
Wnt Signaling Pathways: From Inflammation to Non-Melanoma Skin Cancers

 , ,
, ,  ,
,  ,
, 
Abstract
1. Introduction
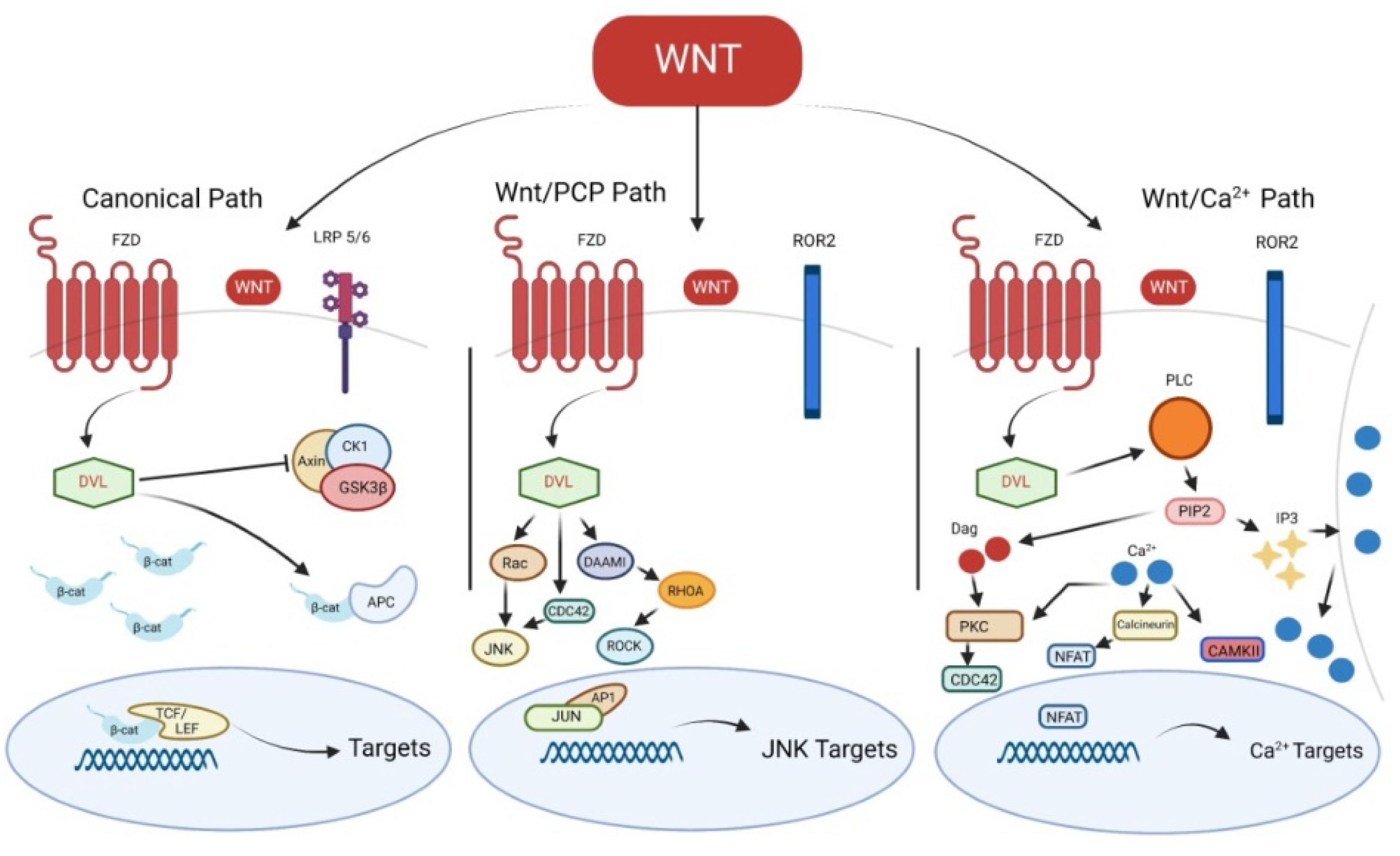
2. Wnt Signaling
3. Wnt Pathway, Inflammation and Carcinogenesis
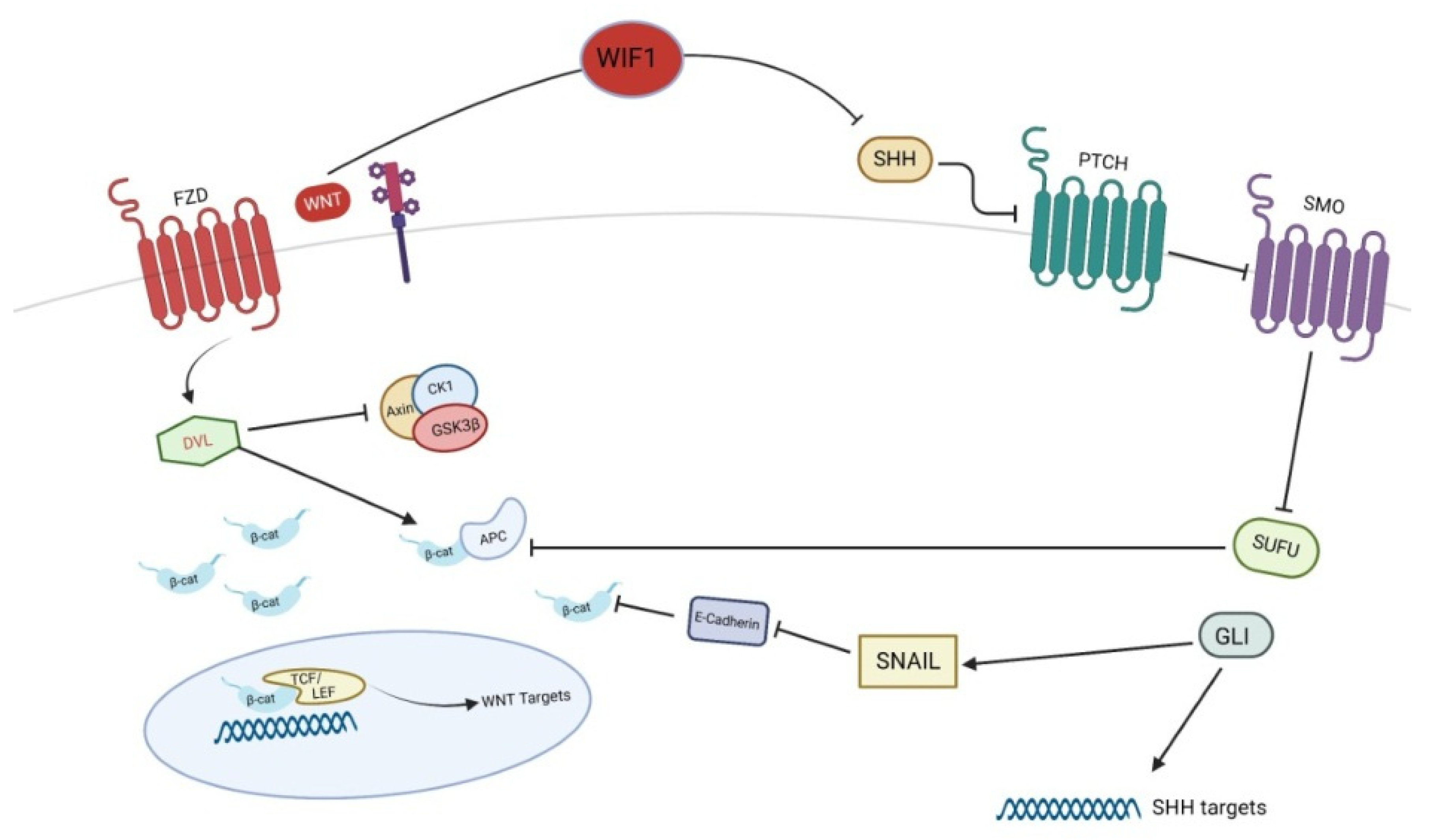
4. Wnt and Basal Cell Carcinoma
5. Wnt and Keratinocyte Carcinomas: From Actinic Keratosis to Cutaneous Squamous Cell Carcinoma
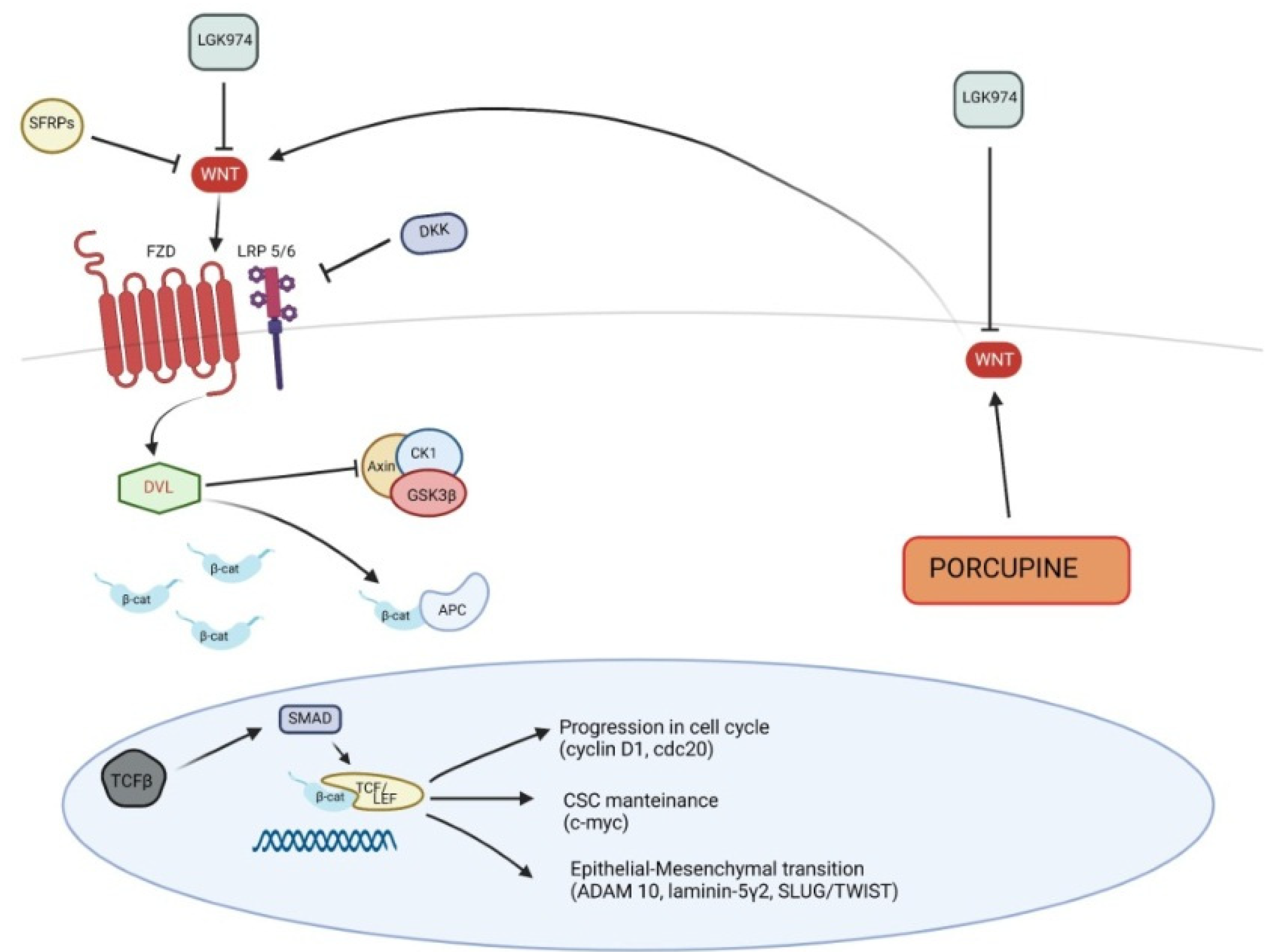
5.1. Canonical Wnt Pathway in Pathogenesis of Cutaneous SCC
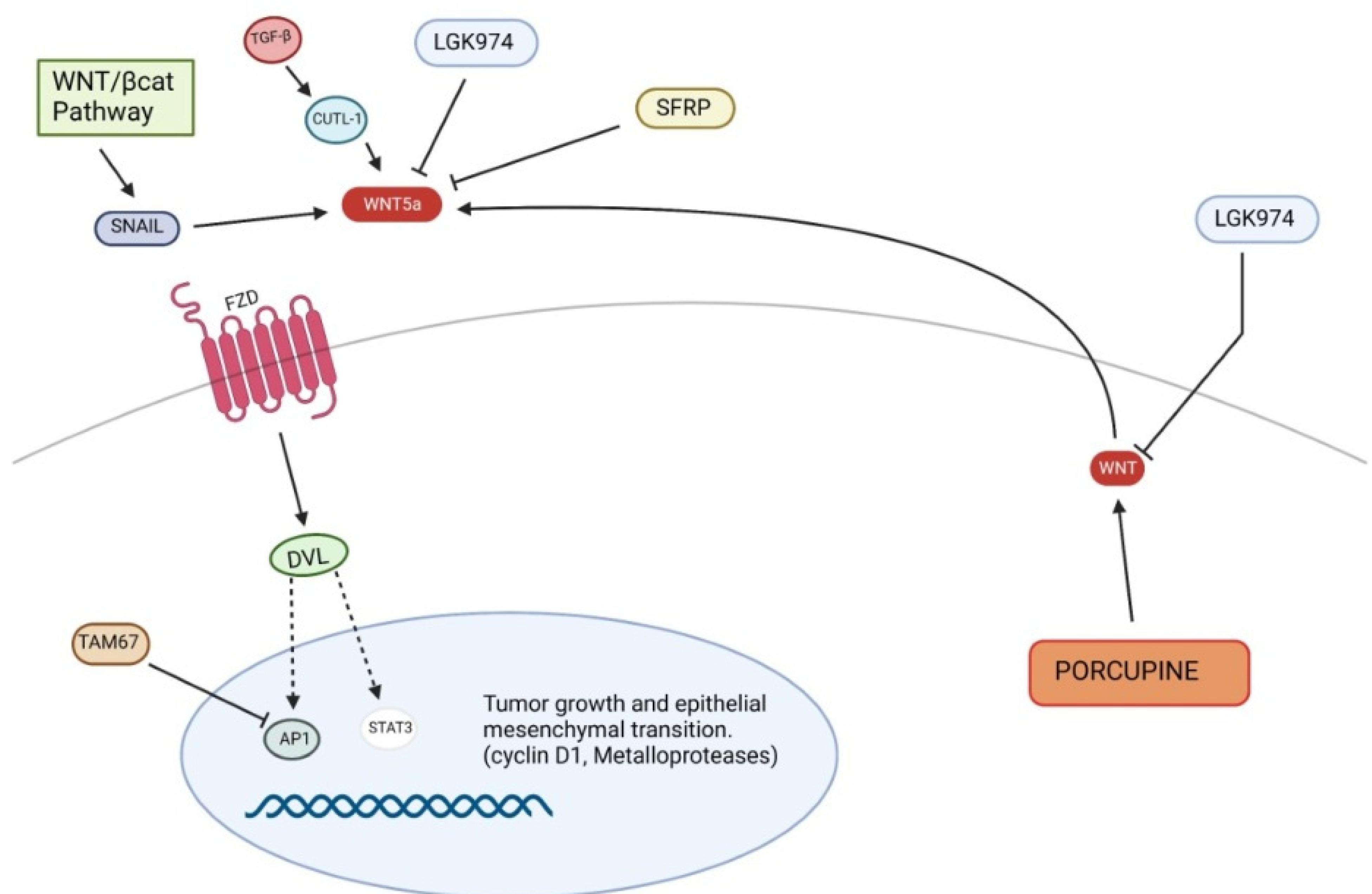
5.2. Non-Canonical Wnt Pathway in Pathogenesis of Cutaneous SCC
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alyoussef, A.; Taha, M. Blocking Wnt as a therapeutic target in mice model of skin cancer. Arch. Dermatol. Res. 2019, 311, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J.D.; Miller, J.R.; Shulman, J.M.; Moon, R.T.; Perrimon, N. Differential recruitment of Dishevelled provides signaling specificity in the planar cell polarity and Wingless signaling pathways. Genes Dev. 1998, 12, 2610–2622. [Google Scholar] [CrossRef] [PubMed]
- Veltri, A.; Lang, C.; Lien, W.H. Concise Review: Wnt Signaling Pathways in Skin Development and Epidermal Stem Cells. Stem Cells 2018, 36, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Koni, M.; Pinnarò, V.; Brizzi, M.F. The Wnt Signalling Pathway: A Tailored Target in Cancer. Int. J. Mol. Sci. 2020, 21, 7697. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Lee, E.; Salic, A.; Krüger, R.; Heinrich, R.; Kirschner, M.W. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003, 1, E10. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell. Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef]
- Sheldahl, L.C.; Slusarski, D.C.; Pandur, P.; Miller, J.R.; Kühl, M.; Moon, R.T. Dishevelled activates Ca2+ flux, PKC, and CamKII in vertebrate embryos. J. Cell Biol. 2003, 161, 769–777. [Google Scholar] [CrossRef]
- Ishitani, T.; Kishida, S.; Hyodo-Miura, J.; Ueno, N.; Yasuda, J.; Waterman, M.; Shibuya, H.; Moon, R.T.; Ninomiya-Tsuji, J.; Matsumoto, K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol. Cell Biol. 2003, 23, 131–139. [Google Scholar] [CrossRef]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef]
- Yamanaka, H.; Moriguchi, T.; Masuyama, N.; Kusakabe, M.; Hanafusa, H.; Takada, R.; Takada, S.; Nishida, E. JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep. 2002, 3, 69–75. [Google Scholar] [CrossRef]
- Habas, R.; Kato, Y.; He, X. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell 2001, 107, 843–854. [Google Scholar] [CrossRef]
- Endo, Y.; Wolf, V.; Muraiso, K.; Kamijo, K.; Soon, L.; Uren, A.; Barshishat-Küpper, M.; Rubin, J.S. Wnt-3a-dependent cell motility involves RhoA activation and is specifically regulated by dishevelled-2. J. Biol. Chem. 2005, 280, 777–786. [Google Scholar] [CrossRef]
