
Synapse Dysfunctions in Multiple Sclerosis



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Primer on Multiple Sclerosis (MS): Multi-Faceted Neuroinflammatory Autoimmune Disease with Pathologies in the White and Grey Matter of the Brain
2. Primer on Brain Synapses: Communication Nano-Machines with Multiple Adjustment “Screws”
3. Communication at Glutamatergic Synapses
4. A More Extended View on Brain Synapses: Contribution of Glial Cells
5. Primer on Astrocytes: A Network of Guardians of Brain Homeostasis with Strong Impact on Synapses
- (1)
- Astrocytes play an important role in synapse development [177,198]. Astrocytes secrete synaptogenic factors that promote synapse formation and maturation, e.g., synapse organizing molecules, such as thrombospondin, hevin, or trophic factors that promote presynaptic differentiation [177,199,200,201,202,203,204,205,206,207,208]. Astrocytes also secrete glypicans that increase the surface expression of postsynaptic AMPA receptors [209,210].
- (2)
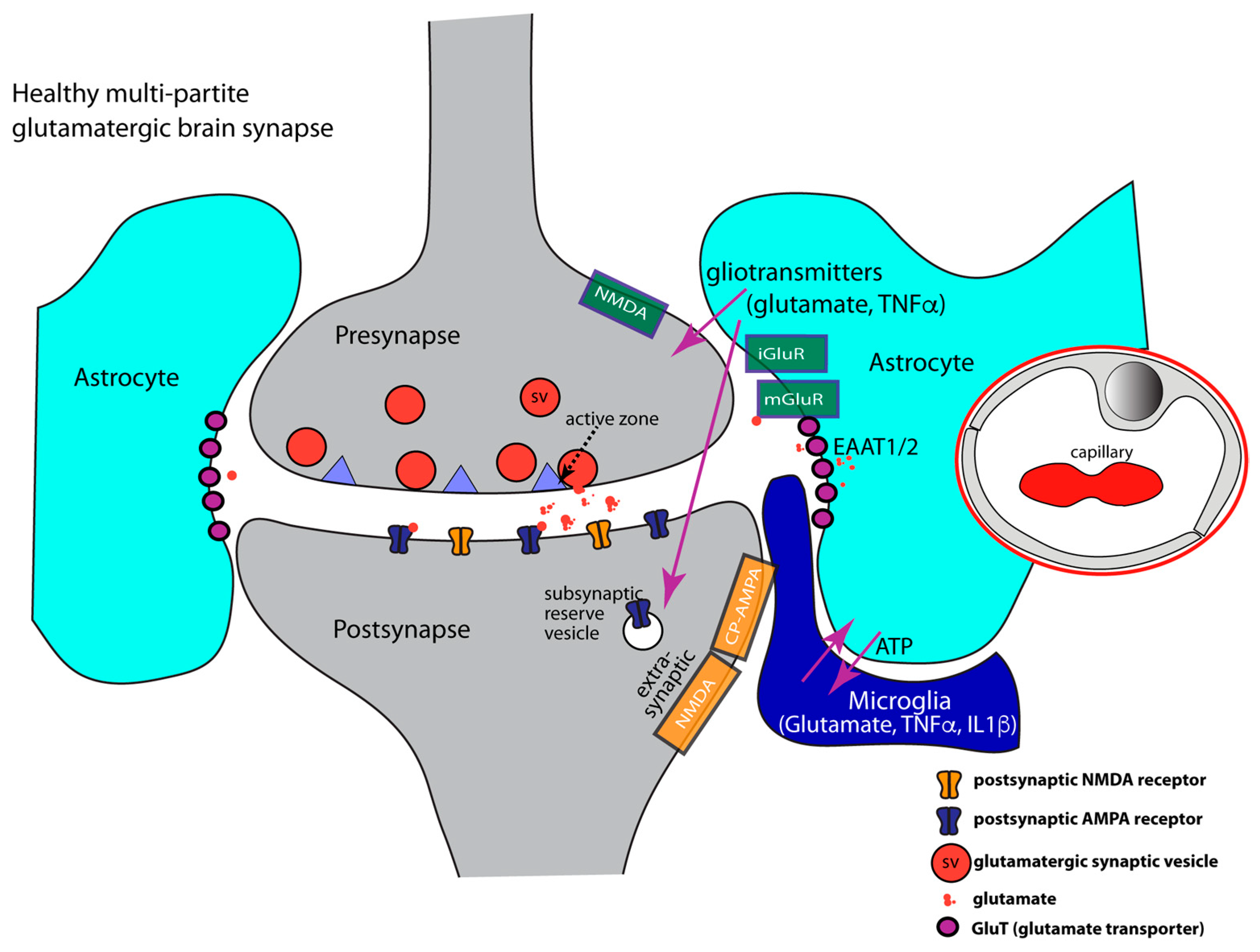
- At glutamatergic synapses, the perisynaptic processes of astrocytes contribute to the uptake of synaptically released glutamate [186,189,192,211,212,213,214,215,216,217,218,219]. Glutamate is taken up by various glutamate transporters [220,221,222]. Glutamate transporters (GluTs), also called excitatory amino acid transporters (EAATs), belong to the solute carrier 1 family (SLC1). Five sodium-dependent glutamate transporters (GluT) of the SLC1 family have been cloned: EAAT1/GLAST1, GLT1/EAAT2, EAAT3/EAAC1, EAAT4, and EAAT5/SLC1A7 [221,223,224,225,226]. GluTs have been localized to different localizations at the synapse. In general, GluTs are present either in the plasma membrane of the presynaptic terminal or in the plasma membrane of perisynaptic astroglial processes [222,227,228,229,230,231,232]. In this way, presynaptic neuronal and glial (astrocytic) glutamate uptake mechanisms collaborate to maintain low resting concentrations of extracellular glutamate and to prevent excitatory over-stimulation/excitatory synapse damage [84,190,216,220,233].
- (3)
- Perisynaptic astrocytes possess different types of neurotransmitter receptors, e.g., metabotropic glutamate receptors (mGluR2, mGluR3, mGluR5), AMPA-type ionotropic glutamate receptors, GABA receptors, and purinoreceptors to sense synaptic activity [177,196,197,269,270,271,272,273,274]. Astrocytes are capable of secreting TNFα and also possess TNFα receptors that serve autocrine effects. TNFα receptors are also important for communication with microglia [82,162,177,275,276,277] and neurons [162,163,173,278,279,280].
6. Primer on Microglia: Never-Resting Brain “Police” with the Mission to Survey and to Take (Strong) Action
7. Multivalent Microglia: Potentiator of Inflammatory Signals with Strong Impact on Synapses
8. Neuroinflammation-Induced Synapse Dysfunctions in MS
- (1)
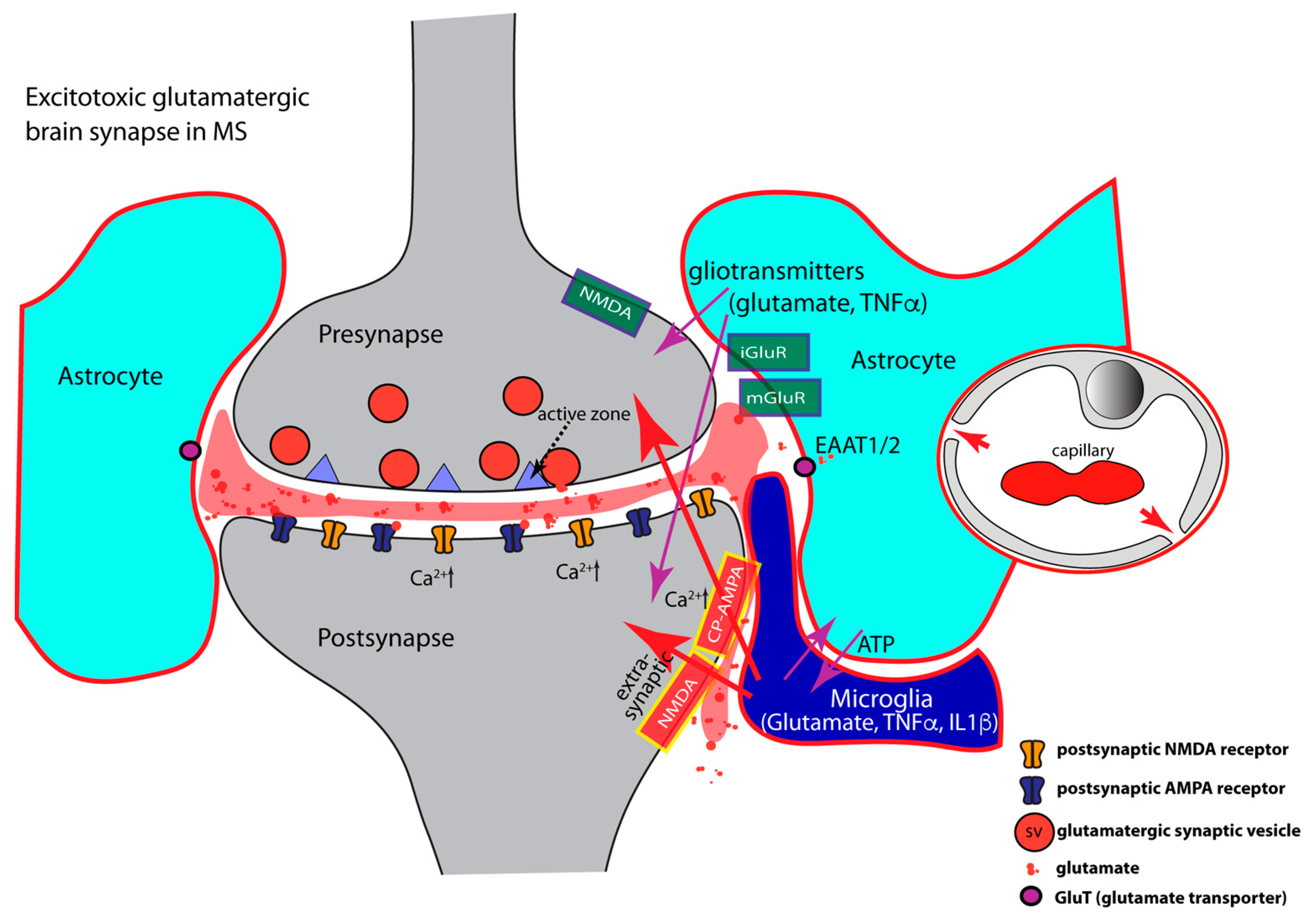
- TNFα regulates AMPA- and GABA- receptor trafficking in an antagonistic manner. The highly elevated levels of inflammatory cytokines released by activated microglia, astrocytes, and inflammatory CD3+ T-cells in MS, inhibit the expression of glial glutamate transporters (EAAT1/2), resulting in a decreased clearance of glutamate from the synaptic cleft [85,128,129,380,381,382,383,384,385] (Figure 3). The decreased glutamate clearance results in increased levels of extrasynaptic glutamate. Extrasynaptic glutamate binds to extrasynaptic glutamate receptors, including Ca2+-permeable NMDA receptors and Ca2+-permeable AMPA receptors. Stimulation of these extrasynaptic glutamate receptors is considered the central mechanism causing glutamate excitotoxicity, neurodegeneration, and neuronal cell death [386,387,388,389]. Many of these mechanisms involve elevated levels of Ca2+. Extrasynaptic NMDA receptor activation will trigger a deleterious signaling cascade that includes structural degeneration of the synapse, mitochondrial damage, and transcriptional shut-off of neuroprotective pathways [386,387,388,389]. Paradoxically, increased extrasynaptic glutamate can further inhibit the expression of astrocytic glutamate transporters [390], thus fostering a vicious cycle that leads to glutamate excitotoxicity.
