improving virtual Kohn–Sham orbitals and eigenvalues: Application to excitation energies and static polarizabilities. J. Chem. Phys. 1998, 109, 10180–10189. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Bader, R.W.F. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Raissi, H.; Khoshbin, Z.; Mollania, F. The analysis of structural and electronic properties for assessment of intramolecular hydrogen bond (IMHB) interaction: A comprehensive study into the effect of substitution on intramolecular hydrogen bond of 4-nitropyridine-3-thiol in ground and electronic excited state. Struct. Chem. 2013, 25, 515–538. [Google Scholar]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptiors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Wang, X.B.; Wang, L.S. Development of a low-temperature photoelectron spectroscopy instrument using an electrospray ion source and a cryogenically controlled ion trap. Rev. Sci. Instrum. 2008, 79, 1957. [Google Scholar] [CrossRef]
- Hanstorp, D.; Gustafsson, M. Determination of the electron affinity of iodine. J. Phys. B At. Mol. Opt. Phys. 1992, 25, 1773. [Google Scholar] [CrossRef]
- Wang, X.-B.; Wang, Y.-L.; Yang, J.; Xing, X.-P.; Li, J.; Wang, L.-S. Evidence of significant covalent bonding in Au(CN)2−. J. Am. Chem. Soc. 2009, 131, 16368–16370. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Papajak, E.; Leverentz, H.R.; Zheng, J.; Truhlar, D.G. Efficient diffuse basis sets: Cc-pVxZ+ and maug-cc-pVxZ. J. Chem. Theo. Comput. 2009, 5, 1197. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- EMSL Basis Set Exchange. Available online: https://bse.pnl.gov/bse/portal (accessed on 10 October 2022).
- Kashinski, D.O.; Chase, G.M.; Nelson, R.G.; Di Nallo, O.E.; Scales, A.N.; VanderLey, D.L.; Byrd, E.F. Harmonic vibrational frequencies: Approximate global scaling factors for TPSS, M06, and M11 functional families using several common basis sets. J. Phys. Chem. A 2017, 121, 2265–2273. [Google Scholar] [CrossRef]
- Qin, Z.B.; Hou, G.L.; Yang, Z.; Valiev, M.; Wang, X.B. Negative ion photoelectron spectra of ISO3−, IS2O3−, and IS2O4− intermediates formed in interfacial reactions of ozone and iodide/sulfite aqueous microdroplets. J. Chem. Phys. 2016, 145, 214310. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conformers. | Relative Energy [kcal/mol] | ADE [eV] | VDE [eV] | ||
|---|---|---|---|---|---|
| Theo. a | Exp. b | Theo. a | Exp. b | ||
| S1 | 0.00 | 4.87 | 4.95 | 5.26 | 5.30 |
| S2 | 0.16 | 4.88 | 5.27 | ||
| S3 | 1.23 | 4.80 | 5.26 | ||
| S4 | 1.60 | 4.82 | 5.29 | ||
| S5 | 3.71 | 4.80 | 5.55 | ||
| S6 | 4.86 | 4.70 | 5.56 | ||
| S7 | 6.46 | 4.90 | 5.55 | ||
| S8 | 7.53 | 4.63 | 4.97 | ||
| S9 | 7.79 | 4.64 | 4.99 | ||
| S10 | 8.12 | 4.83 | 5.54 | ||
| S11 | 8.28 | 4.65 | 5.04 | ||
| S12 | 8.33 | 4.62 | 5.00 | ||
| S13 | 8.37 | 4.64 | 5.03 | ||
| S14 | 8.53 | 4.37 | 5.01 | ||
| S15 | 8.82 | 4.41 | 5.05 | ||
| S16 | 9.04 | 4.65 | 5.06 | ||
| S17 | 9.22 | 4.59 | 4.97 | ||
| S18 | 9.26 | 4.59 | 4.94 | ||
| S19 | 9.57 | 4.60 | 4.99 | ||
| S20 | 9.61 | 4.51 | 4.95 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Qin, Z.; Hou, G.-L.; Yang, Z.; Valiev, M.; Wang, X.-B.; Zheng, X.; Cui, Z. Properties of Gaseous Deprotonated L-Cysteine S-Sulfate Anion [cysS-SO3]−: Intramolecular H-Bond Network, Electron Affinity, Chemically Active Site, and Vibrational Fingerprints. Int. J. Mol. Sci. 2023, 24, 1682. https://doi.org/10.3390/ijms24021682
Wang Q, Qin Z, Hou G-L, Yang Z, Valiev M, Wang X-B, Zheng X, Cui Z. Properties of Gaseous Deprotonated L-Cysteine S-Sulfate Anion [cysS-SO3]−: Intramolecular H-Bond Network, Electron Affinity, Chemically Active Site, and Vibrational Fingerprints. International Journal of Molecular Sciences. 2023; 24(2):1682. https://doi.org/10.3390/ijms24021682
Chicago/Turabian StyleWang, Qiaolin, Zhengbo Qin, Gao-Lei Hou, Zheng Yang, Marat Valiev, Xue-Bin Wang, Xianfeng Zheng, and Zhifeng Cui. 2023. "Properties of Gaseous Deprotonated L-Cysteine S-Sulfate Anion [cysS-SO3]−: Intramolecular H-Bond Network, Electron Affinity, Chemically Active Site, and Vibrational Fingerprints" International Journal of Molecular Sciences 24, no. 2: 1682. https://doi.org/10.3390/ijms24021682
APA StyleWang, Q., Qin, Z., Hou, G.-L., Yang, Z., Valiev, M., Wang, X.-B., Zheng, X., & Cui, Z. (2023). Properties of Gaseous Deprotonated L-Cysteine S-Sulfate Anion [cysS-SO3]−: Intramolecular H-Bond Network, Electron Affinity, Chemically Active Site, and Vibrational Fingerprints. International Journal of Molecular Sciences, 24(2), 1682. https://doi.org/10.3390/ijms24021682