- Schlessinger, K.; McManus, E.J.; Hall, A. Cdc42 and noncanonical Wnt signal transduction pathways cooperate to promote cell polarity. J. Cell Biol. 2007, 178, 355–361. [Google Scholar] [CrossRef]
- Riihilä, P.; Nissinen, L.; Knuutila, J.; Rahmati Nezhad, P.; Viiklepp, K.; Kähäri, V.M. Complement System in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 3550. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-κB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Jridi, I.; Canté-Barrett, K.; Pike-Overzet, K.; Staal, F.J.T. Inflammation and Wnt Signaling: Target for Immunomodulatory Therapy? Front. Cell. Dev. Biol. 2021, 8, 615131. [Google Scholar] [CrossRef]
- Picciolo, G.; Pallio, G.; Altavilla, D.; Vaccaro, M.; Oteri, G.; Irrera, N.; Squadrito, F. β-Caryophyllene Reduces the Inflammatory Phenotype of Periodontal Cells by Targeting CB2 Receptors. Biomedicines 2020, 8, 164. [Google Scholar] [CrossRef]
- Custurone, P.; Di Bartolomeo, L.; Irrera, N.; Borgia, F.; Altavilla, D.; Bitto, A.; Pallio, G.; Squadrito, F.; Vaccaro, M. Role of Cytokines in Vitiligo: Pathogenesis and Possible Targets for Old and New Treatments. Int. J. Mol. Sci. 2021, 22, 11429. [Google Scholar] [CrossRef]
- Irrera, N.; Bitto, A.; Vaccaro, M.; Mannino, F.; Squadrito, V.; Pallio, G.; Arcoraci, V.; Minutoli, L.; Ieni, A.; Lentini, M.; et al. PDRN, a Bioactive Natural Compound, Ameliorates Imiquimod-Induced Psoriasis through NF-κB Pathway Inhibition and Wnt/β-Catenin Signaling Modulation. Int. J. Mol. Sci. 2020, 21, 1215. [Google Scholar] [CrossRef] [PubMed]
- Picciolo, G.; Mannino, F.; Irrera, N.; Altavilla, D.; Minutoli, L.; Vaccaro, M.; Arcoraci, V.; Squadrito, V.; Picciolo, G.; Squadrito, F.; et al. PDRN, a natural bioactive compound, blunts inflammation and positively reprograms healing genes in an "in vitro" model of oral mucositis. Biomed. Pharmacother. 2021, 138, 111538. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Mauro, T.M.; Li, Z. The pathological role of Wnt5a in psoriasis and psoriatic arthritis. J. Cell. Mol. Med. 2019, 23, 5876–5883. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.C.; Collins, G.D.; Vandanmagsar, B.; Patel, K.; Brill, M.; Carter, A.; Lustig, A.; Becker, K.G.; Wood, W.W., 3rd; Emeche, C.D.; et al. Activation of Wnt5A signaling is required for CXC chemokine ligand 12-mediated T-cell migration. Blood 2009, 114, 1366–1373. [Google Scholar] [CrossRef]
- Valencia, J.; Hernández‐López, C.; Martínez, V.G.; Hidalgo, L.; Zapata, A.G.; Vicente, Á.; Varas, A.; Sacedón, R. Wnt5a skews dendritic cell differentiation to an unconventional phenotype with tolerogenic features. J. Immunol. 2011, 187, 4129–4139. [Google Scholar] [CrossRef]