- (2)
- Inflammatory cytokines (TNFα, IL1β) induce an increased surface expression of AMPA receptors [85,127,129,162,163,391,392] (Figure 3). Increased surface expression of AMPA receptors was observed in animal models of MS as well as in MS patients [127]. Of note, the significantly increased levels of TNFα/IL1β in MS/EAE, lead to an increased surface expression of the Ca2+-permeable AMPA receptors that lack the GluA2 subunit and thus lead to an enhancement of excitatory synaptic signaling [85,129,142,162,163,173,292,293,294,392,393]. As mentioned above, the Ca2+-permeability of glutamate-gated receptors is of particular importance for excitotoxic effects. High concentrations of TNFα increase not only synaptic but also non-synaptic AMPA receptor expression that further contributes to inflammation-induced glutamate excitotoxicity [127,278,373,392,394]. NMDA glutamate receptors could also be affected [138,139,395,396,397,398,399,400,401,402,403]. Increased surface expression of synaptic or extrasynaptic NMDA receptors in response to TNFα [397,404,405] aggravates glutamate excitotoxicity. This could occur either via Ca2+ overload of the postsynaptic compartment (Figure 3) or the formation of pathological glutamate receptor complexes [389] that lead to neurodegeneration and neuronal cell death [386,387,388].
- (3)
- Inflammatory cytokines (IL1β, TNFα) induce decreased surface expression of GABA receptors resulting in an imbalance between excitatory and inhibitory signaling [292,406,407,408,409]. TNFα promotes endocytosis of inhibitory GABA receptors thus leading to a decrease in GABA receptor surface expression [292,408]. In the EAE model of multiple sclerosis, inhibitory GABAergic signaling is diminished [84,85,87,128,129,130,407,408,409]. The TNFα effects on AMPA and GABA receptor trafficking are mediated by neuronal TNFR1 receptors [177,292]. IL1β is also involved in the downregulation of synaptic GABA receptors [128,129,407,408,410,411,412]. On the other hand, IL-1β enhances the surface expression of GABA transporters (GATs), thus promoting increased GABA clearance from the synaptic cleft [413,414,415]. In MS patients, GABA levels are significantly reduced and correlated with increasing physical disability in progressive multiple sclerosis [416].
- (4)
- Elevated levels of TNFα increase glutaminase activity in microglia and induce significant release of glutamate from microglia [108,417,418,419]. Activated astrocytes and invading T cells also contribute to increased levels of glutamate in neuroinflammation [259]. These mechanisms will lead to strong activation of extrasynaptic N-methyl-D-aspartate (NMDA) and non-NMDA glutamate receptors. Activation of extrasynaptic glutamate receptors results in neurotoxic effects and ultimately leads to neuronal cell death via various mechanisms [386,387,388,389]. The underlying mechanisms are still under intense investigation, and likely include dysfunctional Ca2+ homeostasis, molecular and structural alterations of the synapse, malfunctional pre- and postsynaptic signaling cascades, mitochondrial dysfunctions, and dysregulation of synapse-dependent transcriptional programs [386,387,388,389].
9. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MS | multiple sclerosis |
| EAE | experimental autoimmune encephalo-myelitis |
| CNS | central nervous system |
| OL | oligodendrocyte |
| GluT | glutamate transporter |
| Cav-channel | voltage-gated Ca2+-channels |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| PKA | protein kinase A |
| CREB | cAMP response element-binding protein |
| pCREB | phospho-CREB |
| PSD | postsynaptic density |
| mGluR | metabotropic glutamate receptor |
| iGluR | ionotropic glutamate receptor |
References
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Gbaguidi, B.; Guillemin, F.; Soudant, M.; Debouverie, M.; Mathey, G.; Epstein, J. Age-period-cohort analysis of the incidence of multiple sclerosis over twenty years in Lorraine, France. Sci. Rep. 2022, 12, 1001. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef]
- Coyle, P.K. What Can We Learn from Sex Differences in MS? J. Pers. Med. 2021, 11, 1006. [Google Scholar] [CrossRef] [PubMed]
- Lulu, S.; Graves, J.; Waubant, E. Menarche increases relapse risk in pediatric multiple sclerosis. Mult. Scler. J. 2016, 22, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Wallin, M.T.; Culpepper, W.J.; Campbell, J.D.; Nelson, L.M.; Langer-Gould, A.; Marrie, R.A.; Cutter, G.R.; Kaye, W.E.; Wagner, L.; Tremlett, H.; et al. The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology 2019, 92, e1029–e1040. [Google Scholar] [CrossRef]
- Ramien, C.; Taenzer, A.; Lupu, A.; Heckmann, N.; Engler, J.B.; Patas, K.; Friese, M.A.; Gold, S.M. Sex effects on inflammatory and neurodegenerative processes in multiple sclerosis. Neurosci. Biobehav. Rev. 2016, 67, 137–146. [Google Scholar] [CrossRef]
- Rosati, G.; Aiello, I.; Pirastru, M.I.; Mannu, L.; Sanna, G.; Sau, G.F.; Sotgiu, S. Epidemiology of multiple sclerosis in Northwestern Sardinia: Further evidence for higher frequency in Sardinians compared to other Italians. Neuroepidemiology 1996, 15, 10–19. [Google Scholar] [CrossRef]
- Bufill, E.; Blesa, R.; Galan, I.; Dean, G. Prevalence of multiple sclerosis in the region of Osona, Catalonia, northern Spain. J. Neurol. Neurosurg. Psychiatry 1995, 58, 577–581. [Google Scholar] [CrossRef][Green Version]
- Compston, A. Risk factors for multiple sclerosis: Race or place? J. Neurol. Neurosurg. Psychiatry 1990, 53, 821–823. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Xiong, Y.; Larsson, S.C. An atlas on risk factors for multiple sclerosis: A Mendelian randomization study. J. Neurol. 2021, 268, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Yamout, B.I.; Alroughani, R. Multiple Sclerosis. Semin. Neurol. 2018, 38, 212–225. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis-a review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef]
- Attfield, K.E.; Jensen, L.T.; Kaufmann, M.; Friese, M.A.; Fugger, L. The immunology of multiple sclerosis. Nat. Rev. Immunol. 2022, 22, 734–750. [Google Scholar] [CrossRef]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann. Neurol. 2007, 61, 504–513. [Google Scholar] [CrossRef]
- Thacker, E.L.; Mirzaei, F.; Ascherio, A. Infectious mononucleosis and risk for multiple sclerosis: A meta-analysis. Ann. Neurol. 2006, 59, 499–503. [Google Scholar] [CrossRef]
- Venkatesan, A.; Johnson, R.T. Infections and multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 151–171. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Kaufmann, M.; Evans, H.; Schaupp, A.L.; Engler, J.B.; Kaur, G.; Willing, A.; Kursawe, N.; Schubert, C.; Attfield, K.E.; Fugger, L.; et al. Identifying CNS-colonizing T cells as potential therapeutic targets to prevent progression of multiple sclerosis. Med 2021, 2, 296–312.e8. [Google Scholar] [CrossRef]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Jacome-Galarza, C.E.; Blank, T.; Lazarov, T.; Durham, B.H.; Ozkaya, N.; Pastore, A.; Schwabenland, M.; Chung, Y.R.; Rosenblum, M.K.; et al. A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature 2017, 549, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Muller, P.F.; Wolf, Y.; Varol, D.; Yona, S.; Brendecke, S.M.; Kierdorf, K.; Staszewski, O.; Datta, M.; et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 2013, 16, 1618–1626. [Google Scholar] [CrossRef]
- Ruiz, F.; Vigne, S.; Pot, C. Resolution of inflammation during multiple sclerosis. Semin. Immunopathol. 2019, 41, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R. The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2019, 137, 757–783. [Google Scholar] [CrossRef] [PubMed]
- das Neves, S.P.; Sousa, J.C.; Sousa, N.; Cerqueira, J.J.; Marques, F. Altered astrocytic function in experimental neuroinflammation and multiple sclerosis. Glia 2021, 69, 1341–1368. [Google Scholar] [CrossRef]
- Seals, M.R.; Moran, M.M.; Leavenworth, J.D.; Leavenworth, J.W. Contribution of Dysregulated B-Cells and IgE Antibody Responses to Multiple Sclerosis. Front. Immunol. 2022, 13, 900117. [Google Scholar] [CrossRef]
- Mathias, A.; Perriard, G.; Canales, M.; Soneson, C.; Delorenzi, M.; Schluep, M.; Du Pasquier, R.A. Increased ex vivo antigen presentation profile of B cells in multiple sclerosis. Mult. Scler. J. 2017, 23, 802–809. [Google Scholar] [CrossRef]
- Bar-Or, A.; Fawaz, L.; Fan, B.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.; et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann. Neurol. 2010, 67, 452–461. [Google Scholar] [CrossRef]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Calabresi, P.A.; Arnold, D.; Markowitz, C.; Shafer, S.; Kasper, L.H.; Waubant, E.; Gazda, S.; Fox, R.J.; Panzara, M.; et al. Rituximab in relapsing-remitting multiple sclerosis: A 72-week, open-label, phase I trial. Ann. Neurol. 2008, 63, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Myhr, K.M.; Torkildsen, O.; Lossius, A.; Bo, L.; Holmoy, T. B cell depletion in the treatment of multiple sclerosis. Expert Opin. Biol. Ther. 2019, 19, 261–271. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Sakai, R.E.; Feller, D.J.; Galetta, K.M.; Galetta, S.L.; Balcer, L.J. Vision in multiple sclerosis: The story, structure-function correlations, and models for neuroprotection. J. Neuroophthalmol. 2011, 31, 362–373. [Google Scholar] [CrossRef]
- Ford, H. Clinical presentation and diagnosis of multiple sclerosis. Clin. Med. 2020, 20, 380–383. [Google Scholar] [CrossRef]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Wilkins, A. Cerebellar Dysfunction in Multiple Sclerosis. Front. Neurol. 2017, 8, 312. [Google Scholar] [CrossRef]
- Mross, K.; Jankowska, M.; Meller, A.; Machowska-Sempruch, K.; Nowacki, P.; Masztalewicz, M.; Pawlukowska, W. Sensory Integration Disorders in Patients with Multiple Sclerosis. J. Clin. Med. 2022, 11, 5183. [Google Scholar] [CrossRef] [PubMed]
- Biname, F.; Pham-Van, L.D.; Bagnard, D. Manipulating oligodendrocyte intrinsic regeneration mechanism to promote remyelination. Cell. Mol. Life Sci. 2021, 78, 5257–5273. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Vukusic, S.; Moreau, T.; Adeleine, P. Relapses and progression of disability in multiple sclerosis. N. Engl. J. Med. 2000, 343, 1430–1438. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Schlager, C.; Korner, H.; Krueger, M.; Vidoli, S.; Haberl, M.; Mielke, D.; Brylla, E.; Issekutz, T.; Cabanas, C.; Nelson, P.J.; et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 2016, 530, 349–353. [Google Scholar] [CrossRef]
- Quinn, J.L.; Kumar, G.; Agasing, A.; Ko, R.M.; Axtell, R.C. Role of TFH Cells in Promoting T Helper 17-Induced Neuroinflammation. Front. Immunol. 2018, 9, 382. [Google Scholar] [CrossRef]
- Galicia, G.; Boulianne, B.; Pikor, N.; Martin, A.; Gommerman, J.L. Secondary B cell receptor diversification is necessary for T cell mediated neuro-inflammation during experimental autoimmune encephalomyelitis. PLoS ONE 2013, 8, e61478. [Google Scholar] [CrossRef]