- van Loosdregt, J.; Fleskens, V.; Tiemessen, M.M.; Mokry, M.; van Boxtel, R.; Meerding, J.; Pals, C.E.; Kurek, D.; Baert, M.R.; Delemarre, E.M.; et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity 2013, 39, 298–310. [Google Scholar] [CrossRef]
- Svegliati, S.; Marrone, G.; Pezone, A.; Spadoni, T.; Grieco, A.; Moroncini, G.; Grieco, D.; Vinciguerra, M.; Agnese, S.; Jüngel, A.; et al. Oxidative DNA damage induces the ATM-mediated transcriptional suppression of the Wnt inhibitor WIF-1 in systemic sclerosis and fibrosis. Sci. Signal 2014, 7, ra84. [Google Scholar] [CrossRef]
- Bagnato, G.L.; Irrera, N.; Pizzino, G.; Santoro, D.; Roberts, W.N.; Bagnato, G.; Pallio, G.; Vaccaro, M.; Squadrito, F.; Saitta, A.; et al. Dual αvβ3 and αvβ5 blockade attenuates fibrotic and vascular alterations in a murine model of systemic sclerosis. Clin. Sci. 2018, 132, 231–242. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Cho, M.S.; Rupaimoole, R.; Choi, H.J.; Noh, K.; Chen, J.; Hu, Q.; Sood, A.K.; Afshar-Kharghan, V. Complement Component 3 Is Regulated by TWIST1 and Mediates Epithelial-Mesenchymal Transition. J. Immunol. 2016, 196, 1412–1418. [Google Scholar] [CrossRef]
- Li, J.; Zhou, B.P. Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 2011, 11, 49. [Google Scholar] [CrossRef]
- Fan, Z.; Qin, J.; Wang, D.; Geng, S. Complement C3a promotes proliferation, migration and stemness in cutaneous squamous cell carcinoma. J. Cell. Mol. Med. 2019, 23, 3097–3107. [Google Scholar] [CrossRef]
- Di Piazza, M.; Nowell, C.S.; Koch, U.; Durham, A.D.; Radtke, F. Loss of cutaneous TSLP-dependent immune responses skews the balance of inflammation from tumor protective to tumor promoting. Cancer Cell. 2012, 22, 479–493. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. A new paradigm for tumor immune escape: β-catenin-driven immune exclusion. J. Immunother. Cancer 2015, 3, 43. [Google Scholar] [CrossRef]
- Li, X.; Xiang, Y.; Li, F.; Yin, C.; Li, B.; Ke, X. WNT/β-Catenin Signaling Pathway Regulating T Cell-Inflammation in the Tumor Microenvironment. Front. Immunol. 2019, 10, 2293. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Tanegashima, T.; Sato, E.; Irie, T.; Sai, A.; Itahashi, K.; Kumagai, S.; Tada, Y.; Togashi, Y.; Koyama, S.; et al. Highly immunogenic cancer cells require activation of the WNT pathway for immunological escape. Sci. Immunol. 2021, 6, eabc6424. [Google Scholar] [CrossRef]
- Marzuka, A.G.; Book, S.E. Basal cell carcinoma: Pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J. Biol. Med. 2015, 88, 167–179. [Google Scholar]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T.C.L.S.E. A highlight on Sonic hedgehog pathway. Cell Commun. Signal 2018, 16, 11. [Google Scholar] [CrossRef]
- Duman-Scheel, M.; Weng, L.; Xin, S.; Du, W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 2002, 417, 299–304. [Google Scholar] [CrossRef]
- Nilsson, M.; Undèn, A.B.; Krause, D.; Malmqwist, U.; Raza, K.; Zaphiropoulos, P.G.; Toftgård, R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443. [Google Scholar] [CrossRef]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, Y.S.; Lee, C.; Shin, M.S.; Kim, J.W.; Jang, B.G. Expression profile of sonic hedgehog signaling-related molecules in basal cell carcinoma. PLoS ONE 2019, 14, e0225511. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, F.; Aragane, Y.; Kawada, A.; Tezuka, T. Immunohistochemical detection for nuclear beta-catenin in sporadic basal cell carcinoma. Br. J. Dermatol. 2001, 145, 771–777. [Google Scholar] [CrossRef] [PubMed]
- El-Bahrawy, M.; El-Masry, N.; Alison, M.; Poulsom, R.; Fallowfield, M. Expression of beta-catenin in basal cell carcinoma. Br. J. Dermatol. 2003, 148, 964–970. [Google Scholar] [CrossRef]
- Doglioni, C.; Piccinin, S.; Demontis, S.; Cangi, M.G.; Pecciarini, L.; Chiarelli, C.; Armellin, M.; Vukosavljevic, T.; Boiocchi, M.; Maestro, R. Alterations of beta-catenin pathway in non-melanoma skin tumors: Loss of alpha-ABC nuclear reactivity correlates with the presence of beta-catenin gene mutation. Am. J. Pathol. 2003, 163, 2277–2287. [Google Scholar] [CrossRef]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2018, 9, 5492–5508. [Google Scholar] [CrossRef]
- Brinkhuizen, T.; van den Hurk, K.; Winnepenninckx, V.J.; de Hoon, J.P.; van Marion, A.M.; Veeck, J.; van Engeland, M.; van Steensel, M.A. Epigenetic changes in Basal Cell Carcinoma affect SHH and WNT signaling components. PLoS ONE 2012, 7, e51710. [Google Scholar] [CrossRef]
- Meng, X.; Poon, R.; Zhang, X.; Cheah, A.; Ding, Q.; Hui, C.C.; Alman, B. Suppressor of fused negatively regulates beta-catenin signaling. J. Biol. Chem. 2001, 276, 40113–40119. [Google Scholar] [CrossRef]
- Li, X.; Deng, W.; Lobo-Ruppert, S.M.; Ruppert, J.M. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by beta-catenin. Oncogene 2007, 26, 4489–4498. [Google Scholar] [CrossRef]
- Kerekes, K.; Trexler, M.; Bányai, L.; Patthy, L. Wnt Inhibitory Factor 1 Binds to and Inhibits the Activity of Sonic Hedgehog. Cells 2021, 10, 3496. [Google Scholar] [CrossRef]
- Biehs, B.; Dijkgraaf, G.J.P.; Piskol, R.; Alicke, B.; Boumahdi, S.; Peale, F.; Gould, S.E.; de Sauvage, F.J. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018, 562, 429–433. [Google Scholar] [CrossRef]