- Kuerten, S.; Lanz, T.V.; Lingampalli, N.; Lahey, L.J.; Kleinschnitz, C.; Maurer, M.; Schroeter, M.; Braune, S.; Ziemssen, T.; Ho, P.P.; et al. Autoantibodies against central nervous system antigens in a subset of B cell-dominant multiple sclerosis patients. Proc. Natl. Acad. Sci. USA 2020, 117, 21512–21518. [Google Scholar] [CrossRef]
- Kuerten, S.; Jackson, L.J.; Kaye, J.; Vollmer, T.L. Impact of Glatiramer Acetate on B Cell-Mediated Pathogenesis of Multiple Sclerosis. CNS Drugs 2018, 32, 1039–1051. [Google Scholar] [CrossRef]
- Wanleenuwat, P.; Iwanowski, P. Role of B cells and antibodies in multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 36, 101416. [Google Scholar] [CrossRef] [PubMed]
- Chunder, R.; Weier, A.; Maurer, H.; Luber, N.; Enders, M.; Luber, G.; Heider, T.; Spitzer, A.; Tacke, S.; Becker-Gotot, J.; et al. Antibody cross-reactivity between casein and myelin-associated glycoprotein results in central nervous system demyelination. Proc. Natl. Acad. Sci. USA 2022, 119, e2117034119. [Google Scholar] [CrossRef] [PubMed]
- Chunder, R.; Schropp, V.; Kuerten, S. B Cells in Multiple Sclerosis and Virus-Induced Neuroinflammation. Front. Neurol. 2020, 11, 591894. [Google Scholar] [CrossRef]
- Tengvall, K.; Huang, J.; Hellstrom, C.; Kammer, P.; Bistrom, M.; Ayoglu, B.; Lima Bomfim, I.; Stridh, P.; Butt, J.; Brenner, N.; et al. Molecular mimicry between Anoctamin 2 and Epstein-Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc. Natl. Acad. Sci. USA 2019, 116, 16955–16960. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, L.; Schafer, D.P.; Bartels, T.; Rowitch, D.H.; Calabresi, P.A. Diversity and Function of Glial Cell Types in Multiple Sclerosis. Trends Immunol. 2021, 42, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.K.; Mahns, D.A.; Coorssen, J.R.; Shortland, P.J. The roles of microglia and astrocytes in phagocytosis and myelination: Insights from the cuprizone model of multiple sclerosis. Glia 2022, 70, 1215–1250. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.M.; Stratton, J.A.; Kuhlmann, T.; Antel, J. The role of glial cells in multiple sclerosis disease progression. Nat. Rev. Neurol. 2022, 18, 237–248. [Google Scholar] [CrossRef]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Bruck, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128 Pt 11, 2705–2712. [Google Scholar] [CrossRef]
- DeLuca, G.C.; Williams, K.; Evangelou, N.; Ebers, G.C.; Esiri, M.M. The contribution of demyelination to axonal loss in multiple sclerosis. Brain 2006, 129 Pt 6, 1507–1516. [Google Scholar] [CrossRef]
- Nikic, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Bruck, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, T.; Metz, I.; Dallenga, T.; Konig, F.B.; Muller, S.; Stadelmann, C.; Bruck, W. Wallerian degeneration: A major component of early axonal pathology in multiple sclerosis. Brain Pathol. 2010, 20, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Salapa, H.E.; Lee, S.; Shin, Y.; Levin, M.C. Contribution of the Degeneration of the Neuro-Axonal Unit to the Pathogenesis of Multiple Sclerosis. Brain Sci. 2017, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Carassiti, D.; Altmann, D.R.; Petrova, N.; Pakkenberg, B.; Scaravilli, F.; Schmierer, K. Neuronal loss, demyelination and volume change in the multiple sclerosis neocortex. Neuropathol. Appl. Neurobiol. 2018, 44, 377–390. [Google Scholar] [CrossRef]
- Luchicchi, A.; Hart, B.; Frigerio, I.; van Dam, A.M.; Perna, L.; Offerhaus, H.L.; Stys, P.K.; Schenk, G.J.; Geurts, J.J.G. Axon-Myelin Unit Blistering as Early Event in MS Normal Appearing White Matter. Ann. Neurol. 2021, 89, 711–725. [Google Scholar] [CrossRef]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef]
- Derfuss, T.; Parikh, K.; Velhin, S.; Braun, M.; Mathey, E.; Krumbholz, M.; Kumpfel, T.; Moldenhauer, A.; Rader, C.; Sonderegger, P.; et al. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc. Natl. Acad. Sci. USA 2009, 106, 8302–8307. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Chang, A.; Doud, M.K.; Kidd, G.J.; Ribaudo, M.V.; Young, E.A.; Fox, R.J.; Staugaitis, S.M.; Trapp, B.D. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann. Neurol. 2011, 69, 445–454. [Google Scholar] [CrossRef]
- Rossi, S.; Motta, C.; Studer, V.; Barbieri, F.; Buttari, F.; Bergami, A.; Sancesario, G.; Bernardini, S.; De Angelis, G.; Martino, G.; et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult. Scler. J. 2014, 20, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; Gentile, A.; Musella, A.; Fresegna, D.; De Vito, F.; Bullitta, S.; Sepman, H.; Marfia, G.A.; Centonze, D. Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nat. Rev. Neurol. 2015, 11, 711–724. [Google Scholar] [CrossRef]
- Calabrese, M.; Magliozzi, R.; Ciccarelli, O.; Geurts, J.J.; Reynolds, R.; Martin, R. Exploring the origins of grey matter damage in multiple sclerosis. Nat. Rev. Neurosci. 2015, 16, 147–158. [Google Scholar] [CrossRef]
- Louapre, C.; Perlbarg, V.; Garcia-Lorenzo, D.; Urbanski, M.; Benali, H.; Assouad, R.; Galanaud, D.; Freeman, L.; Bodini, B.; Papeix, C.; et al. Brain networks disconnection in early multiple sclerosis cognitive deficits: An anatomofunctional study. Hum. Brain Mapp. 2014, 35, 4706–4717. [Google Scholar] [CrossRef] [PubMed]
- Steenwijk, M.D.; Geurts, J.J.; Daams, M.; Tijms, B.M.; Wink, A.M.; Balk, L.J.; Tewarie, P.K.; Uitdehaag, B.M.; Barkhof, F.; Vrenken, H.; et al. Cortical atrophy patterns in multiple sclerosis are non-random and clinically relevant. Brain 2016, 139 Pt 1, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Bellingacci, L.; Mancini, A.; Gaetani, L.; Tozzi, A.; Parnetti, L.; Di Filippo, M. Synaptic Dysfunction in Multiple Sclerosis: A Red Thread from Inflammation to Network Disconnection. Int. J. Mol. Sci. 2021, 22, 9753. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Mancini, A.; Bellingacci, L.; Gaetani, L.; Mazzocchetti, P.; Zelante, T.; La Barbera, L.; De Luca, A.; Tantucci, M.; Tozzi, A.; et al. Interleukin-17 affects synaptic plasticity and cognition in an experimental model of multiple sclerosis. Cell Rep. 2021, 37, 110094. [Google Scholar] [CrossRef]
- Feuillet, L.; Reuter, F.; Audoin, B.; Malikova, I.; Barrau, K.; Cherif, A.A.; Pelletier, J. Early cognitive impairment in patients with clinically isolated syndrome suggestive of multiple sclerosis. Mult. Scler. J. 2007, 13, 124–127. [Google Scholar] [CrossRef]
- Mandolesi, G.; Grasselli, G.; Musumeci, G.; Centonze, D. Cognitive deficits in experimental autoimmune encephalomyelitis: Neuroinflammation and synaptic degeneration. Neurol. Sci. 2010, 31 (Suppl. S2), S255–S259. [Google Scholar] [CrossRef]
- Haji, N.; Mandolesi, G.; Gentile, A.; Sacchetti, L.; Fresegna, D.; Rossi, S.; Musella, A.; Sepman, H.; Motta, C.; Studer, V.; et al. TNF-α-mediated anxiety in a mouse model of multiple sclerosis. Exp. Neurol. 2012, 237, 296–303. [Google Scholar] [CrossRef]
- Tarasiuk, J.; Kapica-Topczewska, K.; Czarnowska, A.; Chorazy, M.; Kochanowicz, J.; Kulakowska, A. Co-occurrence of Fatigue and Depression in People with Multiple Sclerosis: A Mini-Review. Front. Neurol. 2021, 12, 817256. [Google Scholar] [CrossRef]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef]
- Acharjee, S.; Nayani, N.; Tsutsui, M.; Hill, M.N.; Ousman, S.S.; Pittman, Q.J. Altered cognitive-emotional behavior in early experimental autoimmune encephalitis-cytokine and hormonal correlates. Brain Behav. Immun. 2013, 33, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Osso, L.A.; Chan, J.R. Astrocytes Underlie Neuroinflammatory Memory Impairment. Cell 2015, 163, 1574–1576. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Welsh, C.A.; Barres, B.A.; Stevens, B. Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci. 2015, 18, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Stampanoni Bassi, M.; Mori, F.; Buttari, F.; Marfia, G.A.; Sancesario, A.; Centonze, D.; Iezzi, E. Neurophysiology of synaptic functioning in multiple sclerosis. Clin. Neurophysiol. 2017, 128, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Muzio, L.; Rossi, S.; Cavasinni, F.; De Chiara, V.; Bergami, A.; Musella, A.; D’Amelio, M.; Cavallucci, V.; Martorana, A.; et al. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J. Neurosci. 2009, 29, 3442–3452. [Google Scholar] [CrossRef]
- Audoin, B.; Zaaraoui, W.; Reuter, F.; Rico, A.; Malikova, I.; Confort-Gouny, S.; Cozzone, P.J.; Pelletier, J.; Ranjeva, J.P. Atrophy mainly affects the limbic system and the deep grey matter at the first stage of multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 690–695. [Google Scholar] [CrossRef]
- Di Filippo, M.; de Iure, A.; Durante, V.; Gaetani, L.; Mancini, A.; Sarchielli, P.; Calabresi, P. Synaptic plasticity and experimental autoimmune encephalomyelitis: Implications for multiple sclerosis. Brain Res. 2015, 1621, 205–213. [Google Scholar] [CrossRef]
- Shi, J.; Baxter, L.C.; Kuniyoshi, S.M. Pathologic and imaging correlates of cognitive deficits in multiple sclerosis: Changing the paradigm of diagnosis and prognosis. Cogn. Behav. Neurol. 2014, 27, 1–7. [Google Scholar] [CrossRef]
- DeLuca, G.C.; Yates, R.L.; Beale, H.; Morrow, S.A. Cognitive impairment in multiple sclerosis: Clinical, radiologic and pathologic insights. Brain Pathol. 2015, 25, 79–98. [Google Scholar] [CrossRef]
- Eshaghi, A.; Marinescu, R.V.; Young, A.L.; Firth, N.C.; Prados, F.; Jorge Cardoso, M.; Tur, C.; De Angelis, F.; Cawley, N.; Brownlee, W.J.; et al. Progression of regional grey matter atrophy in multiple sclerosis. Brain 2018, 141, 1665–1677. [Google Scholar] [CrossRef] [PubMed]
- Solana, E.; Martinez-Heras, E.; Montal, V.; Vilaplana, E.; Lopez-Soley, E.; Radua, J.; Sola-Valls, N.; Montejo, C.; Blanco, Y.; Pulido-Valdeolivas, I.; et al. Regional grey matter microstructural changes and volume loss according to disease duration in multiple sclerosis patients. Sci. Rep. 2021, 11, 16805. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, T.; Jafari, M.; Kreutzfeldt, M.; Bahn, E.; Bruck, W.; Kerschensteiner, M.; Merkler, D. Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain 2016, 139 Pt 1, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A. Widespread synaptic loss in multiple sclerosis. Brain 2016, 139 Pt 1, 2–4. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gentile, A.; De Vito, F.; Fresegna, D.; Rizzo, F.R.; Bullitta, S.; Guadalupi, L.; Vanni, V.; Buttari, F.; Stampanoni Bassi, M.; Leuti, A.; et al. Peripheral T cells from multiple sclerosis patients trigger synaptotoxic alterations in central neurons. Neuropathol. Appl. Neurobiol. 2020, 46, 160–170. [Google Scholar] [CrossRef]
- Schattling, B.; Engler, J.B.; Volkmann, C.; Rothammer, N.; Woo, M.S.; Petersen, M.; Winkler, I.; Kaufmann, M.; Rosenkranz, S.C.; Fejtova, A.; et al. Bassoon proteinopathy drives neurodegeneration in multiple sclerosis. Nat. Neurosci. 2019, 22, 887–896. [Google Scholar] [CrossRef]
- Yang, G.; Parkhurst, C.N.; Hayes, S.; Gan, W.B. Peripheral elevation of TNF-α leads to early synaptic abnormalities in the mouse somatosensory cortex in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2013, 110, 10306–10311. [Google Scholar] [CrossRef]