- Corchado-Cobos, R.; García-Sancha, N.; González-Sarmiento, R.; Pérez-Losada, J.; Cañueto, J. Cutaneous Squamous Cell Carcinoma: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 2956. [Google Scholar] [CrossRef]
- Ortonne, J.P. From actinic keratosis to squamous cell carcinoma. Br. J. Dermatol. 2002, 146, 20–23. [Google Scholar] [CrossRef]
- Willenbrink, T.J.; Ruiz, E.S.; Cornejo, C.M.; Schmults, C.D.; Arron, S.T.; Jambusaria-Pahlajani, A. Field cancerization: Definition, epidemiology, risk factors, and outcomes. J. Am. Acad. Dermatol. 2020, 83, 709–717. [Google Scholar] [CrossRef]
- Piipponen, M.; Riihilä, P.; Nissinen, L.; Kähäri, V.M. The Role of p53 in Progression of Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 4507. [Google Scholar] [CrossRef]
- Hedberg, M.L.; Berry, C.T.; Moshiri, A.S.; Xiang, Y.; Yeh, C.J.; Attilasoy, C.; Capell, B.C.; Seykora, J.T. Molecular Mechanisms of Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 3478. [Google Scholar] [CrossRef]
- Azazmeh, N.; Assouline, B.; Winter, E.; Ruppo, S.; Nevo, Y.; Maly, A.; Meir, K.; Witkiewicz, A.K.; Cohen, J.; Rizou, S.V.; et al. Chronic expression of p16INK4a in the epidermis induces Wnt-mediated hyperplasia and promotes tumor initiation. Nat. Commun. 2020, 11, 2711. [Google Scholar] [CrossRef]
- Haider, A.S.; Peters, S.B.; Kaporis, H.; Cardinale, I.; Fei, J.; Ott, J.; Blumenberg, M.; Bowcock, A.M.; Krueger, J.G.; Carucci, J.A. Genomic analysis defines a cancer-specific gene expression signature for human squamous cell carcinoma and distinguishes malignant hyperproliferation from benign hyperplasia. J. Invest. Dermatol. 2006, 126, 869–881. [Google Scholar] [CrossRef]
- Ra, S.H.; Li, X.; Binder, S. Molecular discrimination of cutaneous squamous cell carcinoma from actinic keratosis and normal skin. Mod. Pathol. 2011, 24, 963–973. [Google Scholar] [CrossRef]
- He, X.; Li, S.; Luo, X.; Hu, D.; Cai, T.; Huang, K.; Zhou, W.; Chen, J. Expression of DKK1 and β-catenin in epidermal neoplasms and their correlation. Int. J. Clin. Exp. Med. 2015, 8, 18843–18848. [Google Scholar] [PubMed]
- Malanchi, I.; Peinado, H.; Kassen, D.; Hussenet, T.; Metzger, D.; Chambon, P.; Huber, M.; Hohl, D.; Cano, A.; Birchmeier, W.; et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 2008, 452, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Tsuji, G.; Ohno, F.; Nakahara, T.; Uchi, H.; Furue, M. Potential role of the OVOL1-OVOL2 axis and c-Myc in the progression of cutaneous squamous cell carcinoma. Mod. Pathol. 2017, 30, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Beronja, S.; Janki, P.; Heller, E.; Lien, W.H.; Keyes, B.E.; Oshimori, N.; Fuchs, E. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature 2013, 501, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Piao, Y.J.; Sohn, K.C.; Ha, J.M.; Im, M.; Seo, Y.J.; Whang, K.U.; Lee, J.H.; Lee, Y.; Kim, C.D. Sox9 is a β-catenin-regulated transcription factor that enhances the colony-forming activity of squamous cell carcinoma cells. Mol. Med. Rep. 2016, 14, 337–342. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef]