- Huang, L.; Lafaille, J.J.; Yang, G. Learning-dependent dendritic spine plasticity is impaired in spontaneous autoimmune encephalomyelitis. Dev. Neurobiol. 2021, 81, 736–745. [Google Scholar] [CrossRef]
- Mendel, I.; Kerlero de Rosbo, N.; Ben-Nun, A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: Fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur. J. Immunol. 1995, 25, 1951–1959. [Google Scholar] [CrossRef]
- Baxter, A.G. The origin and application of experimental autoimmune encephalomyelitis. Nat. Rev. Immunol. 2007, 7, 904–912. [Google Scholar] [CrossRef]
- Procaccini, C.; De Rosa, V.; Pucino, V.; Formisano, L.; Matarese, G. Animal models of Multiple Sclerosis. Eur. J. Pharmacol. 2015, 759, 182–191. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Mix, E.; Meyer-Rienecker, H.; Hartung, H.P.; Zettl, U.K. Animal models of multiple sclerosis--potentials and limitations. Prog. Neurobiol. 2010, 92, 386–404. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.P.; Harp, C.T.; Noronha, A.; Miller, S.D. The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handb. Clin. Neurol. 2014, 122, 173–189. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.W.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Serafini, B.; Aloisi, F.; Reynolds, R. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 2010, 68, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.R.; Howell, O.W.; Carassiti, D.; Magliozzi, R.; Gveric, D.; Muraro, P.A.; Nicholas, R.; Roncaroli, F.; Reynolds, R. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012, 135 Pt 10, 2925–2937. [Google Scholar] [CrossRef]
- Rossi, S.; Furlan, R.; De Chiara, V.; Motta, C.; Studer, V.; Mori, F.; Musella, A.; Bergami, A.; Muzio, L.; Bernardi, G.; et al. Interleukin-1beta causes synaptic hyperexcitability in multiple sclerosis. Ann. Neurol. 2012, 71, 76–83. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Sood, A.; Preeti, K.; Fernandes, V.; Khatri, D.K.; Singh, S.B. Glia: A major player in glutamate-GABA dysregulation-mediated neurodegeneration. J. Neurosci. Res. 2021, 99, 3148–3189. [Google Scholar] [CrossRef]
- Sudhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef]
- Brunger, A.T.; Leitz, J. The Core Complex of the Ca(2+)-Triggered Presynaptic Fusion Machinery. J. Mol. Biol. 2023, 435, 167853. [Google Scholar] [CrossRef]
- Südhof, T.C. The Molecular Machinery of Neurotransmitter Release (Nobel Lecture). Angew. Chem. Int. Ed. 2014, 53, 12696–12717. [Google Scholar] [CrossRef]
- Sudhof, T.C. The presynaptic active zone. Neuron 2012, 75, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Piao, C.; Sigrist, S.J. (M)Unc13s in Active Zone Diversity: A Drosophila Perspective. Front. Synaptic Neurosci. 2021, 13, 798204. [Google Scholar] [CrossRef] [PubMed]
- Nanou, E.; Catterall, W.A. Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron 2018, 98, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C.; Lee, A. Presynaptic calcium channels: Specialized control of synaptic neurotransmitter release. Nat. Rev. Neurosci. 2020, 21, 213–229. [Google Scholar] [CrossRef]
- Moser, T.; Grabner, C.P.; Schmitz, F. Sensory Processing at Ribbon Synapses in the Retina and the Cochlea. Physiol. Rev. 2020, 100, 103–144. [Google Scholar] [CrossRef]
- Williams, B.; Maddox, J.W.; Lee, A. Calcium Channels in Retinal Function and Disease. Annu. Rev. Vis. Sci. 2022, 8, 53–77. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, J.B.; Yuan, H.L.; Chen, S.Y.; Singer, J.H.; Ke, J.B. Multiple Calcium Channel Types with Unique Expression Patterns Mediate Retinal Signaling at Bipolar Cell Ribbon Synapses. J. Neurosci. 2022, 42, 6487–6505. [Google Scholar] [CrossRef]
- Azarnia Tehran, D.; Maritzen, T. Endocytic proteins: An expanding repertoire of presynaptic functions. Curr. Opin. Neurobiol. 2022, 73, 102519. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, H.; Li, C. SAPAP Scaffold Proteins: From Synaptic Function to Neuropsychiatric Disorders. Cells 2022, 11, 3815. [Google Scholar] [CrossRef]
- Levy, A.M.; Gomez-Puertas, P.; Tumer, Z. Neurodevelopmental Disorders Associated with PSD-95 and Its Interaction Partners. Int. J. Mol. Sci. 2022, 23, 4390. [Google Scholar] [CrossRef] [PubMed]
- Krueger-Burg, D.; Papadopoulos, T.; Brose, N. Organizers of inhibitory synapses come of age. Curr. Opin. Neurobiol. 2017, 45, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. Towards an Understanding of Synapse Formation. Neuron 2018, 100, 276–293. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef]
- Biederer, T.; Kaeser, P.S.; Blanpied, T.A. Transcellular Nanoalignment of Synaptic Function. Neuron 2017, 96, 680–696. [Google Scholar] [CrossRef]
- Sudhof, T.C. The cell biology of synapse formation. J. Cell. Biol. 2021, 220, e202103052. [Google Scholar] [CrossRef]
- Newcombe, J.; Uddin, A.; Dove, R.; Patel, B.; Turski, L.; Nishizawa, Y.; Smith, T. Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol. 2008, 18, 52–61. [Google Scholar] [CrossRef]
- Mandolesi, G.; Gentile, A.; Musella, A.; Centonze, D. IL-1beta dependent cerebellar synaptopathy in a mouse mode of multiple sclerosis. Cerebellum 2015, 14, 19–22. [Google Scholar] [CrossRef]
- Mandolesi, G.; Musella, A.; Gentile, A.; Grasselli, G.; Haji, N.; Sepman, H.; Fresegna, D.; Bullitta, S.; De Vito, F.; Musumeci, G.; et al. Interleukin-1beta alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J. Neurosci. 2013, 33, 12105–12121. [Google Scholar] [CrossRef]
- Mori, F.; Nistico, R.; Nicoletti, C.G.; Zagaglia, S.; Mandolesi, G.; Piccinin, S.; Martino, G.; Finardi, A.; Rossini, P.M.; Marfia, G.A.; et al. RANTES correlates with inflammatory activity and synaptic excitability in multiple sclerosis. Mult. Scler. J. 2016, 22, 1405–1412. [Google Scholar] [CrossRef]
- Watkins, J.C.; Evans, R.H. Excitatory amino acid transmitters. Annu. Rev. Pharmacol. Toxicol. 1981, 21, 165–204. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural. Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol. Rev. 2021, 73, 298–487. [Google Scholar] [CrossRef] [PubMed]
- Stover, J.F.; Pleines, U.E.; Morganti-Kossmann, M.C.; Kossmann, T.; Lowitzsch, K.; Kempski, O.S. Neurotransmitters in cerebrospinal fluid reflect pathological activity. Eur. J. Clin. Investig. 1997, 27, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Sarchielli, P.; Greco, L.; Floridi, A.; Floridi, A.; Gallai, V. Excitatory amino acids and multiple sclerosis: Evidence from cerebrospinal fluid. Arch. Neurol. 2003, 60, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Sailasuta, N.; Hurd, R.; Nelson, S.; Pelletier, D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain 2005, 128 Pt 5, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, G.; Dabrowska-Bouta, B.; Salinska, E.; Struzynska, L. Modulation of glutamate transport and receptor binding by glutamate receptor antagonists in EAE rat brain. PLoS ONE 2014, 9, e113954. [Google Scholar] [CrossRef] [PubMed]
- Levite, M. Glutamate, T cells and multiple sclerosis. J. Neural. Transm. 2017, 124, 775–798. [Google Scholar] [CrossRef]
- Gentile, A.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Rizzo, F.R.; Centonze, D.; Mandolesi, G. Laquinimod ameliorates excitotoxic damage by regulating glutamate re-uptake. J. Neuroinflamm. 2018, 15, 5. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Beattie, M.S.; Ferguson, A.R.; Bresnahan, J.C. AMPA-receptor trafficking and injury-induced cell death. Eur. J. Neurosci. 2010, 32, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Stroebel, D.; Casado, M.; Paoletti, P. Triheteromeric NMDA receptors: From structure to synaptic physiology. Curr. Opin. Physiol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Miyashita, T.; Oda, Y.; Horiuchi, J.; Yin, J.C.; Morimoto, T.; Saitoe, M. Mg2+ block of Drosophila NMDA receptors is required for long-term memory formation and CREB-dependent gene expression. Neuron 2012, 74, 887–898. [Google Scholar] [CrossRef]
- Granger, A.J.; Nicoll, R.A. Expression mechanisms underlying long-term potentiation: A postsynaptic view, 10 years on. Philos. Trans. R. Soc. B 2014, 369, 20130136. [Google Scholar] [CrossRef]
- Malinow, R. AMPA receptor trafficking and long-term potentiation. Philos. Trans. R. Soc. B 2003, 358, 707–714. [Google Scholar] [CrossRef]
- Lisman, J.; Raghavachari, S. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Sci. STKE 2006, 2006, re11. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. Expression of NMDA receptor-dependent LTP in the hippocampus: Bridging the divide. Mol. Brain 2013, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Luscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef]
- Huganir, R.L.; Nicoll, R.A. AMPARs and synaptic plasticity: The last 25 years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef]
- Chater, T.E.; Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci. 2014, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Herring, B.E.; Nicoll, R.A. Long-Term Potentiation: From CaMKII to AMPA Receptor Trafficking. Annu. Rev. Physiol. 2016, 78, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J. Glutamatergic synapses are structurally and biochemically complex because of multiple plasticity processes: Long-term potentiation, long-term depression, short-term potentiation and scaling. Philos. Trans. R. Soc. B 2017, 372, 20160260. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, R.; Hayashi, Y.; Hell, J.W. CaMKII: A central molecular organizer of synaptic plasticity, learning and memory. Nat. Rev. Neurosci. 2022, 23, 666–682. [Google Scholar] [CrossRef]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef]
- Kessels, H.W.; Malinow, R. Synaptic AMPA receptor plasticity and behavior. Neuron 2009, 61, 340–350. [Google Scholar] [CrossRef]
- Nabavi, S.; Fox, R.; Proulx, C.D.; Lin, J.Y.; Tsien, R.Y.; Malinow, R. Engineering a memory with LTD and LTP. Nature 2014, 511, 348–352. [Google Scholar] [CrossRef]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Derkach, V.A.; Oh, M.C.; Guire, E.S.; Soderling, T.R. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 2007, 8, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFα. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovacs, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Faria-Pereira, A.; Morais, V.A. Synapses: The Brain’s Energy-Demanding Sites. Int. J. Mol. Sci. 2022, 23, 3627. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E., Jr.; Perkins, G.A.; Giddabasappa, A.; Chaney, S.; Xiao, W.; White, A.D.; Brown, J.M.; Waggoner, J.; Ellisman, M.H.; Fox, D.A. Spatiotemporal regulation of ATP and Ca2+ dynamics in vertebrate rod and cone ribbon synapses. Mol. Vis. 2007, 13, 887–919. [Google Scholar] [PubMed]
- Huang, L.; Jin, J.; Chen, K.; You, S.; Zhang, H.; Sideris, A.; Norcini, M.; Recio-Pinto, E.; Wang, J.; Gan, W.B.; et al. BDNF produced by cerebral microglia promotes cortical plasticity and pain hypersensitivity after peripheral nerve injury. PLoS Biol. 2021, 19, e3001337. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E. Presynaptic LTP and LTD of excitatory and inhibitory synapses. Cold Spring Harb. Perspect. Biol. 2012, 4, a005728. [Google Scholar] [CrossRef] [PubMed]