- Xia, X.; Qian, S.; Soriano, S.; Wu, Y.; Fletcher, A.M.; Wang, X.J.; Koo, E.H.; Wu, X.; Zheng, H. Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10863–10868. [Google Scholar] [CrossRef]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef]
- Chu, Z.; Zhang, X.; Li, Q.; Hu, G.; Lian, C.G.; Geng, S. CDC20 contributes to the development of human cutaneous squamous cell carcinoma through the Wnt/β catenin signaling pathway. Int. J. Oncol. 2019, 54, 1534–1544. [Google Scholar] [CrossRef]
- Zimmerli, D.; Cecconi, V.; Valenta, T.; Hausmann, G.; Cantù, C.; Restivo, G.; Hafner, J.; Basler, K.; van den Broek, M. WNT ligands control initiation and progression of human papillomavirus-driven squamous cell carcinoma. Oncogene 2018, 37, 3753–3762. [Google Scholar] [CrossRef]
- Cheng, Y.; Phoon, Y.P.; Jin, X.; Chong, S.Y.; Ip, J.C.; Wong, B.W.; Lung, M.L. Wnt-C59 arrests stemness and suppresses growth of nasopharyngeal carcinoma in mice by inhibiting the Wnt pathway in the tumor microenvironment. Oncotarget 2015, 6, 14428–14439. [Google Scholar] [CrossRef]
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634. [Google Scholar] [CrossRef]
- Sharma, G.; Sharma, A.R.; Seo, E.M.; Nam, J.S. Genetic polymorphism in extracellular regulators of Wnt signaling pathway. Biomed. Res. Int. 2015, 2015, 847529. [Google Scholar] [CrossRef]
- Liang, J.; Kang, X.; Halifu, Y.; Zeng, X.; Jin, T.; Zhang, M.; Luo, D.; Ding, Y.; Zhou, Y.; Yakeya, B.; et al. Secreted frizzled-related protein promotors are hypermethylated in cutaneous squamous carcinoma compared with normal epidermis. BMC Cancer 2015, 15, 641. [Google Scholar] [CrossRef]
- Fujii, M.; Katase, N.; Lefeuvre, M.; Gunduz, M.; Buery, R.R.; Tamamura, R.; Tsujigiwa, H.; Nagatsuka, H. Dickkopf (Dkk)-3 and β-catenin expressions increased in the transition from normal oral mucosal to oral squamous cell carcinoma. J. Mol. Histol. 2011, 42, 499–504. [Google Scholar] [CrossRef]
- Shin, J.M.; Choi, D.K.; Kang, H.Y.; Sohn, K.C.; Lee, Y.; Kim, C.D.; Lee, J.H.; Park, B.C. The expression pattern and functional role of REIC/Dkk-3 in the development of cutaneous squamous cell carcinoma. J. Dermatol. Sci. 2016, 84, 88–96. [Google Scholar] [CrossRef]
- Bhatia, N.; Spiegelman, V.S. Activation of Wnt/beta-catenin/Tcf signaling in mouse skin carcinogenesis. Mol. Carcinog. 2005, 42, 213–221. [Google Scholar] [CrossRef]
- Zanfi, E.D.; Fantini, S.; Lotti, R.; Bertesi, M.; Marconi, A.; Grande, A.; Manfredini, R.; Pincelli, C.; Zanocco-Marani, T. Wnt/CTNNB1 Signal Transduction Pathway Inhibits the Expression of ZFP36 in Squamous Cell Carcinoma, by Inducing Transcriptional Repressors SNAI1, SLUG and TWIST. Int. J. Mol. Sci. 2020, 21, 5692. [Google Scholar] [CrossRef]
- Li, X.; Zhang, C.; Yuan, Y.; Wang, Y.; Lu, S.; Zhou, Z.; Zhen, P.; Zhou, M. Downregulation of ARMC8 promotes tumorigenesis through activating Wnt/β-catenin pathway and EMT in cutaneous squamous cell carcinomas. J. Dermatol. Sci. 2021, 102, 184–192. [Google Scholar] [CrossRef]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef]
- Margulis, A.; Zhang, W.; Alt-Holland, A.; Crawford, H.C.; Fusenig, N.E.; Garlick, J.A. E-cadherin suppression accelerates squamous cell carcinoma progression in three-dimensional, human tissue constructs. Cancer Res. 2005, 65, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; Simcha, I.; Ben-Yedidia, T.; Blechman, J.; Savagner, P.; Ben-Ze'ev, A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: The roles of beta-catenin signaling, Slug, and MAPK. J. Cell Biol. 2003, 163, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Chen, H.C.; Zhang, D.; Fu, J. Twist: A molecular target in cancer therapeutics. Tumour Biol 2013, 34, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Hlubek, F.; Spaderna, S.; Jung, A.; Kirchner, T.; Brabletz, T. Beta-catenin activates a coordinated expression of the proinvasive factors laminin-5 gamma2 chain and MT1-MMP in colorectal carcinomas. Int. J. Cancer 2004, 108, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.I.; Baker, A.R.; Dextras, C.R.; Cabarcas, S.M.; Young, M.R.; Colburn, N.H. Targeting of Noncanonical Wnt5a Signaling by AP-1 Blocker Dominant-Negative Jun When It Inhibits Skin Carcinogenesis. Genes Cancer 2012, 3, 37–50. [Google Scholar] [CrossRef]
- Bachelder, R.E.; Yoon, S.O.; Franci, C.; de Herreros, A.G.; Mercurio, A.M. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: Implications for the epithelial-mesenchymal transition. J. Cell Biol. 2005, 168, 29–33. [Google Scholar] [CrossRef]
- Yook, J.I.; Li, X.Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt–Axin2–GSK3b cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef]
- Ren, D.; Minami, Y.; Nishita, M. Critical role of Wnt5a-Ror2 signaling in motility and invasiveness of carcinoma cells following Snail-mediated epithelial-mesenchymal transition. Genes Cells 2011, 16, 304–315. [Google Scholar] [CrossRef]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Labbé, E.; Letamendia, A.; Attisano, L. Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-beta and wnt pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 8358–8363. [Google Scholar] [CrossRef]