- Turrigiano, G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef] [PubMed]
- Turrigiano, G.G. The self-tuning neuron: Synaptic scaling of excitatory synapses. Cell 2008, 135, 422–435. [Google Scholar] [CrossRef]
- Vitureira, N.; Goda, Y. Cell biology in neuroscience: The interplay between Hebbian and homeostatic synaptic plasticity. J. Cell Biol. 2013, 203, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Vitureira, N.; Letellier, M.; Goda, Y. Homeostatic synaptic plasticity: From single synapses to neural circuits. Curr. Opin. Neurobiol. 2012, 22, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Pribiag, H.; Stellwagen, D. Neuroimmune regulation of homeostatic synaptic plasticity. Neuropharmacology 2014, 78, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, D.; Carvalho, A.L. Mechanisms of homeostatic plasticity in the excitatory synapse. J. Neurochem. 2016, 139, 973–996. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The human brain in numbers: A linearly scaled-up primate brain. Front. Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Chung, W.S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896. [Google Scholar] [CrossRef]
- Deitmer, J.W. pH regulation and acid/base-mediated transport in glial cells. In Glial ⇔ Neuronal Signaling; Hatton, G.I., Parpura, V., Eds.; Springer: Boston, MA, USA, 2004; pp. 263–277. [Google Scholar] [CrossRef]
- Brown, A.M.; Ransom, B.R. Astrocyte glycogen and brain energy metabolism. Glia 2007, 55, 1263–1271. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M. Garbage truck of the brain. Science 2013, 340, 1529–1530. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. The homeostatic astroglia emerges from evolutionary specialization of neural cells. Philos. Trans. R. Soc. B 2016, 371, 20150428. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Barres, B.A. Glia in mammalian development and disease. Development 2015, 142, 3805–3809. [Google Scholar] [CrossRef]
- Chao, T.; Rickmann, M.; Wolff, J. The synapse-astrocyte boundary: Anatomical basis for an integrative role of glia in synaptic transmission. In Tripertite Synapses: Synaptic Transmission with Glia; Volterra, A., Magistretti, P., Haydon, P., Eds.; Oxford University Press: New York, NY, USA, 2002; pp. 3–23. [Google Scholar]
- Farmer, W.T.; Murai, K. Resolving Astrocyte Heterogeneity in the CNS. Front. Cell. Neurosci. 2017, 11, 300. [Google Scholar] [CrossRef]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron 2017, 95, 531–549.e9. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Ventura, R.; Harris, K.M. Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 1999, 19, 6897–6906. [Google Scholar] [CrossRef]
- Perea, G.; Araque, A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science 2007, 317, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Astroglial cradle in the life of the synapse. Philos. Trans. R. Soc. B 2014, 369, 20130595. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Volterra, A. A neuron-glia signalling network in the active brain. Curr. Opin. Neurobiol. 2001, 11, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Panatier, A.; Robitaille, R. Astrocytic mGluR5 and the tripartite synapse. Neuroscience 2016, 323, 29–34. [Google Scholar] [CrossRef]
- De Pitta, M.; Brunel, N.; Volterra, A. Astrocytes: Orchestrating synaptic plasticity? Neuroscience 2016, 323, 43–61. [Google Scholar] [CrossRef]
- Eroglu, C.; Barres, B.A. Regulation of synaptic connectivity by glia. Nature 2010, 468, 223–231. [Google Scholar] [CrossRef]
- Colon-Ramos, D.A.; Margeta, M.A.; Shen, K. Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science 2007, 318, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, C.; Allen, N.J.; Susman, M.W.; O’Rourke, N.A.; Park, C.Y.; Ozkan, E.; Chakraborty, C.; Mulinyawe, S.B.; Annis, D.S.; Huberman, A.D.; et al. Gabapentin receptor α2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 2009, 139, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.C.; Jiang, X.; Taylor, A.; Mennerick, S. Astrocyte-derived thrombospondins mediate the development of hippocampal presynaptic plasticity in vitro. J. Neurosci. 2012, 32, 13100–13110. [Google Scholar] [CrossRef]
- Fuentes-Medel, Y.; Ashley, J.; Barria, R.; Maloney, R.; Freeman, M.; Budnik, V. Integration of a retrograde signal during synapse formation by glia-secreted TGF-beta ligand. Curr. Biol. 2012, 22, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Risher, W.C.; Eroglu, C. Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix Biol. 2012, 31, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Stogsdill, J.A.; Pulimood, N.S.; Dingsdale, H.; Kim, Y.H.; Pilaz, L.J.; Kim, I.H.; Manhaes, A.C.; Rodrigues, W.S., Jr.; Pamukcu, A.; et al. Astrocytes Assemble Thalamocortical Synapses by Bridging NRX1α and NL1 via Hevin. Cell 2016, 164, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.J.; Lopez-Soto, E.J.; Li, L.; Liu, H.; Nedelcu, D.; Lipscombe, D.; Hu, Z.; Kaplan, J.M. Retrograde Synaptic Inhibition Is Mediated by α-Neurexin Binding to the α2δ Subunits of N-Type Calcium Channels. Neuron 2017, 95, 326–340.e325. [Google Scholar] [CrossRef] [PubMed]
- Song, I.; Dityatev, A. Crosstalk between glia, extracellular matrix and neurons. Brain Res. Bull. 2018, 136, 101–108. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; van Casteren, A.C.M.; Lee, A.; Chang, V.T.; Aricescu, A.R.; Allen, N.J. Astrocyte-Secreted Glypican 4 Regulates Release of Neuronal Pentraxin 1 from Axons to Induce Functional Synapse Formation. Neuron 2017, 96, 428–445.e413. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Allen, N.J. Astrocyte regulation of synaptic behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 439–463. [Google Scholar] [CrossRef]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Oliet, S.H.; Piet, R.; Poulain, D.A. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 2001, 292, 923–926. [Google Scholar] [CrossRef]
- Conti, F.; Minelli, A.; Melone, M. GABA transporters in the mammalian cerebral cortex: Localization, development and pathological implications. Brain Res. Rev. 2004, 45, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Beenhakker, M.P.; Huguenard, J.R. Astrocytes as gatekeepers of GABAB receptor function. J. Neurosci. 2010, 30, 15262–15276. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Shih, P.Y.; Gomi, H.; Yoshida, T.; Nakai, J.; Ando, R.; Furuichi, T.; Mikoshiba, K.; Semyanov, A.; Itohara, S. Astrocytic Ca2+ signals are required for the functional integrity of tripartite synapses. Mol. Brain 2013, 6, 6. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.; Barres, B. SnapShot: Astrocytes in Health and Disease. Cell 2015, 162, 1170–1170.e1. [Google Scholar] [CrossRef]
- Lopez-Colome, A.M.; Lopez, E.; Mendez-Flores, O.G.; Ortega, A. Glutamate Receptor Stimulation Up-Regulates Glutamate Uptake in Human Muller Glia Cells. Neurochem. Res. 2016, 41, 1797–1805. [Google Scholar] [CrossRef]
- Danbolt, N.C.; Furness, D.N.; Zhou, Y. Neuronal vs. glial glutamate uptake: Resolving the conundrum. Neurochem. Int. 2016, 98, 29–45. [Google Scholar] [CrossRef]
- Grewer, C.; Gameiro, A.; Rauen, T. SLC1 glutamate transporters. Pflügers Arch. 2014, 466, 3–24. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Pines, G.; Danbolt, N.C.; Bjoras, M.; Zhang, Y.; Bendahan, A.; Eide, L.; Koepsell, H.; Storm-Mathisen, J.; Seeberg, E.; Kanner, B.I. Cloning and expression of a rat brain L-glutamate transporter. Nature 1992, 360, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Storck, T.; Schulte, S.; Hofmann, K.; Stoffel, W. Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc. Natl. Acad. Sci. USA 1992, 89, 10955–10959. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Hediger, M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 1992, 360, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Fairman, W.A.; Vandenberg, R.J.; Arriza, J.L.; Kavanaugh, M.P.; Amara, S.G. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 1995, 375, 599–603. [Google Scholar] [CrossRef]
- Palmer, M.J.; Taschenberger, H.; Hull, C.; Tremere, L.; von Gersdorff, H. Synaptic activation of presynaptic glutamate transporter currents in nerve terminals. J. Neurosci. 2003, 23, 4831–4841. [Google Scholar] [CrossRef]
- Hasegawa, J.; Obara, T.; Tanaka, K.; Tachibana, M. High-density presynaptic transporters are required for glutamate removal from the first visual synapse. Neuron 2006, 50, 63–74. [Google Scholar] [CrossRef]
- Furness, D.N.; Dehnes, Y.; Akhtar, A.Q.; Rossi, D.J.; Hamann, M.; Grutle, N.J.; Gundersen, V.; Holmseth, S.; Lehre, K.P.; Ullensvang, K.; et al. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: New insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 2008, 157, 80–94. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. New functions of Muller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef]
- Rimmele, T.S.; Rosenberg, P.A. GLT-1: The elusive presynaptic glutamate transporter. Neurochem. Int. 2016, 98, 19–28. [Google Scholar] [CrossRef]
- Murphy-Royal, C.; Dupuis, J.; Groc, L.; Oliet, S.H.R. Astroglial glutamate transporters in the brain: Regulating neurotransmitter homeostasis and synaptic transmission. J. Neurosci. Res. 2017, 95, 2140–2151. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Takano, H.; Dong, J.H.; Haydon, P.G. Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 2007, 27, 6473–6477. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Asan, E.; Lesch, K.P.; Kugler, P. A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: Cloning and localization in rat nervous system. Neuroscience 2002, 109, 45–61. [Google Scholar] [CrossRef]
- Reye, P.; Sullivan, R.; Fletcher, E.L.; Pow, D.V. Distribution of two splice variants of the glutamate transporter GLT1 in the retinas of humans, monkeys, rabbits, rats, cats, and chickens. J. Comp. Neurol. 2002, 445, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kugler, P.; Schmitt, A. Complementary neuronal and glial expression of two high-affinity glutamate transporter GLT1/EAAT2 forms in rat cerebral cortex. Histochem. Cell Biol. 2003, 119, 425–435. [Google Scholar] [CrossRef]
- Tang, F.S.; Yuan, H.L.; Liu, J.B.; Zhang, G.; Chen, S.Y.; Ke, J.B. Glutamate Transporters EAAT2 and EAAT5 Differentially Shape Synaptic Transmission from Rod Bipolar Cell Terminals. eNeuro 2022, 9. [Google Scholar] [CrossRef]
- Dembla, M.; Kesharwani, A.; Natarajan, S.; Fecher-Trost, C.; Fairless, R.; Williams, S.K.; Flockerzi, V.; Diem, R.; Schwarz, K.; Schmitz, F. Early auto-immune targeting of photoreceptor ribbon synapses in mouse models of multiple sclerosis. EMBO Mol. Med. 2018, 10, e8926. [Google Scholar] [CrossRef]
- Mukherjee, A.; Katiyar, R.; Dembla, E.; Dembla, M.; Kumar, P.; Belkacemi, A.; Jung, M.; Beck, A.; Flockerzi, V.; Schwarz, K.; et al. Disturbed Presynaptic Ca2+ Signaling in Photoreceptors in the EAE Mouse Model of Multiple Sclerosis. iScience 2020, 23, 101830. [Google Scholar] [CrossRef]
- Kesharwani, A.; Schwarz, K.; Dembla, E.; Dembla, M.; Schmitz, F. Early Changes in Exo- and Endocytosis in the EAE Mouse Model of Multiple Sclerosis Correlate with Decreased Synaptic Ribbon Size and Reduced Ribbon-Associated Vesicle Pools in Rod Photoreceptor Synapses. Int. J. Mol. Sci. 2021, 22, 10789. [Google Scholar] [CrossRef]
- Bennett, J.L.; Costello, F.; Chen, J.J.; Petzold, A.; Biousse, V.; Newman, N.J.; Galetta, S.L. Optic neuritis and autoimmune optic neuropathies: Advances in diagnosis and treatment. Lancet Neurol. 2023, 22, 89–100. [Google Scholar] [CrossRef]
- Lehre, K.P.; Davanger, S.; Danbolt, N.C. Localization of the glutamate transporter protein GLAST in rat retina. Brain Res. 1997, 744, 129–137. [Google Scholar] [CrossRef]
- Derouiche, A.; Rauen, T. Coincidence of L-glutamate/L-aspartate transporter (GLAST) and glutamine synthetase (GS) immunoreactions in retinal glia: Evidence for coupling of GLAST and GS in transmitter clearance. J. Neurosci. Res. 1995, 42, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Rauen, T.; Rothstein, J.D.; Wassle, H. Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res. 1996, 286, 325–336. [Google Scholar] [CrossRef]
- Rauen, T.; Taylor, W.R.; Kuhlbrodt, K.; Wiessner, M. High-affinity glutamate transporters in the rat retina: A major role of the glial glutamate transporter GLAST-1 in transmitter clearance. Cell Tissue Res. 1998, 291, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Harada, C.; Watanabe, M.; Inoue, Y.; Sakagawa, T.; Nakayama, N.; Sasaki, S.; Okuyama, S.; Watase, K.; Wada, K.; et al. Functions of the two glutamate transporters GLAST and GLT-1 in the retina. Proc. Natl. Acad. Sci. USA 1998, 95, 4663–4666. [Google Scholar] [CrossRef] [PubMed]
- Pow, D.V.; Barnett, N.L. Changing patterns of spatial buffering of glutamate in developing rat retinae are mediated by the Muller cell glutamate transporter GLAST. Cell Tissue Res. 1999, 297, 57–66. [Google Scholar] [CrossRef]
- Kugler, P.; Beyer, A. Expression of glutamate transporters in human and rat retina and rat optic nerve. Histochem. Cell Biol. 2003, 120, 199–212. [Google Scholar] [CrossRef]
- Fyk-Kolodziej, B.; Qin, P.; Dzhagaryan, A.; Pourcho, R.G. Differential cellular and subcellular distribution of glutamate transporters in the cat retina. Vis. Neurosci. 2004, 21, 551–565. [Google Scholar] [CrossRef]
- Sarthy, V.P.; Pignataro, L.; Pannicke, T.; Weick, M.; Reichenbach, A.; Harada, T.; Tanaka, K.; Marc, R. Glutamate transport by retinal Muller cells in glutamate/aspartate transporter-knockout mice. Glia 2005, 49, 184–196. [Google Scholar] [CrossRef]
- Broer, S.; Brookes, N. Transfer of glutamine between astrocytes and neurons. J. Neurochem. 2001, 77, 705–719. [Google Scholar] [CrossRef]
- Wersinger, E.; Schwab, Y.; Sahel, J.A.; Rendon, A.; Pow, D.V.; Picaud, S.; Roux, M.J. The glutamate transporter EAAT5 works as a presynaptic receptor in mouse rod bipolar cells. J. Physiol. 2006, 577, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Veruki, M.L.; Morkve, S.H.; Hartveit, E. Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat. Neurosci. 2006, 9, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Gehlen, J.; Aretzweiler, C.; Mataruga, A.; Fahlke, C.; Muller, F. Excitatory Amino Acid Transporter EAAT5 Improves Temporal Resolution in the Retina. eNeuro 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Barbour, B.; Brew, H.; Attwell, D. Electrogenic glutamate uptake in glial cells is activated by intracellular potassium. Nature 1988, 335, 433–435. [Google Scholar] [CrossRef]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef]
- Grewer, C.; Gameiro, A.; Zhang, Z.; Tao, Z.; Braams, S.; Rauen, T. Glutamate forward and reverse transport: From molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 2008, 60, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Malarkey, E.B.; Parpura, V. Mechanisms of glutamate release from astrocytes. Neurochem. Int. 2008, 52, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Jackman, N.A.; Uliasz, T.F.; Hewett, J.A.; Hewett, S.J. Regulation of system x(c)(-)activity and expression in astrocytes by interleukin-1beta: Implications for hypoxic neuronal injury. Glia 2010, 58, 1806–1815. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Park, H.; Oh, S.J.; Han, K.S.; Woo, D.H.; Park, H.; Mannaioni, G.; Traynelis, S.F.; Lee, C.J. Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. J. Neurosci. 2009, 29, 13063–13073. [Google Scholar] [CrossRef]
- Park, H.; Han, K.S.; Seo, J.; Lee, J.; Dravid, S.M.; Woo, J.; Chun, H.; Cho, S.; Bae, J.Y.; An, H.; et al. Channel-mediated astrocytic glutamate modulates hippocampal synaptic plasticity by activating postsynaptic NMDA receptors. Mol. Brain 2015, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Woo, D.H.; Han, K.S.; Shim, J.W.; Yoon, B.E.; Kim, E.; Bae, J.Y.; Oh, S.J.; Hwang, E.M.; Marmorstein, A.D.; Bae, Y.C.; et al. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 2012, 151, 25–40. [Google Scholar] [CrossRef]
- Satarker, S.; Bojja, S.L.; Gurram, P.C.; Mudgal, J.; Arora, D.; Nampoothiri, M. Astrocytic Glutamatergic Transmission and Its Implications in Neurodegenerative Disorders. Cells 2022, 11, 1139. [Google Scholar] [CrossRef] [PubMed]
- Junankar, P.R.; Kirk, K. Organic osmolyte channels: A comparative view. Cell. Physiol. Biochem. 2000, 10, 355–360. [Google Scholar] [CrossRef]
- Eggermont, J.; Trouet, D.; Carton, I.; Nilius, B. Cellular function and control of volume-regulated anion channels. Cell. Biochem. Biophys. 2001, 35, 263–274. [Google Scholar] [CrossRef]
- Beppu, K.; Kubo, N.; Matsui, K. Glial amplification of synaptic signals. J. Physiol. 2021, 599, 2085–2102. [Google Scholar] [CrossRef]
- Jourdain, P.; Bergersen, L.H.; Bhaukaurally, K.; Bezzi, P.; Santello, M.; Domercq, M.; Matute, C.; Tonello, F.; Gundersen, V.; Volterra, A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 2007, 10, 331–339. [Google Scholar] [CrossRef]
- Halassa, M.M.; Haydon, P.G. Integrated brain circuits: Astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 2010, 72, 335–355. [Google Scholar] [CrossRef]
- Sun, W.; McConnell, E.; Pare, J.F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; Nedergaard, M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 2013, 339, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef]
- Ceprian, M.; Fulton, D. Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. Int. J. Mol. Sci. 2019, 20, 2450. [Google Scholar] [CrossRef] [PubMed]
- Durkee, C.A.; Araque, A. Diversity and Specificity of Astrocyte-neuron Communication. Neuroscience 2019, 396, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFα: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef]
- Pascual, O.; Ben Achour, S.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2012, 109, E197–E205. [Google Scholar] [CrossRef]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Klaus, F.R.; Kollias, G.; Fontana, A.; Pryce, C.R.; et al. Neuroinflammatory TNFα Impairs Memory via Astrocyte Signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Santello, M.; Volterra, A. TNFα in synaptic function: Switching gears. Trends Neurosci. 2012, 35, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, A.; Damasceno, B.P.; Cendes, F. The clinical impact of cerebellar grey matter pathology in multiple sclerosis. PLoS ONE 2014, 9, e96193. [Google Scholar] [CrossRef]
- Planche, V.; Ruet, A.; Coupe, P.; Lamargue-Hamel, D.; Deloire, M.; Pereira, B.; Manjon, J.V.; Munsch, F.; Moscufo, N.; Meier, D.S.; et al. Hippocampal microstructural damage correlates with memory impairment in clinically isolated syndrome suggestive of multiple sclerosis. Mult. Scler. J. 2017, 23, 1214–1224. [Google Scholar] [CrossRef]
- Newman, E.A. New roles for astrocytes: Regulation of synaptic transmission. Trends Neurosci. 2003, 26, 536–542. [Google Scholar] [CrossRef]
- Hamilton, N.B.; Attwell, D. Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 2010, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Sofroniew, M.V. Seducing astrocytes to the dark side. Cell Res. 2017, 27, 726–727. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Scarlata, C.; Brambilla, L.; Rossi, D. Tumor Necrosis Factor Alpha in Amyotrophic Lateral Sclerosis: Friend or Foe? Cells 2021, 10, 518. [Google Scholar] [CrossRef]
- Fiacco, T.A.; McCarthy, K.D. Multiple Lines of Evidence Indicate that Gliotransmission does not Occur under Physiological Conditions. J. Neurosci. 2018, 38, 3–13. [Google Scholar] [CrossRef]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef]
- Schwarz, Y.; Zhao, N.; Kirchhoff, F.; Bruns, D. Astrocytes control synaptic strength by two distinct v-SNARE-dependent release pathways. Nat. Neurosci. 2017, 20, 1529–1539. [Google Scholar] [CrossRef]
- Santello, M.; Bezzi, P.; Volterra, A. TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 2011, 69, 988–1001. [Google Scholar] [CrossRef]
- Petrelli, F.; Bezzi, P. Novel insights into gliotransmitters. Curr. Opin. Pharmacol. 2016, 26, 138–145. [Google Scholar] [CrossRef]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef]
- Heir, R.; Stellwagen, D. TNF-Mediated Homeostatic Synaptic Plasticity: From in vitro to in vivo Models. Front. Cell. Neurosci. 2020, 14, 565841. [Google Scholar] [CrossRef] [PubMed]
- Galanis, C.; Vlachos, A. Hebbian and Homeostatic Synaptic Plasticity-Do Alterations of One Reflect Enhancement of the Other? Front. Cell. Neurosci. 2020, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Vesce, S.; Rossi, D.; Brambilla, L.; Volterra, A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int. Rev. Neurobiol. 2007, 82, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S. Pathophysiology of depression and innovative treatments: Remodeling glutamatergic synaptic connections. Dialogues Clin. Neurosci. 2014, 16, 11–27. [Google Scholar] [CrossRef]
- McEwen, B.S.; Nasca, C.; Gray, J.D. Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology 2016, 41, 3–23. [Google Scholar] [CrossRef]
- Bocchio, M.; Lukacs, I.P.; Stacey, R.; Plaha, P.; Apostolopoulos, V.; Livermore, L.; Sen, A.; Ansorge, O.; Gillies, M.J.; Somogyi, P.; et al. Group II Metabotropic Glutamate Receptors Mediate Presynaptic Inhibition of Excitatory Transmission in Pyramidal Neurons of the Human Cerebral Cortex. Front. Cell. Neurosci. 2018, 12, 508. [Google Scholar] [CrossRef]
- del Rey, A.; Balschun, D.; Wetzel, W.; Randolf, A.; Besedovsky, H.O. A cytokine network involving brain-borne IL-1β, IL-1ra, IL-18, IL-6, and TNFα operates during long-term potentiation and learning. Brain Behav. Immun. 2013, 33, 15–23. [Google Scholar] [CrossRef]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the Central Nervous System. Cell. Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef]
- Hyvarinen, T.; Hagman, S.; Ristola, M.; Sukki, L.; Veijula, K.; Kreutzer, J.; Kallio, P.; Narkilahti, S. Co-stimulation with IL-1beta and TNF-α induces an inflammatory reactive astrocyte phenotype with neurosupportive characteristics in a human pluripotent stem cell model system. Sci. Rep. 2019, 9, 16944. [Google Scholar] [CrossRef]