- Ripka, S.; König, A.; Buchholz, M.; Wagner, M.; Sipos, B.; Klöppel, G.; Downward, J.; Gress, T.; Michl, P. WNT5A—Target of CUTL1 and potent modulator of tumor cell migration and invasion in pancreatic cancer. Carcinogenesis 2007, 28, 1178–1187. [Google Scholar] [CrossRef]
- Pourreyron, C.; Reilly, L.; Proby, C.; Panteleyev, A.; Fleming, C.; McLean, K.; South, A.P.; Foerster, J. Wnt5a is strongly expressed at the leading edge in non-melanoma skin cancer, forming active gradients, while canonical Wnt signalling is repressed. PLoS ONE 2012, 7, e31827. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Effect | |
|---|---|---|
| Basal cell carcinoma | ||
| Up-regulated | GLI | promotion of β-catenin activation |
| Down-regulated | SUFU | negative regulation of β-catenin |
| WIF1 | inhibition of SHH signaling | |
| Canonical Wnt pathway in SCC | ||
| Up-regulated | β-catenin | overexpression of canonical Wnt pathway |
| Porcuspine | secretion of Wnt from endoplasmic reticulum | |
| Lef/Tcf | transcription of Wnt target genes | |
| c-myc | maintenance of cutaneous CSCs | |
| TGF-β | activation of Lef/Tcf | |
| Down-regulated | SFRPs | inhibition of Wnt ligands |
| WIF1 | inhibition of Wnt ligands | |
| Dkk1-3 | inhibition of Wnt receptor complex | |
| Non-canonical Wnt pathway in SCC | ||
| Up-regulated | STAT3 | transcription of Wnt target genes |
| Snail | inhibition of E-cadherin and promotion of EMT | |
| CUTL-1 | up-regulation of Wnt5a | |
| Down-regulated | SFRPs | inhibition of Wnt ligands |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Bartolomeo, L.; Vaccaro, F.; Irrera, N.; Borgia, F.; Li Pomi, F.; Squadrito, F.; Vaccaro, M. Wnt Signaling Pathways: From Inflammation to Non-Melanoma Skin Cancers. Int. J. Mol. Sci. 2023, 24, 1575. https://doi.org/10.3390/ijms24021575
Di Bartolomeo L, Vaccaro F, Irrera N, Borgia F, Li Pomi F, Squadrito F, Vaccaro M. Wnt Signaling Pathways: From Inflammation to Non-Melanoma Skin Cancers. International Journal of Molecular Sciences. 2023; 24(2):1575. https://doi.org/10.3390/ijms24021575
Chicago/Turabian StyleDi Bartolomeo, Luca, Federico Vaccaro, Natasha Irrera, Francesco Borgia, Federica Li Pomi, Francesco Squadrito, and Mario Vaccaro. 2023. "Wnt Signaling Pathways: From Inflammation to Non-Melanoma Skin Cancers" International Journal of Molecular Sciences 24, no. 2: 1575. https://doi.org/10.3390/ijms24021575
APA StyleDi Bartolomeo, L., Vaccaro, F., Irrera, N., Borgia, F., Li Pomi, F., Squadrito, F., & Vaccaro, M. (2023). Wnt Signaling Pathways: From Inflammation to Non-Melanoma Skin Cancers. International Journal of Molecular Sciences, 24(2), 1575. https://doi.org/10.3390/ijms24021575