- Spurgat, M.S.; Tang, S.J. Single-Cell RNA-Sequencing: Astrocyte and Microglial Heterogeneity in Health and Disease. Cells 2022, 11, 2021. [Google Scholar] [CrossRef]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.K.; Kuipers, H.F. She Doesn’t Even Go Here: The Role of Inflammatory Astrocytes in CNS Disorders. Front. Cell. Neurosci. 2021, 15, 704884. [Google Scholar] [CrossRef] [PubMed]
- Ingram, G.; Hakobyan, S.; Robertson, N.P.; Morgan, B.P. Complement in multiple sclerosis: Its role in disease and potential as a biomarker. Clin. Exp. Immunol. 2009, 155, 128–139. [Google Scholar] [CrossRef]
- Michailidou, I.; Naessens, D.M.; Hametner, S.; Guldenaar, W.; Kooi, E.J.; Geurts, J.J.; Baas, F.; Lassmann, H.; Ramaglia, V. Complement C3 on microglial clusters in multiple sclerosis occur in chronic but not acute disease: Implication for disease pathogenesis. Glia 2017, 65, 264–277. [Google Scholar] [CrossRef]
- Michailidou, I.; Willems, J.G.; Kooi, E.J.; van Eden, C.; Gold, S.M.; Geurts, J.J.; Baas, F.; Huitinga, I.; Ramaglia, V. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann. Neurol. 2015, 77, 1007–1026. [Google Scholar] [CrossRef] [PubMed]
- Watkins, L.M.; Neal, J.W.; Loveless, S.; Michailidou, I.; Ramaglia, V.; Rees, M.I.; Reynolds, R.; Robertson, N.P.; Morgan, B.P.; Howell, O.W. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J. Neuroinflamm. 2016, 13, 161. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef]
- Gomez-Nicola, D.; Perry, V.H. Microglial dynamics and role in the healthy and diseased brain: A paradigm of functional plasticity. Neuroscientist 2015, 21, 169–184. [Google Scholar] [CrossRef]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [CrossRef]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Jordao, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar, G.; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar] [CrossRef] [PubMed]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Amann, L.; Monaco, G.; Sankowski, R.; Staszewski, O.; Krueger, M.; Del Gaudio, F.; He, L.; Paterson, N.; Nent, E.; et al. Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature 2022, 604, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Bilimoria, P.M.; Stevens, B. Microglia function during brain development: New insights from animal models. Brain Res. 2015, 1617, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Itriago, A.; Radford, R.A.W.; Aramideh, J.A.; Maurel, C.; Scherer, N.M.; Don, E.K.; Lee, A.; Chung, R.S.; Graeber, M.B.; Morsch, M. Microglia morphophysiological diversity and its implications for the CNS. Front. Immunol. 2022, 13, 997786. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef]
- Tremblay, M.E.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, L.; Zhang, J.; Fariss, R.N.; Ma, W.; Kretschmer, F.; Wang, M.; Qian, H.H.; Badea, T.C.; Diamond, J.S.; et al. Requirement for Microglia for the Maintenance of Synaptic Function and Integrity in the Mature Retina. J. Neurosci. 2016, 36, 2827–2842. [Google Scholar] [CrossRef]
- Hong, S.; Stevens, B. Microglia: Phagocytosing to Clear, Sculpt, and Eliminate. Dev. Cell 2016, 38, 126–128. [Google Scholar] [CrossRef]
- Ohgomori, T.; Iinuma, K.; Yamada, J.; Jinno, S. A unique subtype of ramified microglia associated with synapses in the rat hippocampus. Eur. J. Neurosci. 2021, 54, 4740–4754. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Hristovska, I.; Pascual, O. Deciphering Resting Microglial Morphology and Process Motility from a Synaptic Prospect. Front. Integr. Neurosci. 2015, 9, 73. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, R.; Wake, H.; Kato, D.; Horiuchi, H.; Ono, R.; Ikegami, A.; Haruwaka, K.; Omori, T.; Tachibana, Y.; Moorhouse, A.J.; et al. Microglia Enhance Synapse Activity to Promote Local Network Synchronization. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Fontainhas, A.M.; Wang, M.; Liang, K.J.; Chen, S.; Mettu, P.; Damani, M.; Fariss, R.N.; Li, W.; Wong, W.T. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS ONE 2011, 6, e15973. [Google Scholar] [CrossRef]
- Dissing-Olesen, L.; LeDue, J.M.; Rungta, R.L.; Hefendehl, J.K.; Choi, H.B.; MacVicar, B.A. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J. Neurosci. 2014, 34, 10511–10527. [Google Scholar] [CrossRef]
- Eyo, U.B.; Peng, J.; Swiatkowski, P.; Mukherjee, A.; Bispo, A.; Wu, L.J. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J. Neurosci. 2014, 34, 10528–10540. [Google Scholar] [CrossRef]
- Campagno, K.E.; Mitchell, C.H. The P2X(7) Receptor in Microglial Cells Modulates the Endolysosomal Axis, Autophagy, and Phagocytosis. Front. Cell. Neurosci. 2021, 15, 645244. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Kyrargyri, V.; Madry, C.; Rifat, A.; Arancibia-Carcamo, I.L.; Jones, S.P.; Chan, V.T.T.; Xu, Y.; Robaye, B.; Attwell, D. P2Y(13) receptors regulate microglial morphology, surveillance, and resting levels of interleukin 1beta release. Glia 2020, 68, 328–344. [Google Scholar] [CrossRef]
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 2020, 9, 1108. [Google Scholar] [CrossRef]
- Lalo, U.; Palygin, O.; Verkhratsky, A.; Grant, S.G.; Pankratov, Y. ATP from synaptic terminals and astrocytes regulates NMDA receptors and synaptic plasticity through PSD-95 multi-protein complex. Sci. Rep. 2016, 6, 33609. [Google Scholar] [CrossRef] [PubMed]
- Boue-Grabot, E.; Pankratov, Y. Modulation of Central Synapses by Astrocyte-Released ATP and Postsynaptic P2X Receptors. Neural. Plast. 2017, 2017, 9454275. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Suh, H.S.; Zhao, M.L.; Derico, L.; Choi, N.; Lee, S.C. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: Differential regulation by inflammatory mediators. J. Neuroinflamm. 2013, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Masuda, T.; Prinz, M. Microglia: A Unique Versatile Cell in the Central Nervous System. ACS Chem. Neurosci. 2016, 7, 428–434. [Google Scholar] [CrossRef]
- Zhou, L.J.; Peng, J.; Xu, Y.N.; Zeng, W.J.; Zhang, J.; Wei, X.; Mai, C.L.; Lin, Z.J.; Liu, Y.; Murugan, M.; et al. Microglia Are Indispensable for Synaptic Plasticity in the Spinal Dorsal Horn and Chronic Pain. Cell Rep. 2019, 27, 3844–3859.e3846. [Google Scholar] [CrossRef]
- Li, Y.; Du, X.F.; Liu, C.S.; Wen, Z.L.; Du, J.L. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev. Cell 2012, 23, 1189–1202. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Are “resting” microglia more “m2”? Front. Immunol. 2014, 5, 594. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Fernandez-Suarez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar, G.; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Tan, Y.L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e3. [Google Scholar] [CrossRef]
- Chhatbar, C.; Prinz, M. The roles of microglia in viral encephalitis: From sensome to therapeutic targeting. Cell. Mol. Immunol. 2021, 18, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Farber, K.; Kettenmann, H. Purinergic signaling and microglia. Pflügers Arch. 2006, 452, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Koizumi, S.; Tsuda, M. The role of nucleotides in the neuron-glia communication responsible for the brain functions. J. Neurochem. 2007, 102, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Hoshiko, M.; Arnoux, I.; Avignone, E.; Yamamoto, N.; Audinat, E. Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 2012, 32, 15106–15111. [Google Scholar] [CrossRef]
- Cheadle, L.; Rivera, S.A.; Phelps, J.S.; Ennis, K.A.; Stevens, B.; Burkly, L.C.; Lee, W.A.; Greenberg, M.E. Sensory Experience Engages Microglia to Shape Neural Connectivity through a Non-Phagocytic Mechanism. Neuron 2020, 108, 451–468.e459. [Google Scholar] [CrossRef]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed]
- de Jong, B.A.; Huizinga, T.W.; Bollen, E.L.; Uitdehaag, B.M.; Bosma, G.P.; van Buchem, M.A.; Remarque, E.J.; Burgmans, A.C.; Kalkers, N.F.; Polman, C.H.; et al. Production of IL-1β and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J. Neuroimmunol. 2002, 126, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate immunity and neuroinflammation. Mediat. Inflamm. 2013, 2013, 342931. [Google Scholar] [CrossRef] [PubMed]
- Levesque, S.A.; Pare, A.; Mailhot, B.; Bellver-Landete, V.; Kebir, H.; Lecuyer, M.A.; Alvarez, J.I.; Prat, A.; de Rivero Vaccari, J.P.; Keane, R.W.; et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1β-driven neuroinflammation during autoimmune encephalomyelitis in mice. J. Exp. Med. 2016, 213, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.R.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Vanni, V.; Guadalupi, L.; Stampanoni Bassi, M.; Buttari, F.; Mandolesi, G.; et al. Tumor Necrosis Factor and Interleukin-1β Modulate Synaptic Plasticity during Neuroinflammation. Neural. Plast. 2018, 2018, 8430123. [Google Scholar] [CrossRef] [PubMed]
- Rovaris, M.; Barnes, D.; Woodrofe, N.; du Boulay, G.H.; Thorpe, J.W.; Thompson, A.J.; McDonald, W.I.; Miller, D.H. Patterns of disease activity in multiple sclerosis patients: A study with quantitative gadolinium-enhanced brain MRI and cytokine measurement in different clinical subgroups. J. Neurol. 1996, 243, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Baraczka, K.; Pozsonyi, T.; Szuts, I.; Ormos, G.; Nekam, K. Increased levels of tumor necrosis alpha and soluble vascular endothelial adhesion molecule-1 in the cerebrospinal fluid of patients with connective tissue diseases and multiple sclerosis. Acta Microbiol. Immunol. Hung. 2003, 50, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef]
- Szymocha, R.; Akaoka, H.; Dutuit, M.; Malcus, C.; Didier-Bazes, M.; Belin, M.F.; Giraudon, P. Human T-cell lymphotropic virus type 1-infected T lymphocytes impair catabolism and uptake of glutamate by astrocytes via Tax-1 and tumor necrosis factor alpha. J. Virol. 2000, 74, 6433–6441. [Google Scholar] [CrossRef]
- Ohgoh, M.; Hanada, T.; Smith, T.; Hashimoto, T.; Ueno, M.; Yamanishi, Y.; Watanabe, M.; Nishizawa, Y. Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002, 125, 170–178. [Google Scholar] [CrossRef]
- Mitosek-Szewczyk, K.; Sulkowski, G.; Stelmasiak, Z.; Struzynska, L. Expression of glutamate transporters GLT-1 and GLAST in different regions of rat brain during the course of experimental autoimmune encephalomyelitis. Neuroscience 2008, 155, 45–52. [Google Scholar] [CrossRef]
- Prow, N.A.; Irani, D.N. The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J. Neurochem. 2008, 105, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; De Vito, F.; Musella, A.; Gentile, A.; Bullitta, S.; Fresegna, D.; Sepman, H.; Di Sanza, C.; Haji, N.; Mori, F.; et al. miR-142-3p Is a Key Regulator of IL-1β-Dependent Synaptopathy in Neuroinflammation. J. Neurosci. 2017, 37, 546–561. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Hardingham, G.E. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem. Soc. Trans. 2009, 37 Pt 6, 1147–1160. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Bengtson, C.P.; Buchthal, B.; Hagenston, A.M.; Bading, H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Takaki, J.; Fujimori, K.; Miura, M.; Suzuki, T.; Sekino, Y.; Sato, K. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: The ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J. Neuroinflamm. 2012, 9, 275. [Google Scholar] [CrossRef]
- Yu, Z.; Cheng, G.; Wen, X.; Wu, G.D.; Lee, W.T.; Pleasure, D. Tumor necrosis factor alpha increases neuronal vulnerability to excitotoxic necrosis by inducing expression of the AMPA-glutamate receptor subunit GluR1 via an acid sphingomyelinase- and NF-kappaB-dependent mechanism. Neurobiol. Dis. 2002, 11, 199–213. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Zhao, P.; Beattie, E.C. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J. Neurosci. 2008, 28, 2119–2130. [Google Scholar] [CrossRef]
- Ogoshi, F.; Yin, H.Z.; Kuppumbatti, Y.; Song, B.; Amindari, S.; Weiss, J.H. Tumor necrosis-factor-alpha (TNF-α) induces rapid insertion of Ca2+-permeable alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp. Neurol. 2005, 193, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.R.; Christensen, R.N.; Gensel, J.C.; Miller, B.A.; Sun, F.; Beattie, E.C.; Bresnahan, J.C.; Beattie, M.S. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J. Neurosci. 2008, 28, 11391–11400. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.; Paul, C. MK-801 limits neurovascular dysfunction during experimental allergic encephalomyelitis. J. Pharmacol. Exp. Ther. 1997, 282, 397–402. [Google Scholar]
- Paul, C.; Bolton, C. Modulation of blood-brain barrier dysfunction and neurological deficits during acute experimental allergic encephalomyelitis by the N-methyl-D-aspartate receptor antagonist memantine. J. Pharmacol. Exp. Ther. 2002, 302, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.; Knapp, E.; Bandaru, V.V.; Wang, Y.; Knorr, D.; Poirier, C.; Mattson, M.P.; Geiger, J.D.; Haughey, N.J. Tumor necrosis factor-alpha-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J. Neurochem. 2009, 109, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, G.; Rossi, S.; Musella, A.; Gentile, A.; Loizzo, S.; Muzio, L.; Di Sanza, C.; Errico, F.; Musumeci, G.; Haji, N.; et al. Abnormal NMDA receptor function exacerbates experimental autoimmune encephalomyelitis. Br. J. Pharmacol. 2013, 168, 502–517. [Google Scholar] [CrossRef]
- Sulkowski, G.; Dabrowska-Bouta, B.; Chalimoniuk, M.; Struzynska, L. Effects of antagonists of glutamate receptors on pro-inflammatory cytokines in the brain cortex of rats subjected to experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 261, 67–76. [Google Scholar] [CrossRef]
- Bolton, C.; Wood, E.G.; Ayoub, S.S. N-Methyl-D-aspartate (NMDA) receptor involvement in central nervous system prostaglandin production during the relapse phase of chronic relapsing experimental autoimmune encephalomyelitis (CR EAE). Fundam. Clin. Pharmacol. 2013, 27, 535–543. [Google Scholar] [CrossRef]
- Suhs, K.W.; Fairless, R.; Williams, S.K.; Heine, K.; Cavalie, A.; Diem, R. N-methyl-D-aspartate receptor blockade is neuroprotective in experimental autoimmune optic neuritis. J. Neuropathol. Exp. Neurol. 2014, 73, 507–518. [Google Scholar] [CrossRef]
- Dabrowska-Bouta, B.; Struzynska, L.; Chalimoniuk, M.; Frontczak-Baniewicz, M.; Sulkowski, G. The influence of glutamatergic receptor antagonists on biochemical and ultrastructural changes in myelin membranes of rats subjected to experimental autoimmune encephalomyelitis. Folia Neuropathol. 2015, 53, 317–326. [Google Scholar] [CrossRef]
- Fairless, R.; Bading, H.; Diem, R. Pathophysiological Ionotropic Glutamate Signalling in Neuroinflammatory Disease as a Therapeutic Target. Front. Neurosci. 2021, 15, 741280. [Google Scholar] [CrossRef]
- Floden, A.M.; Li, S.; Combs, C.K. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J. Neurosci. 2005, 25, 2566–2575. [Google Scholar] [CrossRef]
- Han, P.; Whelan, P.J. Tumor necrosis factor alpha enhances glutamatergic transmission onto spinal motoneurons. J. Neurotrauma. 2010, 27, 287–292. [Google Scholar] [CrossRef]
- Rossi, S.; Muzio, L.; De Chiara, V.; Grasselli, G.; Musella, A.; Musumeci, G.; Mandolesi, G.; De Ceglia, R.; Maida, S.; Biffi, E.; et al. Impaired striatal GABA transmission in experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2011, 25, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; Grasselli, G.; Musella, A.; Gentile, A.; Musumeci, G.; Sepman, H.; Haji, N.; Fresegna, D.; Bernardi, G.; Centonze, D. GABAergic signaling and connectivity on Purkinje cells are impaired in experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2012, 46, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Pribiag, H.; Stellwagen, D. TNF-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J. Neurosci. 2013, 33, 15879–15893. [Google Scholar] [CrossRef] [PubMed]
- Nistico, R.; Mango, D.; Mandolesi, G.; Piccinin, S.; Berretta, N.; Pignatelli, M.; Feligioni, M.; Musella, A.; Gentile, A.; Mori, F.; et al. Inflammation subverts hippocampal synaptic plasticity in experimental multiple sclerosis. PLoS ONE 2013, 8, e54666. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.S.; Zurek, A.A.; Lecker, I.; Yu, J.; Abramian, A.M.; Avramescu, S.; Davies, P.A.; Moss, S.J.; Lu, W.Y.; Orser, B.A. Memory deficits induced by inflammation are regulated by alpha5-subunit-containing GABAA receptors. Cell Rep. 2012, 2, 488–496. [Google Scholar] [CrossRef]
- Yan, X.; Jiang, E.; Weng, H.R. Activation of toll like receptor 4 attenuates GABA synthesis and postsynaptic GABA receptor activities in the spinal dorsal horn via releasing interleukin-1 beta. J. Neuroinflamm. 2015, 12, 222. [Google Scholar] [CrossRef]
- Roseti, C.; van Vliet, E.A.; Cifelli, P.; Ruffolo, G.; Baayen, J.C.; Di Castro, M.A.; Bertollini, C.; Limatola, C.; Aronica, E.; Vezzani, A.; et al. GABAA currents are decreased by IL-1beta in epileptogenic tissue of patients with temporal lobe epilepsy: Implications for ictogenesis. Neurobiol. Dis. 2015, 82, 311–320. [Google Scholar] [CrossRef]
- Paul, A.M.; Branton, W.G.; Walsh, J.G.; Polyak, M.J.; Lu, J.Q.; Baker, G.B.; Power, C. GABA transport and neuroinflammation are coupled in multiple sclerosis: Regulation of the GABA transporter-2 by ganaxolone. Neuroscience 2014, 273, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Yin, J.; Qin, W.; Sha, S.; Xu, J.; Jiang, C. Role for pro-inflammatory cytokines in regulating expression of GABA transporter type 1 and 3 in specific brain regions of kainic acid-induced status epilepticus. Neurochem. Res. 2015, 40, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.Y.; He, X.Y.; Li, X.F.; Zhang, X.; Huang, Z.W.; Li, J.; Chen, M.; Duan, C.Z. Nefiracetam Attenuates Pro-Inflammatory Cytokines and GABA Transporter in Specific Brain Regions of Rats with Post-Ischemic Seizures. Cell. Physiol. Biochem. 2015, 37, 2023–2031. [Google Scholar] [CrossRef]
- Cawley, N.; Solanky, B.S.; Muhlert, N.; Tur, C.; Edden, R.A.; Wheeler-Kingshott, C.A.; Miller, D.H.; Thompson, A.J.; Ciccarelli, O. Reduced gamma-aminobutyric acid concentration is associated with physical disability in progressive multiple sclerosis. Brain 2015, 138 Pt 9, 2584–2595. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Jin, S.; Suzuki, H.; Doi, Y.; Liang, J.; Kawanokuchi, J.; Mizuno, T.; Sawada, M.; Suzumura, A. Blockade of microglial glutamate release protects against ischemic brain injury. Exp. Neurol. 2008, 214, 144–146. [Google Scholar] [CrossRef]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Sicotte, N.L.; Kern, K.C.; Giesser, B.S.; Arshanapalli, A.; Schultz, A.; Montag, M.; Wang, H.; Bookheimer, S.Y. Regional hippocampal atrophy in multiple sclerosis. Brain 2008, 131 Pt 4, 1134–1141. [Google Scholar] [CrossRef]
- Geurts, J.J.; Calabrese, M.; Fisher, E.; Rudick, R.A. Measurement and clinical effect of grey matter pathology in multiple sclerosis. Lancet Neurol. 2012, 11, 1082–1092. [Google Scholar] [CrossRef]
- Colasanti, A.; Guo, Q.; Giannetti, P.; Wall, M.B.; Newbould, R.D.; Bishop, C.; Onega, M.; Nicholas, R.; Ciccarelli, O.; Muraro, P.A.; et al. Hippocampal Neuroinflammation, Functional Connectivity, and Depressive Symptoms in Multiple Sclerosis. Biol. Psychiatry 2016, 80, 62–72. [Google Scholar] [CrossRef]
- Chiaravalloti, N.D.; DeLuca, J. Cognitive impairment in multiple sclerosis. Lancet Neurol. 2008, 7, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Ziehn, M.O.; Avedisian, A.A.; Tiwari-Woodruff, S.; Voskuhl, R.R. Hippocampal CA1 atrophy and synaptic loss during experimental autoimmune encephalomyelitis, EAE. Lab. Investig. 2010, 90, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Prochnow, N.; Gold, R.; Haghikia, A. An electrophysiologic approach to quantify impaired synaptic transmission and plasticity in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 264, 48–53. [Google Scholar] [CrossRef]
- Planche, V.; Panatier, A.; Hiba, B.; Ducourneau, E.G.; Raffard, G.; Dubourdieu, N.; Maitre, M.; Leste-Lasserre, T.; Brochet, B.; Dousset, V.; et al. Selective dentate gyrus disruption causes memory impairment at the early stage of experimental multiple sclerosis. Brain Behav. Immun. 2017, 60, 240–254. [Google Scholar] [CrossRef]
- Crombe, A.; Planche, V.; Raffard, G.; Bourel, J.; Dubourdieu, N.; Panatier, A.; Fukutomi, H.; Dousset, V.; Oliet, S.; Hiba, B.; et al. Deciphering the microstructure of hippocampal subfields with in vivo DTI and NODDI: Applications to experimental multiple sclerosis. Neuroimage 2018, 172, 357–368. [Google Scholar] [CrossRef]
- Rocca, M.A.; Barkhof, F.; De Luca, J.; Frisen, J.; Geurts, J.J.G.; Hulst, H.E.; Sastre-Garriga, J.; Filippi, M.; Group, M.S. The hippocampus in multiple sclerosis. Lancet Neurol. 2018, 17, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Portaccio, E.; Mancini, A.; Calabresi, P. Multiple sclerosis and cognition: Synaptic failure and network dysfunction. Nat. Rev. Neurosci. 2018, 19, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Greter, M.; Marino, D.; Falsig, J.; Raivich, G.; Hovelmeyer, N.; Waisman, A.; Rulicke, T.; Prinz, M.; Priller, J.; et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 2005, 11, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G.; Beecham, A.H.; James, T.; Replogle, J.; Vlachos, I.S.; et al. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarz, K.; Schmitz, F. Synapse Dysfunctions in Multiple Sclerosis. Int. J. Mol. Sci. 2023, 24, 1639. https://doi.org/10.3390/ijms24021639
Schwarz K, Schmitz F. Synapse Dysfunctions in Multiple Sclerosis. International Journal of Molecular Sciences. 2023; 24(2):1639. https://doi.org/10.3390/ijms24021639
Chicago/Turabian StyleSchwarz, Karin, and Frank Schmitz. 2023. "Synapse Dysfunctions in Multiple Sclerosis" International Journal of Molecular Sciences 24, no. 2: 1639. https://doi.org/10.3390/ijms24021639
APA StyleSchwarz, K., & Schmitz, F. (2023). Synapse Dysfunctions in Multiple Sclerosis. International Journal of Molecular Sciences, 24(2), 1639. https://doi.org/10.3390/ijms24021639